The Efficacy of Iron Chelators for Removing Iron from Specific Brain Regions and the Pituitary—Ironing out the Brain


{kind=link}
Abstract
:1. Introduction
2. Iron chelation
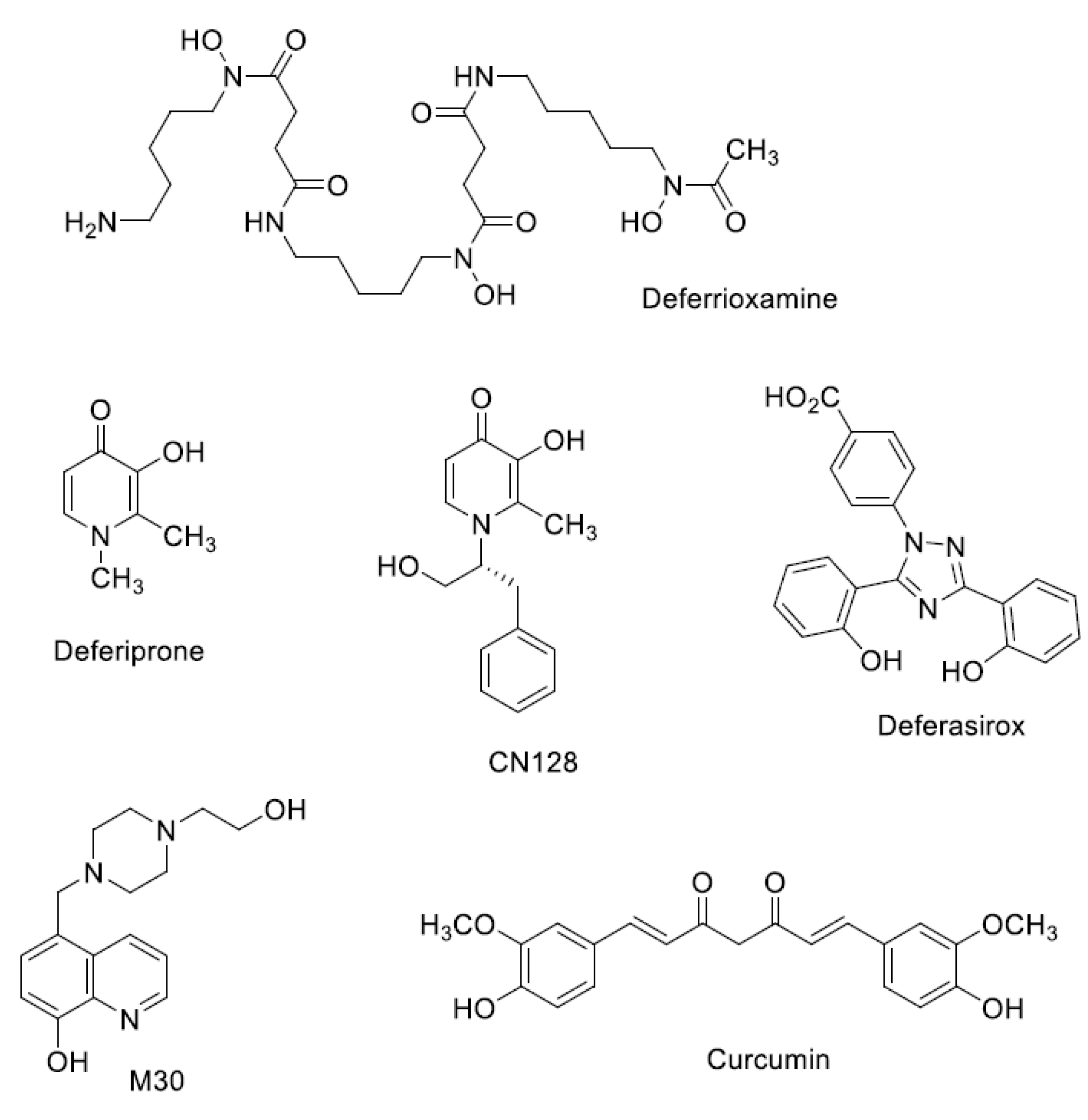
Iron Chelators in Current Clinical Use
3. Thalassemia, Sickle-Cell Anaemia, and Haemoglobinopathies
3.1. Thalassemia-Iron Accumulation in the Pituitary
3.2. Iron Chelation from the Pituitary Gland
4. Parkinson’s Disease—Iron Accumulation in the Substantia Nigra
Chelation of Iron in the Brains of Parkinson’s Disease Patients
5. Alzheimer’s Disease
Alzheimer’s Disease—Iron Chelation from the Cortex and Hippocampus
6. Friederich’s Ataxia
Iron Chelation from the Dentate Nucleus in Friederich’s Ataxia Patients
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s Disease |
| Ca++ | Calcium |
| CSF | Cerebrospinal fluid |
| DFO | Desferioxamine |
| DFP | Deferiprone |
| DFX | Deferasirox |
| Fe2++ | Ferrous |
| FDRA | Friederich’s Ataxia |
| FSH | Follicle stimulating hormone |
| GAA | Trinucleotide repeat |
| GnRH | Gonadotropin-releasing hormone |
| GnRH stimulation | Gonadotropin-releasing hormone test. |
| ICARS | International Co-operative Ataxia Rating Scale |
| IL-6 | Interleukin-6 |
| ISC | Iron sulphur protein |
| LH | Luteinizing hormone |
| LSPD | Late stage Parkinson’s Disease |
| MDS-UPDRS | Movement Disorder Society Unified Parkinson’s Disease Rating Scale |
| MRI | Magnetic resonance imaging |
| NMDA | N-methyl-D-aspartate |
| NTBI | Non transferrin bound iron |
| PD | Parkinson’s Disease |
| SN | Substania nigra |
| WM | White matter |
| ZIP14 | Zinc transporter |
References
- Ward, R.J.; Dexter, D.T.; Crichton, R.R. Neurodegenerative diseases and therapeutic strategies using iron chelators. J. Trace Elem. Med. Biol. 2015, 31, 267–273. [Google Scholar] [CrossRef] [Green Version]
- Nathan, D.G. Thalassemia: The continued challenge. Ann. N. Y. Acad. Sci. 2005, 1054, 1–10. [Google Scholar] [CrossRef]
- Nathan, D.G. Thalassemia: A look to the future. Ann. N. Y. Acad. Sci. 2016, 1368, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Hider, R.C.; Hoffbrand, A.V. The role of deferiprone in iron chelation. N. Engl. J. Med. 2018, 379, 2140–2150. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Weatherall, D.J.; Cappellini, M.D. Thalassemia. Lancet 2018, 391, 155–167. [Google Scholar] [CrossRef]
- Propper, R.D.; Cooper, B.; Rufo, R.R.; Nienhuis, A.W.; Anderson, W.F.; Bunn, H.F.; Rosenthal, A.; Nathan, D.G. Continuous subcutaneous administration of deferoxamine in patients with iron overload. N. Engl. J. Med. 1977, 297, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Pippard, M.J.; Callender, S.T.; Weatherall, D.J. Chelation regimens with desferrioxamine. Lancet 1977, 1, 1101. [Google Scholar] [CrossRef]
- Brittenham, G.M.; Griffith, P.M.; Nienhuis, A.W.; McLaren, C.E.; Young, N.S.; Tucker, E.E.; Allen, C.J.; Farrell, D.E.; Harris, J.W. Efficacy of deferoxamine in preventing complications of iron overload in patients with thalassemia major. N. Engl. J. Med. 1994, 331, 567–573. [Google Scholar] [CrossRef]
- Borgna-Pignatti, C.; Rugolotto, S.; De Stefano, P.; Zhao, H.; Cappellini, M.D.; Del Vecchio, G.C.; Romeo, M.A.; Forni, G.L.; Gamberini, M.R.; Ghilardi, R.; et al. Survival and complications in patients with thalassemia major treated with transfusion and deferoxamine. Haematologica 2004, 89, 1187–1193. [Google Scholar] [PubMed]
- Olivieri, N.F.; Nathan, D.G.; MacMillan, J.H.; Wayne, A.S.; Liu, P.P.; McGee, A.; Martin, M.; Koren, G.; Cohen, A.R. Survival in medically treated patients with homozygous beta thalassemia. N. Engl. J. Med. 1994, 331, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Modell, B.; Khan, M.; Darlison, M. Survival in beta-thalassaemia major in the UK: Data from the UK Thalassaemia Register. Lancet 2000, 355, 2051–2052. [Google Scholar] [CrossRef]
- Kontoghiorghes, G.J.; Aldouri, M.A.; Hoffbrand, A.V.; Barr, J.; Wonke, B.; Kourouclaris, T.; Sheppard, L. Effective chelation of iron in beta thalassaemia with the oral chelator 1,2-dimethyl-3-hydroxypyrid-4-one. Br. Med. J. (Clin. Res. Ed.) 1987, 295, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, N.F.; Koren, G.; Hermann, C.; Bentur, Y.; Chung, D.; Klein, J.; St Louis, P.; Freedman, M.H.; McClelland, R.A.; Templeton, D.M. Comparison of oral iron chelator L1 and desferrioxamine in iron-loaded patients. Lancet 1990, 336, 1275–1279. [Google Scholar] [CrossRef]
- Hoyes, K.P.; Porter, J.B. Subcellular distribution of desferrioxamine and hydroxypyridin-4-one chelators in K562 cells affects chelation of intracellular iron pools. Br. J. Haematol. 1993, 85, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.P.; Spino, M.; Thiessen, J.J. Transport kinetics of iron chelators and their chelates in Caco-2 cells. Pharm. Res. 2006, 23, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Botzenhardt, S.; Li, N.; Chan, E.W.; Sing, C.W.; Wong, I.C.; Neubert, A. Safety profiles of iron chelators in young patients with haemoglobinopathies. Eur. J. Haematol. 2017, 98, 198–217. [Google Scholar] [CrossRef] [Green Version]
- Hershko, C.; Link, G.; Pinson, A.; Peter, H.H.; Dobbin, P.; Hider, R.C. Ironmobilization from myocardial cells by 3-hydroxypyridin-4-one chelators: Studies in rat heart cells in culture. Blood 1991, 77, 2049–2053. [Google Scholar] [PubMed]
- Anderson, L.J.; Wonke, B.; Prescott, E.; Holden, S.; Walker, J.M.; Pennell, D.J. Comparison of effects of oral deferiprone and subcutaneous desferrioxamine on myocardial iron concentrations and ventricular function in beta-thalassaemia. Lancet 2002, 9332, 516–520. [Google Scholar] [CrossRef]
- Pepe, A.; Meloni, A.; Rossi, G.; Cuccia, L.; D’Ascola, G.D.; Santodirocco, M.; Cianciulli, P.; Caruso, V.; Romeo, M.A.; Filosa, A.; et al. Cardiac and hepatic iron and ejection fraction in thalassemia major: Multicentre prospective comparison of combined deferiprone and deferoxamine therapy against deferiprone or deferoxamine monotherapy. J. Cardiovasc. Magn. Reson. 2013, 15. [Google Scholar] [CrossRef]
- Nick, H.; Acklin, P.; Lattmann, R.; Buehlmayer, P.; Hauffe, S.; Schupp, J.; Alberti, D. Development of tridentate iron chelators: From desferrithiocin to ICL670. Curr. Med. Chem. 2003, 10, 1065–1076. [Google Scholar] [CrossRef]
- Longueville, A.; Crichton, R.R. An animal model of iron overload and its application to study hepatic ferritin iron mobilization by chelators. Biochem. Pharmacol. 1986, 35, 3669–3678. [Google Scholar] [CrossRef]
- Wolfe, L.C.; Nicolosi, R.J.; Renaud, M.M.; Finger, J.; Hegsted, M.; Peter, H.; Nathan, D.G. A non-human primate model for the study of oral iron chelators. Br. J. Haematol. 1989, 72, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, R.J.; Streiff, R.R.; Wiegand, J.; Vinson, J.R.; Luchetta, G.; Wiegand, J.; Moerker, T.; Peter, H.H. A comparative evaluation of iron clearance models. Ann. N. Y. Acad. Sci. 1990, 612, 378–393. [Google Scholar] [CrossRef] [PubMed]
- Baker, E.; Wong, A.; Peter, H.; Jacobs, A. Desferrithiocin is an effective iron chelator in vivo and in vitro but ferrithiocin is toxic. Br. J. Haematol. 1992, 81, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, R.J.; Wiegand, J.; McManis, J.S.; Bharti, N. Desferrithiocin: A search for clinically effective iron chelators. J. Med. Chem. 2014, 57, 9259–9291. [Google Scholar] [CrossRef] [PubMed]
- Daar, S.; Pathare, A.; Nick, H.; Kriemler-Krahn, U.; Hmissi, A.; Habr, D.; Taher, A. Reduction in labile plasma iron during treatment with deferasirox, a once-daily oral iron chelator, in heavily loaded patients with beta-thalassaemia. Eur. J. Haematol. 2009, 82, 454–457. [Google Scholar] [CrossRef] [PubMed]
- Cappellini, M.D.; Cohen, A.; Piga, A.; Bejaoui, M.; Perrotta, S.; Agaoglu, L.; Aydinok, Y.; Kattamis, A.; Kilinc, Y.; Porter, J.; et al. A phase 3 study of deferasirox (ICL670), a once-daily oral iron cheater in patients with best-thalassemia. Blood 2006, 207, 3455–3462. [Google Scholar] [CrossRef] [PubMed]
- Cappellini, M.D.; Porter, J.D.; El-Beshlawy, A.; Li, C.K.; Seymour, J.F.; Elalfy, M.; Gattermann, N.; Giraudier, S.; Lee, J.W.; Chan, L.L.; et al. Tailoring iron chelation by iron intake and serum ferritin; the perspective EPIC study of deferasirox in 1744 patients with transfusion-dependent anemias. Haematologica 2010, 95, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, E.J. Oral chelators deferasirox and deferiprone for transfusional iron overload in thalassemia major: New data, new questions. Blood 2006, 107, 3436–3441. [Google Scholar] [CrossRef] [PubMed]
- Hershko, C.; Graham, G.; Bates, G.W.; Rachmilewitz, E.A. Non-specific serum iron in thalassaemia: An abnormal serum iron fraction of potential toxicity. Br. J. Haematol. 1978, 40, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Brissot, P.; Ropert, M.; Le Lan, C.; Loréal, O. Non-transferrin bound iron: A key role in iron overload and iron toxicity. Biochim. Biophys. Acta 2012, 1820, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Pinilla-Tenas, J.J.; Sparkman, B.K.; Shawki, A.; Illing, A.C.; Mitchell, C.J.; Zhao, N.; Liuzzi, J.P.; Cousins, R.J.; Knutson, M.D.; Mackenzie, B. Zip14 is a complex broad-scope metal-ion transporter whose functional properties support roles in the cellular uptake of zinc and nontransferrin-bound iron. Am. J. Physiol. Cell Physiol. 2011, 301, C862–C871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkitkasemwong, S.; Wang, C.Y.; Coffey, R.; Zhang, W.; Chan, A.; Biel, T.; Kim, J.S.; Hojyo, S.; Fukada, T.; Knutson, M.D. SLC39A14 Is Required for the Development of Hepatocellular Iron Overload in Murine Models of Hereditary Hemochromatosis. Cell Metab. 2015, 22, 138–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oudit, G.Y.; Sun, H.; Trivieri, M.G.; Koch, S.E.; Dawood, F.; Ackerley, C.; Yazdanpanah, M.; Wilson, G.J.; Schwartz, A.; Liu, P.P.; et al. L-type Ca2+ channels provide a major pathway for iron entry into cardiomyocytes in iron-overload cardiomyopathy. Nat. Med. 2003, 9, 1187–1194. [Google Scholar] [CrossRef] [PubMed]
- Kumfu, S.; Chattipakorn, S.; Srichairatanakool, S.; Settakorn, J.; Fucharoen, S.; Chattipakorn, N. T-type calcium channel as a portal of iron uptake into cardiomyocytes of beta-thalassemic mice. Eur. J. Haematol. 2011, 86, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Noetzli, L.J.; Panigrahy, A.; Mittelman, S.D.; Hyderi, A.; Dongelyan, A.; Coates, T.D.; Wood, J.C. Pituitary iron and volume predict hypogonadism in transfusional iron overload. Am. J. Hematol. 2012, 87, 167–171. [Google Scholar] [CrossRef] [PubMed]
- De Sanctis, V.; Soliman, A.T.; Elsedfy, H.; Soliman, N.A.; Elalaily, R. Late onset male hypogonadism and fertility potential in thalassaemia major: Two emerging patters. Mediterr. J. Haematol. 2015, 7, e2015047. [Google Scholar]
- Çetinçakmak, M.; Hattapoğlu, S.; Menzilcioğlu, S.; Bircan, A.; Uluca, U.; Uçar, A.; S Mura, S.; Söker, M.; Bilic, A.J. MRI-based evaluation of the factors leading to pituitary iron overload in patients with thalassemia. J. Neuroradiol. 2016, 43, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Fischer, R.; Butensky, E.; Neumayr, L.; Singer, S.T.; Lee, P.; Chu, Z.; Hunte, J.V.; Firebaugh, J.; Vichinsky, E.; et al. MRI Assessment of Pituitary Iron and Volume in Thalassemia, and Relation to Hypothalamic-Pituitary-Gonadal Axis Function (HPG): A Feasibility Study. Blood 2006, 108, 1778. [Google Scholar]
- Christoforidis, A.; Haritandi, A.; Tsitouridis, I.; Tsatra, I.; Tsantali, H.; Karyda, S.; Dimitriadis, A.S.; Athanassiou-Metaxa, M. Correlative study of iron accumulation in liver, myocardium, and pituitary assessed with MRI in young thalassemic patients. J. Pediatr. Hematol. Oncol. 2006, 28, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Berkovitch, M.; Bistritzer, T.; Milone, S.D.; Perlman, K.; Kucharczyk, W.; Olivieri, N.F. Iron deposition in the anterior pituitary in homozygous beta thalassemia: MRI evaluation and correlation with gonadal function. J. Pediatr. Endocrinol. Metab. 2000, 13, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Abdulzahra, M.S.; Al-Hakeim, H.K.; Ridha, M.M. Study of the effect of iron overload on the function of endocrine glands in male thalassemia patients. Asian J. Transfus. Sci. 2011, 5, 127–131. [Google Scholar] [PubMed]
- Wood, J.C.; Kang, B.P.; Thompson, A.; Giardina, P.; Harmatz, P.; Glynos, T.; Paley, C.; Coates, T.D. The effect of deferasirox on cardiac iron in thalassemia major: Impact of total body iron stores. Blood 2010, 116, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Duprez, T.; Maite, D.; Cosnard, G. Transfusional haemochromatosis of the choroid plexus in beta thalassemia major. J. Comput. Assist. Tomogr. 2001, 25, 487–488. [Google Scholar] [CrossRef] [PubMed]
- Hasiloglu, Z.I.; Asik, M.; Ure, E.; Ertem, F.; Apak, H.; Albayram, S. The utility of susceptibility-weighted imaging to evaluate the extent of iron accumulation in the choroid plexus of patients with β-thalassaemiamajor. Clin. Radiol. 2017, 72, 903.e1–903.e7. [Google Scholar] [CrossRef] [PubMed]
- Qiu, D.; Chan, G.C.; Chu, J.; Chan, Q.; Ha, S.Y.; Moseley, M.E.; Khong, P.L. MR quantitative susceptibility imaging for the evaluation of iron loading in the brains of patients with β-thalassemia major. AJNR Am. J. Neuroradiol. 2014, 35, 1085–1090. [Google Scholar] [CrossRef] [PubMed]
- Dexter, D.T.; Wells, F.R.; Agid, F.; Agid, Y.; Lees, A.J.; Jenner, P.; Marsden, C.D. Increased nigral iron content in postmortem parkinsonian brain. Lancet 1987, 21, 1219–1220. [Google Scholar] [CrossRef]
- Fernandez, B.; Ferrer, I.; Fernando, G.; Hilfiker, S. Biomonitorization of iron accumulation in the substantia nigra from Lewy body disease patients. Toxicol. Rep. 2017, 4, 188–193. [Google Scholar] [CrossRef]
- Martin-Bastida, A.; Tilley, B.; Alireza, T.; Bansal, S.; Dexter, D.T.; Ward, R.J. Parkinson’s, Iron and Inflammation. Neurobiol. Dis. 2019, in press. [Google Scholar]
- Jellinger, K.; Paulus, W.; Grundke-Iqbal, I.; Riederer, P.; Youdim, M.B. Brain iron and ferritin in Parkinson’s and Alzheimer’s diseases. J. Neural Transm. Park. Dis. Dement. Sect. 1990, 2, 327–340. [Google Scholar] [CrossRef]
- Ben-Shachar, D.; Riederer, P.; Youdim, M.B. Iron-melanin interaction and lipid peroxidation: Implications for Parkinson’s disease. J. Neurochem. 1991, 57, 1609–1614. [Google Scholar] [CrossRef] [PubMed]
- Hadzhieva, M.; Kirches, E.; Mawrin, C. Review: Iron metabolism and the role of iron in neurodegenerative disorders. Neuropathol. Appl. Neurobiol. 2014, 40, 240–257. [Google Scholar] [CrossRef] [PubMed]
- Zecca, L.; Wilms, H.; Geick, S.; Claasen, J.H.; Brandenburg, L.O.; Holzknecht, C.; Panizza, M.L.; Zucca, F.A.; Deuschl, G.; Sievers, J.; et al. Human neuromelanin induces neuroinflammation and neurodegeneration in the rat substantia nigra: Implications for Parkinson’s disease. Acta Neuropathol. 2008, 116, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Phillips, K.; Wielgus, A.R.; Liu, J.; Albertini, A.; Zucca, F.A.; Faust, R.; Qian, S.Y.; Miller, D.S.; Chignell, C.F.; et al. Neuromelanin activates microglia and induces degeneration of dopaminergic neurons: Implications for progression of Parkinson’s disease. Neurotox. Res. 2011, 19, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Nakane, T.; Nihashi, T.; Kawai, H.; Naganawa, S. Visualization of neuromelanin in the Substantia nigra and locus ceruleus at 1.5T using a 3D-gradient echo sequence with magnetization transfer contrast. Magn. Reson. Med. Sci. 2008, 7, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Shibata, E.; Kudo, K.; Tohyama, K. Neuromelanin-Sensitive MRI. Clin. Neuroradiol. 2008, 18, 147–153. [Google Scholar] [CrossRef]
- Prasad, S.; Stezin, A.; Lenka, A.; George, L.; Saini, J.; Yadav, R.; Pal, P.K. Three-dimensional neuromelanin-sensitive magnetic resonance imaging of the substantia nigra in Parkinson’s disease. Eur. J. Neurol. 2018, 25, 680–686. [Google Scholar] [CrossRef]
- Fabbri, M.; Reimão, S.; Carvalho, M.; Nunes, R.G.; Abreu, D.; Guedes, L.C.; Bouça, R.; Lobo, P.P.; Godinho, C.; Coelho, M.; et al. Substantia nigra neuromelanin as an imaging biomarker of disease progression in Parkinson’s Disease. J. Park. Dis. 2017, 7, 491–501. [Google Scholar] [CrossRef]
- Ward, R.J.; Dexter, D.; Florence, A.; Aouad, F.; Hider, R.; Jenner, P.; Crichton, R.R. Brain iron in the ferrocene-loaded rat: Its chelation and influence on dopamine metabolism. Biochem. Pharmacol. 1995, 49, 1821–1826. [Google Scholar] [CrossRef]
- Dexter, D.T.; Ward, R.J.; Florence, A.; Jenner, P.; Crichton, R.R. Effects of desferrithiocin and its derivatives on peripheral iron and striatal dopamine and 5-hydroxytryptamine metabolism in the ferrocene-loaded rat. Biochem. Pharmacol. 1999, 58, 151–155. [Google Scholar] [CrossRef]
- Kwiatkowski, J.L. Current recommendations for chelation for transfusion-dependent thalassemia. Ann. N. Y. Acad. Sci. 2016, 1368, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Devos, D.; Moreau, C.; Devedjian, J.C.; Kluza, J.; Petrault, M.; Laloux, C.; Jonneaux, A.; Ryckewaert, G.; Garçon, G.; Rouaix, N.; et al. Targeting chelatable iron as a therapeutic modality in Parkinson’s disease. Antioxid. Redox Signal 2014, 21, 195–210. [Google Scholar] [CrossRef] [PubMed]
- Martin-Bastida, A.; Ward, R.J.; Newbould, R.; Piccini, P.; Sharp, D.; Kabba, C.; Patel, M.C.; Spino, M.; Connelly, J.; Tricta, F.; et al. Brain iron chelation by deferiprone in a phase 2 randomised double-blinded placebo controlled clinical trial in Parkinson’s disease. Sci. Rep. 2017, 7, 1398. [Google Scholar] [CrossRef] [PubMed]
- Hider, R.; Liu, Z.; Liu, Y.; Li, Z. Chiral 3-Hydroxypyrid-4-one Derivative and Synthesis and Use Thereof. European Patent 2,692,724A1, 5 February 2014. [Google Scholar]
- Tao, Y.; Wang, Y.; Rogers, J.T.; Wang, F. Perturbed iron distribution in Alzheimer’s disease serum, cerebrospinal fluid, and selected brain regions: A systematic review and meta-analysis. J. Alzheimer’s Dis. 2014, 42, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Hare, D.J.; Raven, E.P.; Roberts, B.R.; Bogeski, M.; Portbury, S.D.; McLean, C.A.; Masters, C.L.; Connor, J.R.; Bush, A.I.; Crouch, P.J.; et al. Laser ablation-inductively coupled plasma-mass spectrometry imaging of white and gray matter iron distribution in Alzheimer’s disease frontal cortex. NeuroImage 2016, 137, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Reznichenko, L.; Amit, T.; Zheng, H.; Avarmovitch-Tirosh, Y.; Youdim, M.B.; Weinreb, O.; Mandel, S. Reduction of iron-regulated amyloid precursor protein and beta-amyloid peptide by (-)-epigallocatechin-3-gallate in cell cultures: Implications for iron chelation in Alzheimer’s disease. J. Neurochem. 2006, 97, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Avramovich-Tirosh, Y.; Reznichenko, L.; Mit, T.; Zheng, H.; Fridkin, M.; Weinreb, O.; Mandel, S.; Youdim, M.B. Neurorescue activity, APP regulation and amyloid-beta peptide reduction by novel multi-functional brain permeable iron-chelating-antioxidants, M-30 and green tea polyphenol, EGCG. Curr. Alzheimer Res. 2007, 4, 403–411. [Google Scholar] [CrossRef] [PubMed]
- McLachlan, D.R.; Smith, W.L.; Kruck, T.P. Desferrioxamine and Alzheimer’s disease: Video home behavior assessment of clinical course and measures of brain aluminium. Ther. Drug Monit. 1993, 15, 602–607. [Google Scholar] [CrossRef] [PubMed]
- Hiremathad, A.; Chand, K.; Tolayan, L.; Rajeshwari Keri, R.S.; Esteves, A.R.; Cardoso, S.M.; Chaves, S.; Santos, M.A. Hydroxypyridinone-benzofuran hybrids with potential protective roles for Alzheimer’s diseasetherapy. J. Inorg. Biochem. 2018, 179, 82–96. [Google Scholar] [CrossRef]
- Palanimuthu, D.; Poon, R.; Sahni, S.; Anjum, R.; Hibbs, D.; Lin, H.Y.; Bernhardt, P.V.; Kalinowski, D.S.; Richardson, D.R. A novel class of thiosemicarbazones show multi-functional activity for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2017, 139, 612–632. [Google Scholar] [CrossRef] [PubMed]
- Lupoli, F.; Vannocci, T.; Longo, G.; Niccolai, N.; Pastore, A. The role of oxidative stress in Friedreich’s ataxia. FEBS Lett. 2018, 592, 718–727. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, S.; Iannuzzi, C.; Prischi, F.; Pastore, C.; Iametti, S.; Martin, S.R.; Bonomi, F.; Pastore, A. Bacterial frataxin CyaY is the gatekeeper of iron-sulfur cluster formation catalyzed by IscS. Nat. Struct. Mol. Biol. 2009, 16, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Prischi, F.; Konarev, P.V.; Iannuzzi, C.; Pastore, C.; Adinolfi, S.; Martin, S.R.; Svergun, D.I.; Pastore, A. Structural bases for the interaction of frataxin with the central components of iron-sulphur cluster assembly. Nat. Commun. 2010, 1. [Google Scholar] [CrossRef] [PubMed]
- Prischi, F.; Pastore, A. Hybrid Methods in Iron-Sulfur Cluster Biogenesis. Front. Mol. Biosci. 2017, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, M.L.; Lane, D.J.; Richardson, D.R. Mitochondrial mayhem: The mitochondrion as a modulator of iron metabolism and its role in disease. Antioxid. Redox Signal 2011, 15, 3003–3019. [Google Scholar] [CrossRef] [PubMed]
- Lill, R.; Hoffmann, B.; Molik, S.; Pierik, A.J.; Rietzschel, N.; Stehling, O.; Uzarska, M.A.; Webert, H.; Wilbrecht, C.; Mühlenhoff, U. The role of mitochondria in cellular iron-sulfur protein biogenesis and iron metabolism. Biochim. Biophys. Acta 2012, 1823, 1491–1508. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, R.L.; Qian, J.; Santambrogio, P.; Levi, S.; Koeppen, A.H. Relation of cytosolic iron excess to cardiomyopathy of Friedreich’s ataxia. Am. J. Cardiol. 2012, 110, 1820–1827. [Google Scholar] [CrossRef] [PubMed]
- Boddaert, N.; Le Quan Sang, K.H.; Rötig, A.; Leroy-Willig, A.; Gallet, S.; Brunelle, F.; Sidi, D.; Thalabard, J.C.; Munnich, A.; Cabantchik, Z.I. Selective iron chelation in Friedreich ataxia: Biologic and clinical implications. Blood 2007, 110, 401–408. [Google Scholar] [CrossRef]
- Velasco-Sánchez, D.; Aracil, A.; Montero, R.; Mas, A.; Jiménez, L.; O’Callaghan, M.; Tondo, M.; Capdevila, A.; Blanch, J.; Artuch, R.; et al. Combined therapy with idebenone and deferiprone in patients with Friedreich’s ataxia. Cerebellum 2011, 10, 1–8. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crichton, R.R.; Ward, R.J.; Hider, R.C. The Efficacy of Iron Chelators for Removing Iron from Specific Brain Regions and the Pituitary—Ironing out the Brain. Pharmaceuticals 2019, 12, 138. https://doi.org/10.3390/ph12030138
Crichton RR, Ward RJ, Hider RC. The Efficacy of Iron Chelators for Removing Iron from Specific Brain Regions and the Pituitary—Ironing out the Brain. Pharmaceuticals. 2019; 12(3):138. https://doi.org/10.3390/ph12030138
Chicago/Turabian StyleCrichton, Robert R., Roberta J. Ward, and Robert C. Hider. 2019. "The Efficacy of Iron Chelators for Removing Iron from Specific Brain Regions and the Pituitary—Ironing out the Brain" Pharmaceuticals 12, no. 3: 138. https://doi.org/10.3390/ph12030138
APA StyleCrichton, R. R., Ward, R. J., & Hider, R. C. (2019). The Efficacy of Iron Chelators for Removing Iron from Specific Brain Regions and the Pituitary—Ironing out the Brain. Pharmaceuticals, 12(3), 138. https://doi.org/10.3390/ph12030138