Effects of Treatment with the Hypomethylating Agent 5-aza-2′-deoxycytidine in Murine Type II Collagen-Induced Arthritis

 ,
, _Di_Marco.png) ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Results
2.1. In Silico Study: Prediction of Autoimmune Disease Potentially Targeted by DAC Treatment
2.2. Animal Study
2.2.1. Toxicity
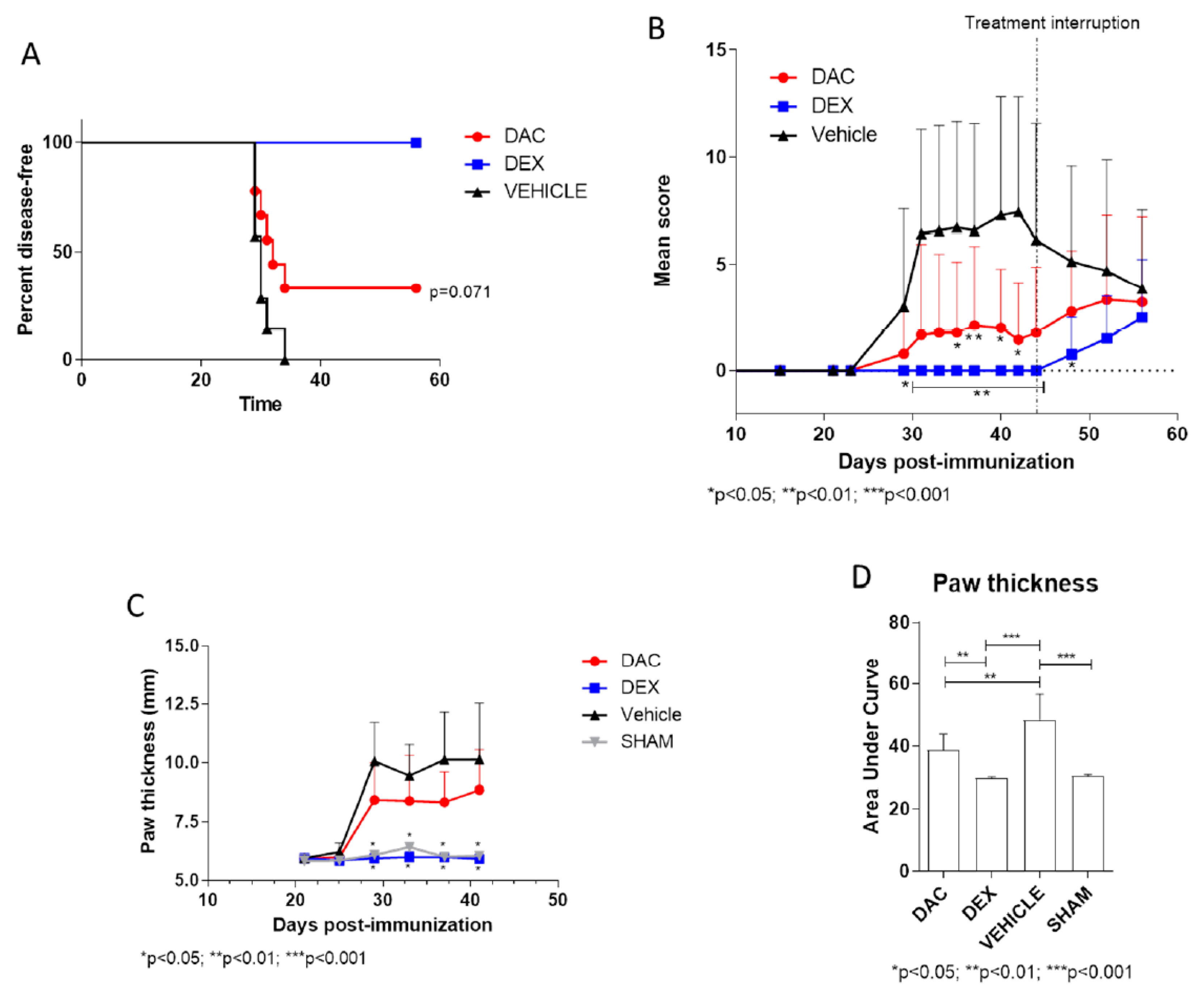
2.2.2. Effect of Late Prophylactic Treatment with DAC on the Arthritic Score and on Paw Thickness
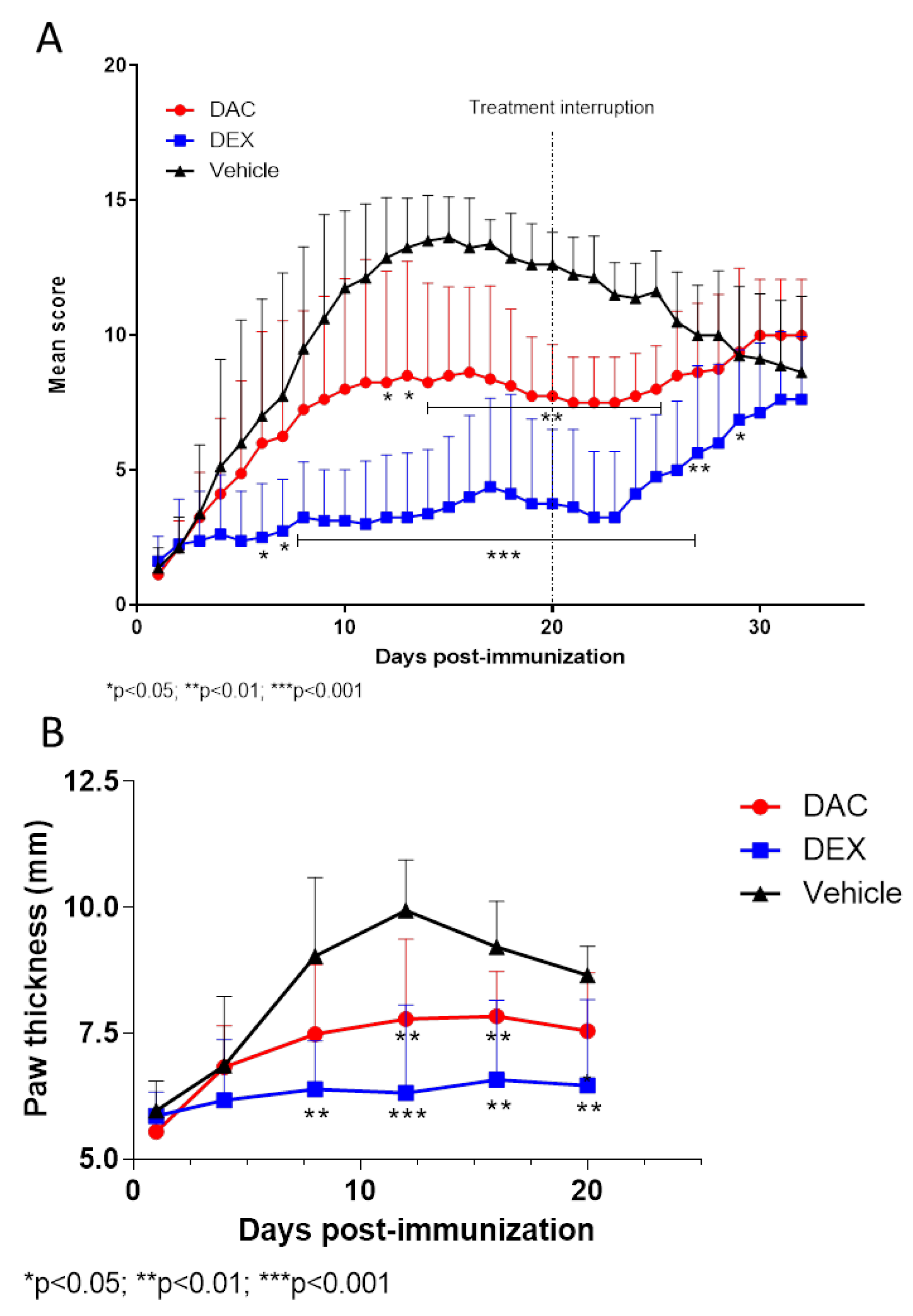
2.2.3. Effect of Therapeutic Treatment with DAC on the Arthritic Score and on Paws Thickness
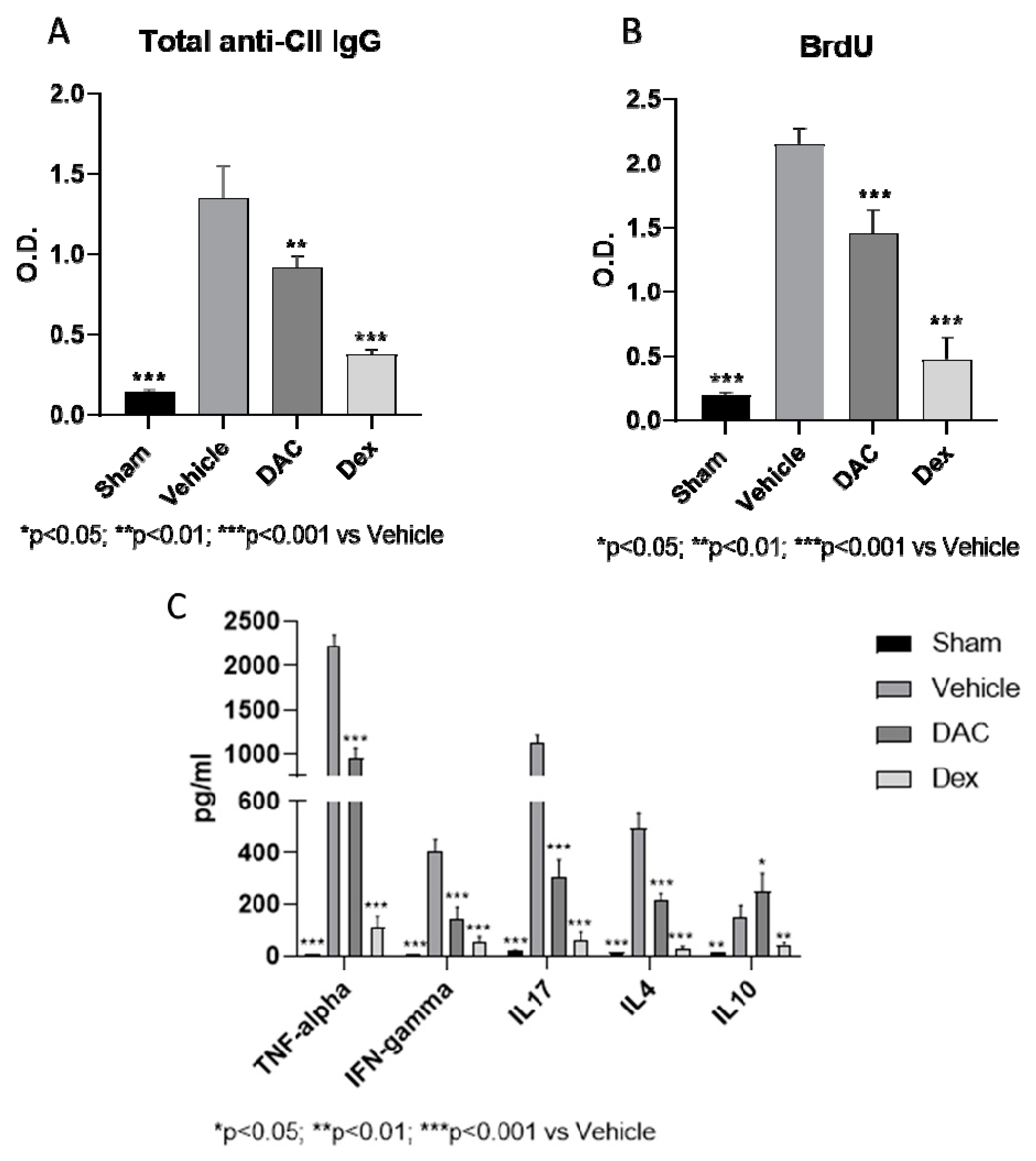
2.2.4. Effects of DAC on Serum Anti-CII Antibodies
2.2.5. DAC Profoundly Modulated Ex Vivo Cytokine Secretion from the Spleens during Type II CIA
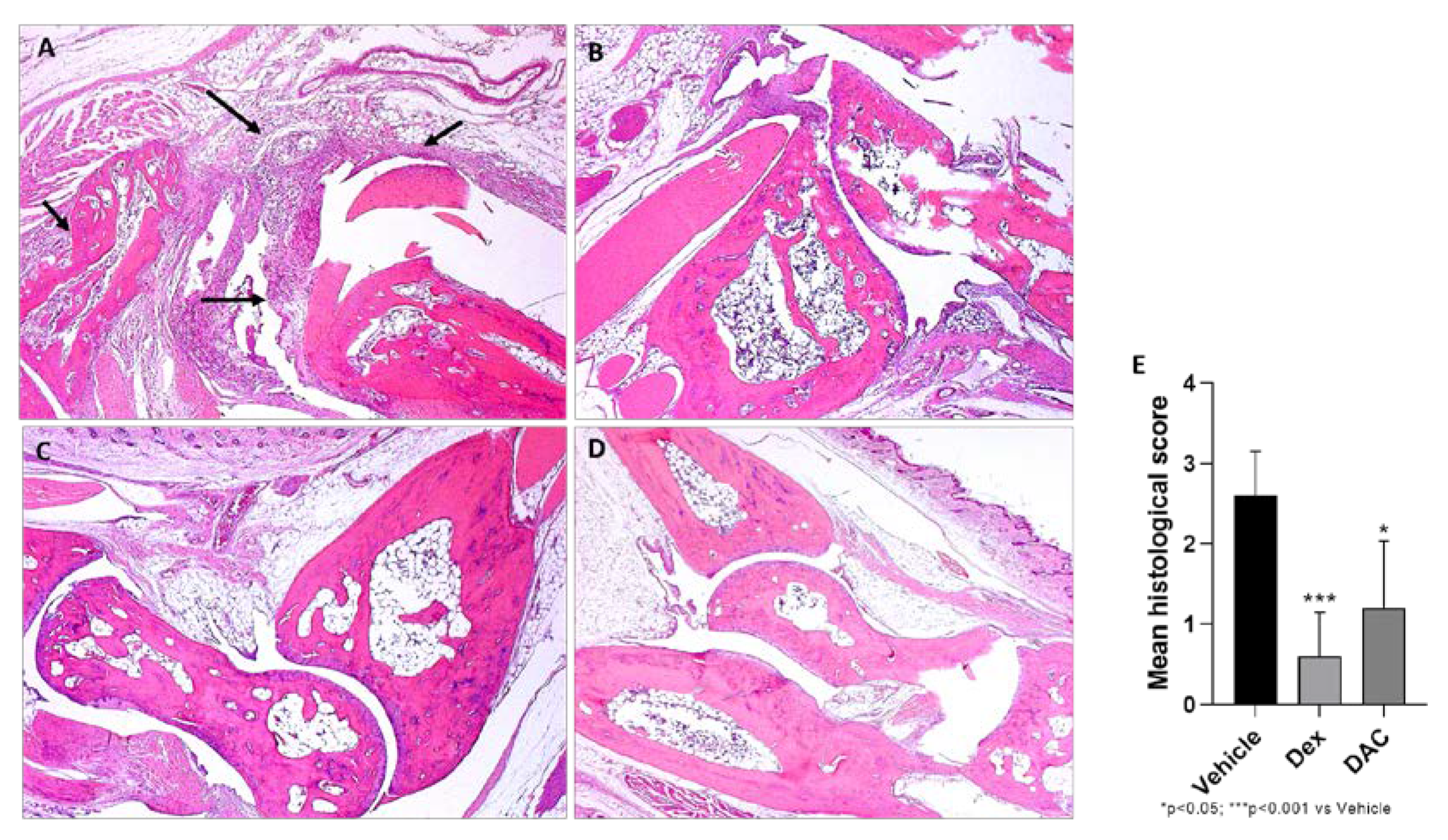
2.2.6. Effect of DAC Treatment on CIA Morphological Changes
3. Discussion
4. Materials and Methods
4.1. In Silico Analysis
4.2. In Vivo Study
4.2.1. Animals
4.2.2. Induction of CIA
4.2.3. Drugs
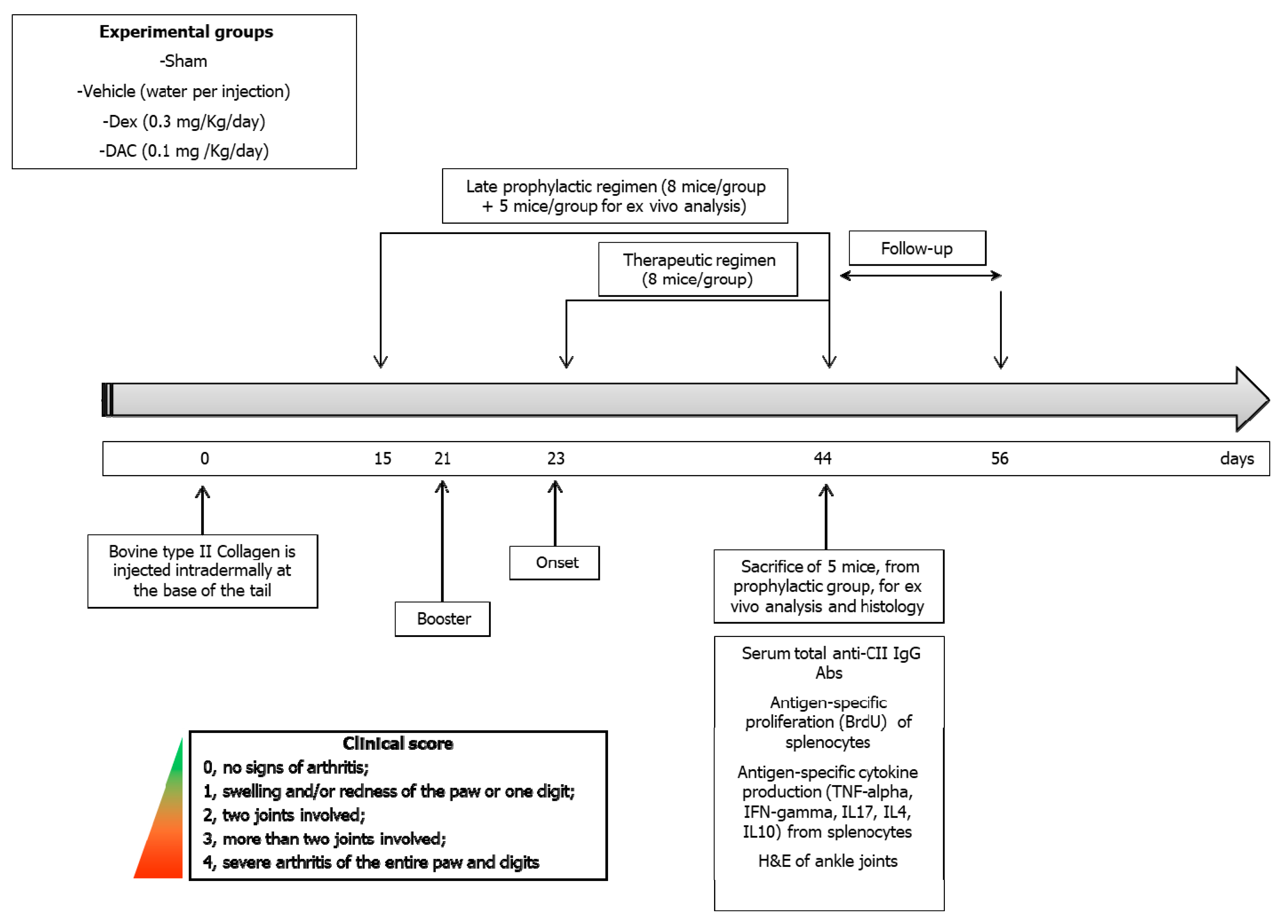
4.2.4. Treatment Regimens
4.2.5. Primary Endpoints
Body Weight
Arthritic Score
Assessment of Paws Thickness
4.3. Ex Vivo Studies
4.3.1. Preparation of Spleen Cell Suspensions from CIA Mice
4.3.2. Ex Vivo Effects of DAC on Splenic Cytokine Secretion
4.3.3. Splenocytes Proliferation, BrdU Incorporation Method
4.3.4. Anti-CII Antibody Levels
4.3.5. Histological Examination
4.4. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- McInnes, I.B.; Schett, G. The Pathogenesis of Rheumatoid Arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef]
- Brennan, F.M.; McInnes, I.B. Evidence that cytokines play a role in rheumatoid arthritis. J. Clin. Investig. 2008, 118, 3537–3545. [Google Scholar] [CrossRef]
- Roeleveld, D.M.; Koenders, M.I. The role of the Th17 cytokines IL-17 and IL-22 in Rheumatoid Arthritis pathogenesis and developments in cytokine immunotherapy. Cytokine 2015, 74, 101–107. [Google Scholar] [CrossRef]
- Zhang, H.-L.; Zheng, X.-Y.; Zhu, J. Th1/Th2/Th17/Treg cytokines in Guillain–Barré syndrome and experimental autoimmune neuritis. Cytokine Growth Factor Rev. 2013, 24, 443–453. [Google Scholar] [CrossRef]
- Ayakannu, R.; Abdullah, N.A.; Radhakrishnan, A.K.; Lechimi Raj, V.; Liam, C.K. Relationship between various cytokines implicated in asthma. Hum. Immunol. 2019, 80, 755–763. [Google Scholar] [CrossRef]
- Nicoletti, F.; Mancuso, G.; Cusumano, V.; Di Marco, R.; Zaccone, P.; Bendtzen, K.; Teti, G. Prevention of endotoxin-induced lethality in neonatal mice by interleukin-13. Eur. J. Immunol. 1997, 27, 1580–1583. [Google Scholar] [CrossRef] [PubMed]
- Nicoletti, F.; Di Marco, R.; Patti, F.; Reggio, E.; Nicoletti, A.; Zaccone, P.; Stivala, F.; Meroni, P.L.; Reggio, A. Blood levels of transforming growth factor-beta 1 (TGF-beta1) are elevated in both relapsing remitting and chronic progressive multiple sclerosis (MS) patients and are further augmented by treatment with interferon-beta 1b (IFN-beta1b). Clin. Exp. Immunol. 1998, 113, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, S.; Xiao, X.; Zhao, Y.; Ding, W.; Li, X.C. IL-9 and Th9 cells in health and diseases—From tolerance to immunopathology. Cytokine Growth Factor Rev. 2017, 37, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front. Immunol. 2014, 5, 514. [Google Scholar] [CrossRef]
- Chemin, K.; Gerstner, C.; Malmström, V. Effector Functions of CD4+ T Cells at the Site of Local Autoimmune Inflammation-Lessons from Rheumatoid Arthritis. Front. Immunol. 2019, 10, 353. [Google Scholar] [CrossRef] [PubMed]
- Senolt, L. Emerging therapies in rheumatoid arthritis: Focus on monoclonal antibodies. F1000Research 2019, 8, 1549. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Banik, S. Pharmacotherapy options in rheumatoid arthritis. Clin. Med. Insights. Arthritis Musculoskelet. Disord. 2013, 6, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Fogdell-Hahn, A. Antidrug Antibodies: B Cell Immunity Against Therapy. Scand. J. Immunol. 2015, 82, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Sardar, S.; Andersson, Å. Old and new therapeutics for Rheumatoid Arthritis: In vivo models and drug development. Immunopharmacol. Immunotoxicol. 2016, 38, 2–13. [Google Scholar] [CrossRef]
- Ozsahin, M.; Dikici, S.; Kocaman, G.; Besir, F.H.; Baltaci, D.; Ataoglu, S. Dual diagnosis: Rheumatoid arthritis and multiple sclerosis. PM R 2014, 6, 96–99. [Google Scholar] [CrossRef]
- Kemanetzoglou, E.; Andreadou, E. CNS Demyelination with TNF-α Blockers. Curr. Neurol. Neurosci. Rep. 2017, 17, 36. [Google Scholar] [CrossRef]
- Caminero, A.; Comabella, M.; Montalban, X. Tumor necrosis factor alpha (TNF-α), anti-TNF-α and demyelination revisited: An ongoing story. J. Neuroimmunol. 2011, 234, 1–6. [Google Scholar] [CrossRef]
- Crow, M.K. Type I interferon in organ-targeted autoimmune and inflammatory diseases. Arthritis Res. Ther. 2010, 12 (Suppl. S5). [Google Scholar] [CrossRef]
- Conigliaro, P.; Perricone, C.; Benson, R.A.; Garside, P.; Brewer, J.M.; Perricone, R.; Valesini, G. The type I IFN system in rheumatoid arthritis. Autoimmunity 2010, 43, 220–225. [Google Scholar] [CrossRef]
- Stresemann, C.; Lyko, F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int. J. Cancer 2008, 123, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Gnyszka, A.; Jastrzebski, Z.; Flis, S. DNA methyltransferase inhibitors and their emerging role in epigenetic therapy of cancer. Anticancer Res. 2013, 33, 2989–2996. [Google Scholar] [PubMed]
- Zheng, Q.; Xu, Y.; Liu, Y.; Zhang, B.; Li, X.; Guo, F.; Zhao, Y. Induction of Foxp3 demethylation increases regulatory CD4+CD25+ T cells and prevents the occurrence of diabetes in mice. J. Mol. Med. (Berl.) 2009, 87, 1191–1205. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, J.; Yu, Y.; Ma, T.; Chen, P.; Zhou, B.; Tao, R. Decitabine inhibits T cell proliferation via a novel TET2-dependent mechanism and exerts potent protective effect in mouse auto- and allo-immunity models. Oncotarget 2017, 8, 56802–56815. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.W.Y.; Chang, C.-B.; Tung, C.-H.; Sun, J.; Suen, J.-L.; Wu, S.-F. Low-dose 5-aza-2′-deoxycytidine pretreatment inhibits experimental autoimmune encephalomyelitis by induction of regulatory T cells. Mol. Med. 2014, 20, 248–256. [Google Scholar] [CrossRef]
- Mangano, K.; Fagone, P.; Bendtzen, K.; Meroni, P.L.; Quattrocchi, C.; Mammana, S.; Di Rosa, M.; Malaguarnera, L.; Coco, M.; Magro, G.; et al. Hypomethylating agent 5-aza-2′-deoxycytidine (DAC) ameliorates multiple sclerosis in mouse models. J. Cell. Physiol. 2014, 229, 1918–1925. [Google Scholar] [CrossRef]
- Fagone, P.; Mazzon, E.; Chikovani, T.; Saraceno, A.; Mammana, S.; Colletti, G.; Mangano, K.; Bramanti, P.; Nicoletti, F. Decitabine induces regulatory T cells, inhibits the production of IFN-gamma and IL-17 and exerts preventive and therapeutic efficacy in rodent experimental autoimmune neuritis. J. Neuroimmunol. 2018, 321, 41–48. [Google Scholar] [CrossRef]
- Li, H.; Tsokos, M.G.; Bickerton, S.; Sharabi, A.; Li, Y.; Moulton, V.R.; Kong, P.; Fahmy, T.M.; Tsokos, G.C. Precision DNA demethylation ameliorates disease in lupus-prone mice. JCI Insight 2018, 3, 120880. [Google Scholar] [CrossRef]
- Malemud, C.J. Defective T-Cell Apoptosis and T-Regulatory Cell Dysfunction in Rheumatoid Arthritis. Cells 2018, 7, 223. [Google Scholar] [CrossRef]
- Rahmanzadeh, R.; Weber, M.S.; Brück, W.; Navardi, S.; Sahraian, M.A. B cells in multiple sclerosis therapy-A comprehensive review. Acta Neurol. Scand. 2018, 137, 544–556. [Google Scholar] [CrossRef]
- Tóth, D.M.; Ocskó, T.; Balog, A.; Markovics, A.; Mikecz, K.; Kovács, L.; Jolly, M.; Bukiej, A.A.; Ruthberg, A.D.; Vida, A.; et al. Amelioration of Autoimmune Arthritis in Mice Treated with the DNA Methyltransferase Inhibitor 5′-Azacytidine. Arthritis Rheumatol. 2019, 71, 1265–1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marquez, V.E.; Kelley, J.A.; Agbaria, R.; Ben-Kasus, T.; Cheng, J.C.; Yoo, C.B.; Jones, P.A. Zebularine: A Unique Molecule for an Epigenetically Based Strategy in Cancer Chemotherapy. Ann. N. Y. Acad. Sci. 2005, 1058, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.-M.; Lin, Y.-T.; Shun, C.-T.; Lin, S.-H.; Wei, T.-T.; Chuang, S.-H.; Wu, M.-S.; Chen, C.-C. Zebularine inhibits tumorigenesis and stemness of colorectal cancer via p53-dependent endoplasmic reticulum stress. Sci. Rep. 2013, 3, 3219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Y.; Hu, X.; Schewitz-Bowers, L.P.; Stimpson, M.; Miao, L.; Ge, X.; Yang, L.; Li, Y.; Bible, P.W.; Wen, X.; et al. The DNA Methylation Inhibitor Zebularine Controls CD4+ T Cell Mediated Intraocular Inflammation. Front. Immunol. 2019, 10, 1950. [Google Scholar] [CrossRef] [Green Version]
- Yokoyama, Y.; Iwasaki, T.; Kitano, S.; Satake, A.; Nomura, S.; Furukawa, T.; Matsui, K.; Sano, H. IL-2–Anti–IL-2 Monoclonal Antibody Immune Complexes Inhibit Collagen-Induced Arthritis by Augmenting Regulatory T Cell Functions. J. Immunol. 2018, 201, 1899–1906. [Google Scholar] [CrossRef]
- Klein, K.; Gay, S. Epigenetics in rheumatoid arthritis. Curr. Opin. Rheumatol. 2015, 27, 76–82. [Google Scholar] [CrossRef] [Green Version]
- Nakano, K.; Whitaker, J.W.; Boyle, D.L.; Wang, W.; Firestein, G.S. DNA methylome signature in rheumatoid arthritis. Ann. Rheum. Dis. 2013, 72, 110–117. [Google Scholar] [CrossRef]
- Tough, D.F.; Tak, P.P.; Tarakhovsky, A.; Prinjha, R.K. Epigenetic drug discovery: Breaking through the immune barrier. Nat. Rev. Drug Discov. 2016, 15, 835–853. [Google Scholar] [CrossRef]
- Ospelt, C.; Gay, S.; Klein, K. Epigenetics in the pathogenesis of RA. Semin. Immunopathol. 2017, 39, 409–419. [Google Scholar] [CrossRef]
- Glossop, J.R.; Emes, R.D.; Nixon, N.B.; Haworth, K.E.; Packham, J.C.; Dawes, P.T.; Fryer, A.A.; Mattey, D.L.; Farrell, W.E.; Packham, J.C. Genome-wide DNA methylation profiling in rheumatoid arthritis identifies disease-associated methylation changes that are distinct to individual T- and B-lymphocyte populations. Epigenetics 2014, 9, 1228–1237. [Google Scholar] [CrossRef] [Green Version]
- Miao, C.; Huang, C.; Huang, Y.; Yang, Y.; He, X.; Zhang, L.; Lv, X.-W.; Jin, Y.; Li, J. MeCP2 modulates the canonical Wnt pathway activation by targeting SFRP4 in rheumatoid arthritis fibroblast-like synoviocytes in rats. Cell. Signal. 2013, 25, 598–608. [Google Scholar] [CrossRef] [PubMed]
- Miao, C.-G.; Qin, D.; Du, C.-L.; Ye, H.; Shi, W.-J.; Xiong, Y.-Y.; Zhang, X.-L.; Yu, H.; Dou, J.-F.; Ma, S.-T.; et al. DNMT1 activates the canonical Wnt signaling in rheumatoid arthritis model rats via a crucial functional crosstalk between miR-152 and the DNMT1, MeCP2. Int. Immunopharmacol. 2015, 28, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Long, L.; Zhou, T.; Tian, J.; Zhou, B. Demethylation of MicroRNA-124a Genes Attenuated Proliferation of Rheumatoid Arthritis Derived Fibroblast-Like Synoviocytes and Synthesis of Tumor Necrosis Factor-α. PLoS ONE 2016, 11, e0164207. [Google Scholar] [CrossRef] [PubMed]
- de la Rica, L.; Urquiza, J.M.; Gómez-Cabrero, D.; Islam, A.B.M.M.K.; López-Bigas, N.; Tegnér, J.; Toes, R.E.M.; Ballestar, E. Identification of novel markers in rheumatoid arthritis through integrated analysis of DNA methylation and microRNA expression. J. Autoimmun. 2013, 41, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-R.; Yang, L.; Xu, Q.-Q.; Lu, X.-Y.; Ma, T.-T.; Huang, C.; Li, J. Long noncoding RNA MEG3 regulates rheumatoid arthritis by targeting NLRC5. J. Cell. Physiol. 2019, 234, 14270–14284. [Google Scholar] [CrossRef]
- Miao, C.; Chang, J.; Dou, J.; Xiong, Y.; Zhou, G. DNA hypermethylation of SFRP2 influences the pathology of rheumatoid arthritis through the canonical Wnt signaling in model rats. Autoimmunity 2018, 51, 1–14. [Google Scholar] [CrossRef]
- Lal, G.; Bromberg, J.S. Epigenetic mechanisms of regulation of Foxp3 expression. Blood 2009, 114, 3727–3735. [Google Scholar] [CrossRef] [Green Version]
- Mazari, L.; Ouarzane, M.; Zouali, M. Subversion of B lymphocyte tolerance by hydralazine, a potential mechanism for drug-induced lupus. Proc. Natl. Acad. Sci. USA 2007, 104, 6317–6322. [Google Scholar] [CrossRef] [Green Version]
- Quddus, J.; Johnson, K.J.; Gavalchin, J.; Amento, E.P.; Chrisp, C.E.; Yung, R.L.; Richardson, B.C. Treating activated CD4+ T cells with either of two distinct DNA methyltransferase inhibitors, 5-azacytidine or procainamide, is sufficient to cause a lupus-like disease in syngeneic mice. J. Clin. Investig. 1993, 92, 38–53. [Google Scholar] [CrossRef] [Green Version]
- Lombardo, S.D.; Mazzon, E.; Basile, M.S.; Campo, G.; Corsico, F.; Presti, M.; Bramanti, P.; Mangano, K.; Petralia, M.C.; Nicoletti, F.; et al. Modulation of Tetraspanin 32 (TSPAN32) Expression in T Cell-Mediated Immune Responses and in Multiple Sclerosis. Int. J. Mol. Sci. 2019, 20, 4323. [Google Scholar] [CrossRef] [Green Version]
- Nicoletti, F.; Mazzon, E.; Fagone, P.; Mangano, K.; Mammana, S.; Cavalli, E.; Basile, M.S.; Bramanti, P.; Scalabrino, G.; Lange, A.; et al. Prevention of clinical and histological signs of MOG-induced experimental allergic encephalomyelitis by prolonged treatment with recombinant human EGF. J. Neuroimmunol. 2019, 332, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Mangano, K.; Cavalli, E.; Mammana, S.; Basile, M.S.; Caltabiano, R.; Pesce, A.; Puleo, S.; Atanasov, A.G.; Magro, G.; Nicoletti, F.; et al. Involvement of the Nrf2/HO-1/CO axis and therapeutic intervention with the CO-releasing molecule CORM-A1, in a murine model of autoimmune hepatitis. J. Cell. Physiol. 2018, 233, 4156–4165. [Google Scholar] [CrossRef] [PubMed]
- Fagone, P.; Mazzon, E.; Cavalli, E.; Bramanti, A.; Petralia, M.C.; Mangano, K.; Al-Abed, Y.; Bramati, P.; Nicoletti, F. Contribution of the macrophage migration inhibitory factor superfamily of cytokines in the pathogenesis of preclinical and human multiple sclerosis: In silico and in vivo evidences. J. Neuroimmunol. 2018, 322, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Mammana, S.; Bramanti, P.; Mazzon, E.; Cavalli, E.; Basile, M.S.; Fagone, P.; Petralia, M.C.; McCubrey, J.A.; Nicoletti, F.; Mangano, K. Preclinical evaluation of the PI3K/Akt/mTOR pathway in animal models of multiple sclerosis. Oncotarget 2018, 9, 8263–8277. [Google Scholar] [CrossRef] [Green Version]
- Fagone, P.; Mazzon, E.; Mammana, S.; Di Marco, R.; Spinasanta, F.; Basile, M.; Petralia, M.; Bramanti, P.; Nicoletti, F.; Mangano, K. Identification of CD4+ T cell biomarkers for predicting the response of patients with relapsing-remitting multiple sclerosis to natalizumab treatment. Mol. Med. Rep. 2019, 20, 678–684. [Google Scholar] [CrossRef] [Green Version]
- Lombardo, S.D.; Mazzon, E.; Basile, M.S.; Cavalli, E.; Bramanti, P.; Nania, R.; Fagone, P.; Nicoletti, F.; Petralia, M.C. Upregulation of IL-1 Receptor Antagonist in a Mouse Model of Migraine. Brain Sci. 2019, 9, 172. [Google Scholar] [CrossRef] [Green Version]
- Falzone, L.; Lupo, G.; Rosa, G.R.M.; Crimi, S.; Anfuso, C.D.; Salemi, R.; Rapisarda, E.; Libra, M.; Candido, S. Identification of Novel MicroRNAs and Their Diagnostic and Prognostic Significance in Oral Cancer. Cancers 2019, 11, 610. [Google Scholar] [CrossRef] [Green Version]
- Presti, M.; Mazzon, E.; Basile, M.; Petralia, M.; Bramanti, A.; Colletti, G.; Bramanti, P.; Nicoletti, F.; Fagone, P. Overexpression of macrophage migration inhibitory factor and functionally-related genes, D-DT, CD74, CD44, CXCR2 and CXCR4, in glioblastoma. Oncol. Lett. 2018, 16, 2881–2886. [Google Scholar] [CrossRef]
- Fagone, P.; Mangano, K.; Mammana, S.; Pesce, A.; Pesce, A.; Caltabiano, R.; Giorlandino, A.; Portale, T.R.; Cavalli, E.; Lombardo, G.A.G.; et al. Identification of novel targets for the diagnosis and treatment of liver fibrosis. Int. J. Mol. Med. 2015, 36, 747–752. [Google Scholar] [CrossRef]
- Basile, M.S.; Mazzon, E.; Russo, A.; Mammana, S.; Longo, A.; Bonfiglio, V.; Fallico, M.; Caltabiano, R.; Fagone, P.; Nicoletti, F.; et al. Differential modulation and prognostic values of immune-escape genes in uveal melanoma. PLoS ONE 2019, 14, e0210276. [Google Scholar] [CrossRef]
- Mangano, K.; Mazzon, E.; Basile, M.S.; Di Marco, R.; Bramanti, P.; Mammana, S.; Petralia, M.C.; Fagone, P.; Nicoletti, F. Pathogenic role for macrophage migration inhibitory factor in glioblastoma and its targeting with specific inhibitors as novel tailored therapeutic approach. Oncotarget 2018, 9, 17951–17970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombardo, S.D.; Presti, M.; Mangano, K.; Petralia, M.C.; Basile, M.S.; Libra, M.; Candido, S.; Fagone, P.; Mazzon, E.; Nicoletti, F.; et al. Prediction of PD-L1 Expression in Neuroblastoma via Computational Modeling. Brain Sci. 2019, 9, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petralia, M.C.; Mazzon, E.; Fagone, P.; Russo, A.; Longo, A.; Avitabile, T.; Nicoletti, F.; Reibaldi, M.; Basile, M.S. Characterization of the Pathophysiological Role of CD47 in Uveal Melanoma. Molecules 2019, 24, 2450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petralia, M.; Mazzon, E.; Fagone, P.; Falzone, L.; Bramanti, P.; Nicoletti, F.; Basile, M. Retrospective follow-up analysis of the transcriptomic patterns of cytokines, cytokine receptors and chemokines at preconception and during pregnancy, in women with post-partum depression. Exp. Ther. Med. 2019, 18, 2055–2062. [Google Scholar] [CrossRef] [Green Version]
- Mammana, S.; Fagone, P.; Cavalli, E.; Basile, M.; Petralia, M.; Nicoletti, F.; Bramanti, P.; Mazzon, E. The Role of Macrophages in Neuroinflammatory and Neurodegenerative Pathways of Alzheimer’s Disease, Amyotrophic Lateral Sclerosis, and Multiple Sclerosis: Pathogenetic Cellular Effectors and Potential Therapeutic Targets. Int. J. Mol. Sci. 2018, 19, 831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paskas, S.; Mazzon, E.; Basile, M.S.; Cavalli, E.; Al-Abed, Y.; He, M.; Rakocevic, S.; Nicoletti, F.; Mijatovic, S.; Maksimovic-Ivanic, D. Lopinavir-NO, a nitric oxide-releasing HIV protease inhibitor, suppresses the growth of melanoma cells in vitro and in vivo. Invest. New Drugs 2019, 37, 1014–1028. [Google Scholar] [CrossRef]
- Basile, M.S.; Mazzon, E.; Krajnovic, T.; Draca, D.; Cavalli, E.; Al-Abed, Y.; Bramanti, P.; Nicoletti, F.; Mijatovic, S.; Maksimovic-Ivanic, D. Anticancer and Differentiation Properties of the Nitric Oxide Derivative of Lopinavir in Human Glioblastoma Cells. Molecules 2018, 23, 2463. [Google Scholar] [CrossRef] [Green Version]
- Maksimovic-Ivanic, D.; Mojic, M.; Bulatovic, M.; Radojkovic, M.; Kuzmanovic, M.; Ristic, S.; Stosic-Grujicic, S.; Miljkovic, D.; Cavalli, E.; Libra, M.; et al. The NO-modified HIV protease inhibitor as a valuable drug for hematological malignancies: Role of p70S6K. Leuk. Res. 2015, 39, 1088–1095. [Google Scholar] [CrossRef] [Green Version]
- Fagone, P.; Mangano, K.; Quattrocchi, C.; Cavalli, E.; Mammana, S.; Lombardo, G.A.G.; Pennisi, V.; Zocca, M.-B.; He, M.; Al-Abed, Y.; et al. Effects of NO-Hybridization on the Immunomodulatory Properties of the HIV Protease Inhibitors Lopinavir and Ritonavir. Basic Clin. Pharmacol. Toxicol. 2015, 117, 306–315. [Google Scholar] [CrossRef]
- Stojanovic, I.; Cuzzocrea, S.; Mangano, K.; Mazzon, E.; Miljkovic, D.; Wang, M.; Donia, M.; Al Abed, Y.; Kim, J.; Nicoletti, F.; et al. In vitro, ex vivo and in vivo immunopharmacological activities of the isoxazoline compound VGX-1027: Modulation of cytokine synthesis and prevention of both organ-specific and systemic autoimmune diseases in murine models. Clin. Immunol. 2007, 123, 311–323. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Significance (p Value) |
|---|---|
| Allergic Contact Dermatitis | <0.00001 |
| Ankylosing Spondylitis | <0.00001 |
| Asthma | <0.00001 |
| Discoid lupus | <0.00001 |
| Multiple Sclerosis | <0.00001 |
| Rheumatoid Arthritis | <0.00001 |
| Ulcerative Colitis | <0.00001 |
| Crohn’s Disease | 0.001701 |
| Atopic Dermatitis | 0.130141 |
| Type 1 Diabetes | 0.141354 |
| Juvenile Rheumatoid Arthritis | 0.423885 |
| Systemic Juvenile Idiopathic Arthritis | 0.45051 |
| Systemic Lupus Erythematosus | 0.819002 |
| Psoriasis | 0.986509 |
| Sjogren’s syndrome | 0.989622 |
| Non-Systemic Juvenile Idiopathic Arthritis | 0.993984 |
| Dermatomyositis | 1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petralia, M.C.; Mazzon, E.; Basile, M.S.; Cutuli, M.; Di Marco, R.; Scandurra, F.; Saraceno, A.; Fagone, P.; Nicoletti, F.; Mangano, K. Effects of Treatment with the Hypomethylating Agent 5-aza-2′-deoxycytidine in Murine Type II Collagen-Induced Arthritis. Pharmaceuticals 2019, 12, 174. https://doi.org/10.3390/ph12040174
Petralia MC, Mazzon E, Basile MS, Cutuli M, Di Marco R, Scandurra F, Saraceno A, Fagone P, Nicoletti F, Mangano K. Effects of Treatment with the Hypomethylating Agent 5-aza-2′-deoxycytidine in Murine Type II Collagen-Induced Arthritis. Pharmaceuticals. 2019; 12(4):174. https://doi.org/10.3390/ph12040174
Chicago/Turabian StylePetralia, Maria Cristina, Emanuela Mazzon, Maria Sofia Basile, Marco Cutuli, Roberto Di Marco, Fabiola Scandurra, Andrea Saraceno, Paolo Fagone, Ferdinando Nicoletti, and Katia Mangano. 2019. "Effects of Treatment with the Hypomethylating Agent 5-aza-2′-deoxycytidine in Murine Type II Collagen-Induced Arthritis" Pharmaceuticals 12, no. 4: 174. https://doi.org/10.3390/ph12040174
APA StylePetralia, M. C., Mazzon, E., Basile, M. S., Cutuli, M., Di Marco, R., Scandurra, F., Saraceno, A., Fagone, P., Nicoletti, F., & Mangano, K. (2019). Effects of Treatment with the Hypomethylating Agent 5-aza-2′-deoxycytidine in Murine Type II Collagen-Induced Arthritis. Pharmaceuticals, 12(4), 174. https://doi.org/10.3390/ph12040174