Electron Microscopy of In-Plaque Phage T3 Assembly: Proposed Analogs of Neurodegenerative Disease Triggers



Abstract
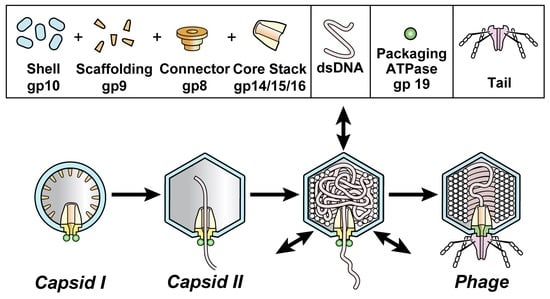
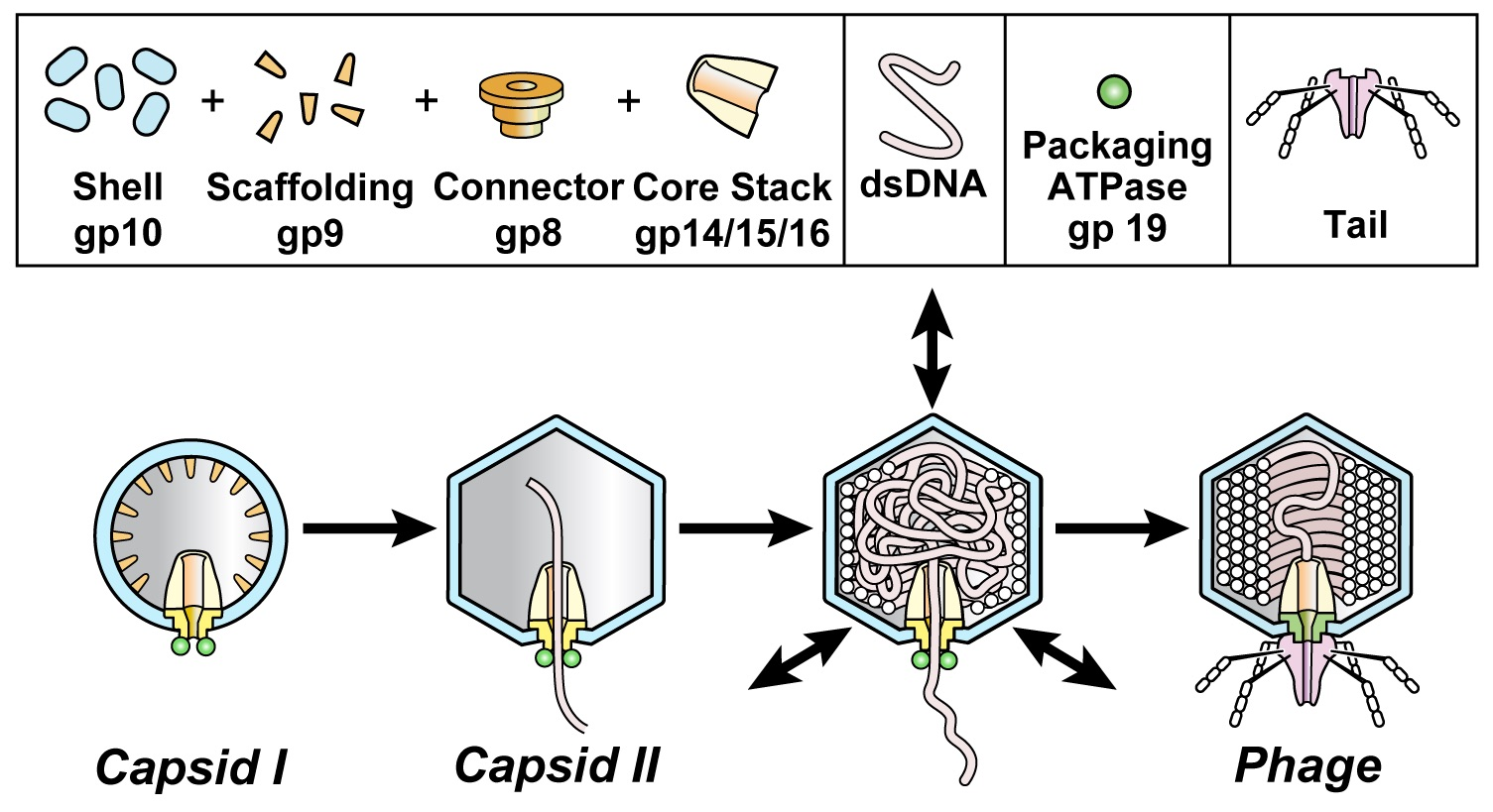
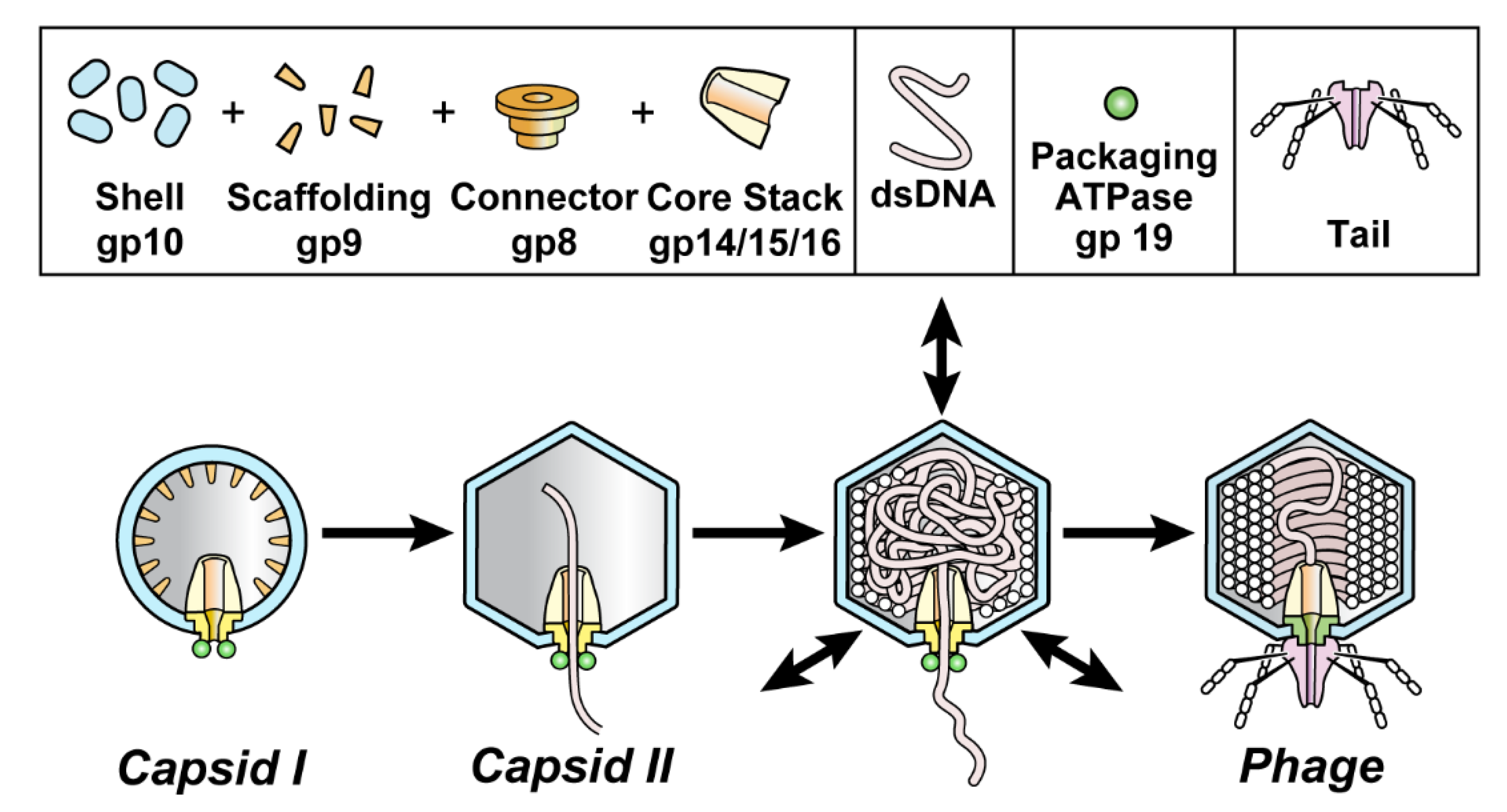
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
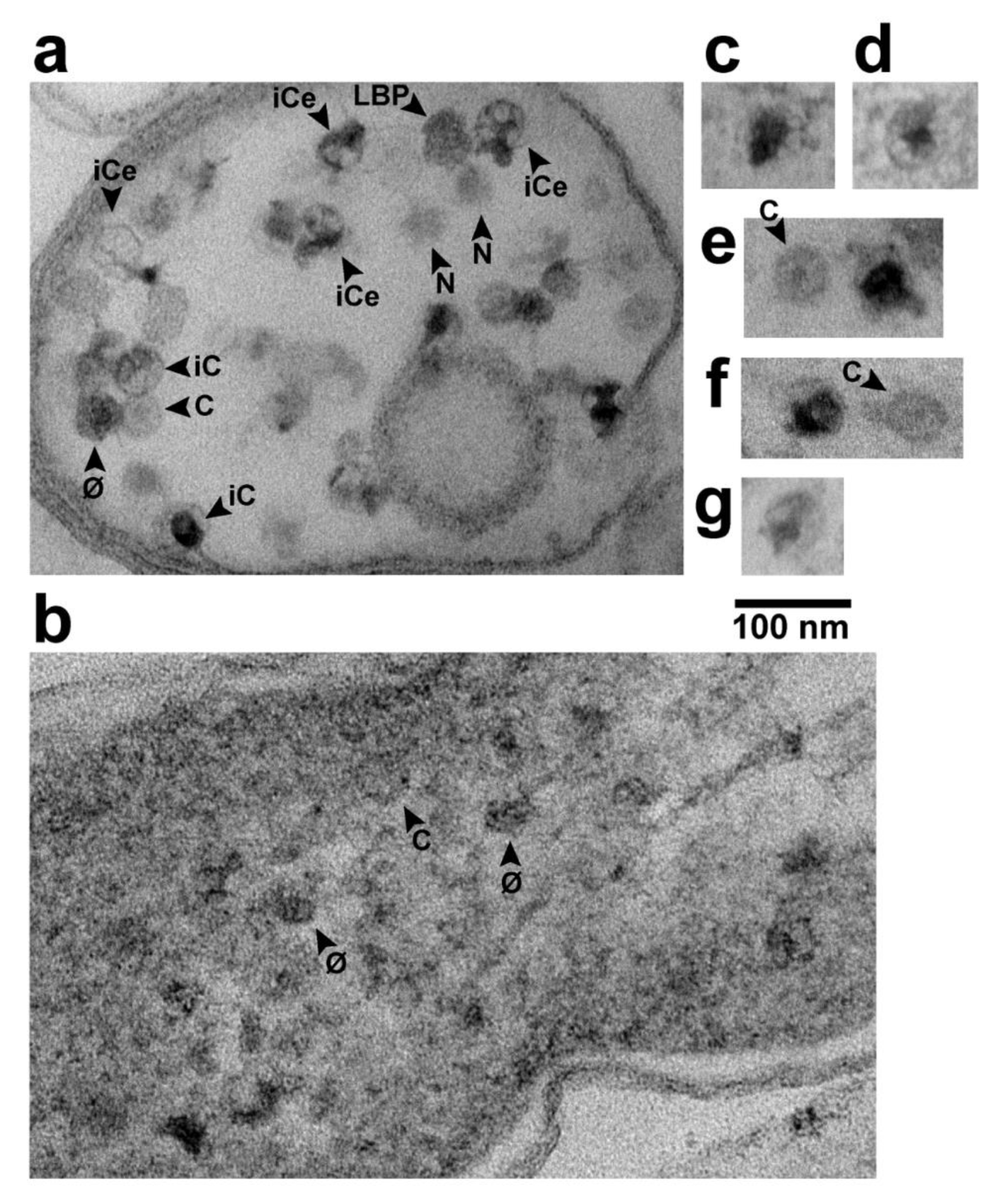
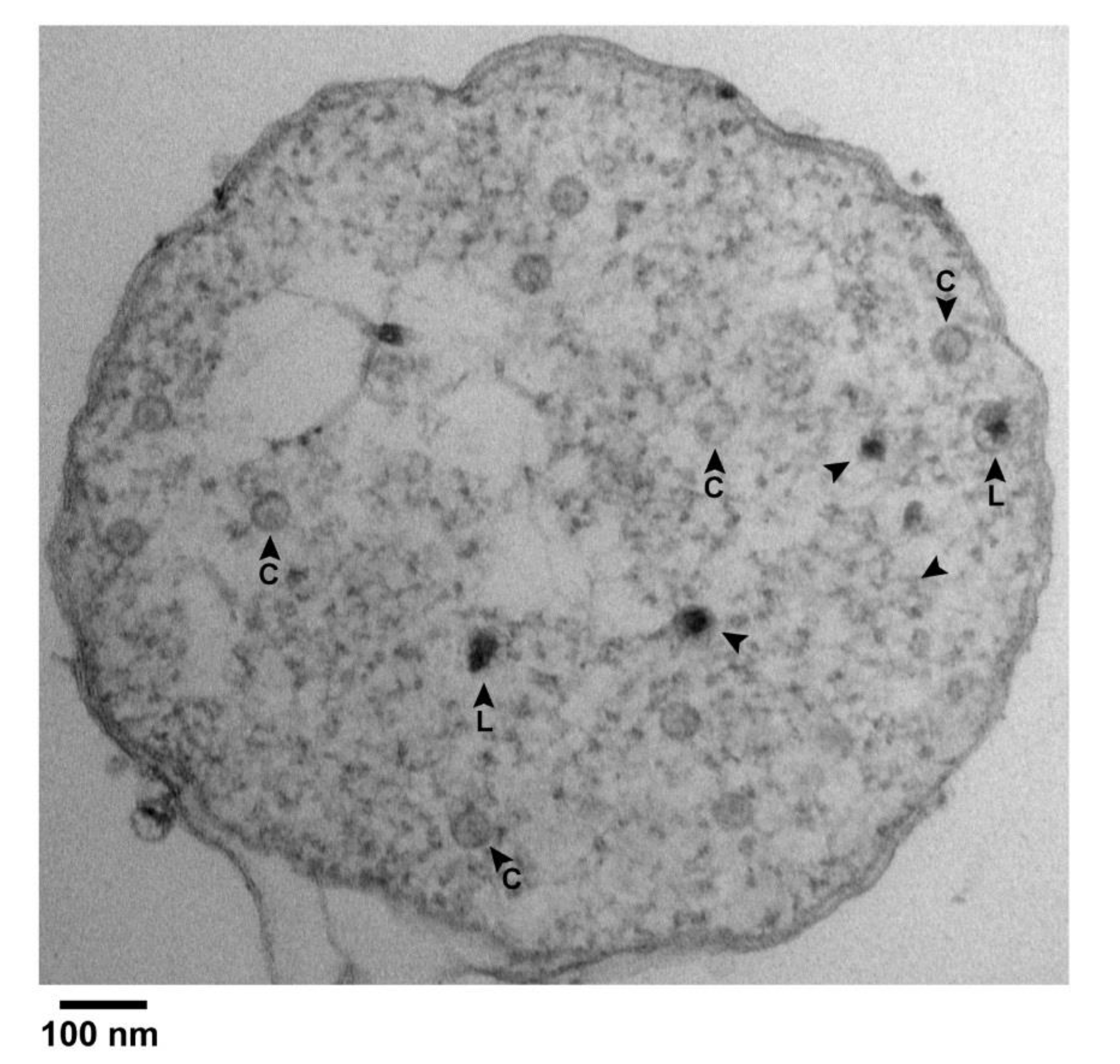
2.1. Large Particles
2.2. Phage-Sized Particles
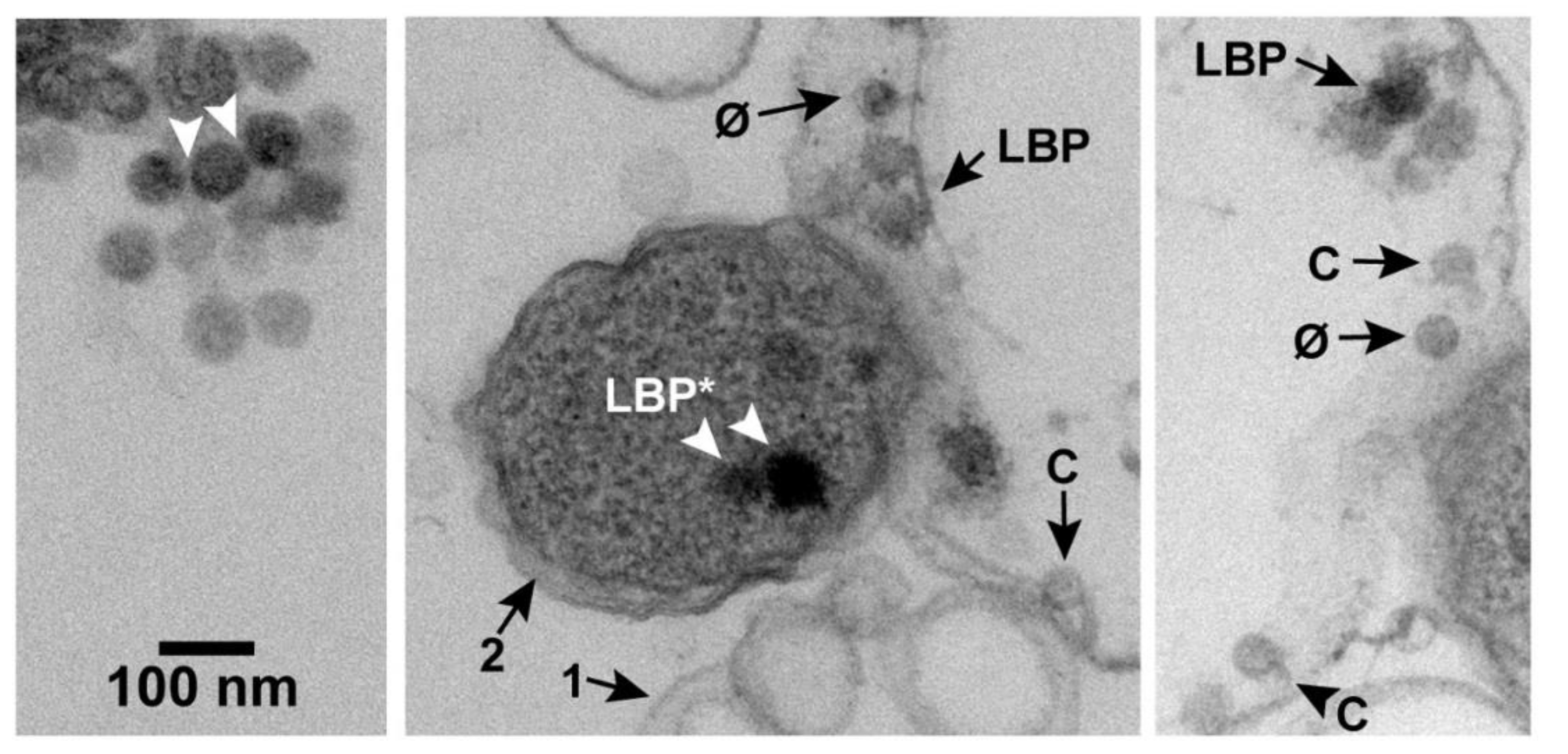
2.3. Intracellular LBPs
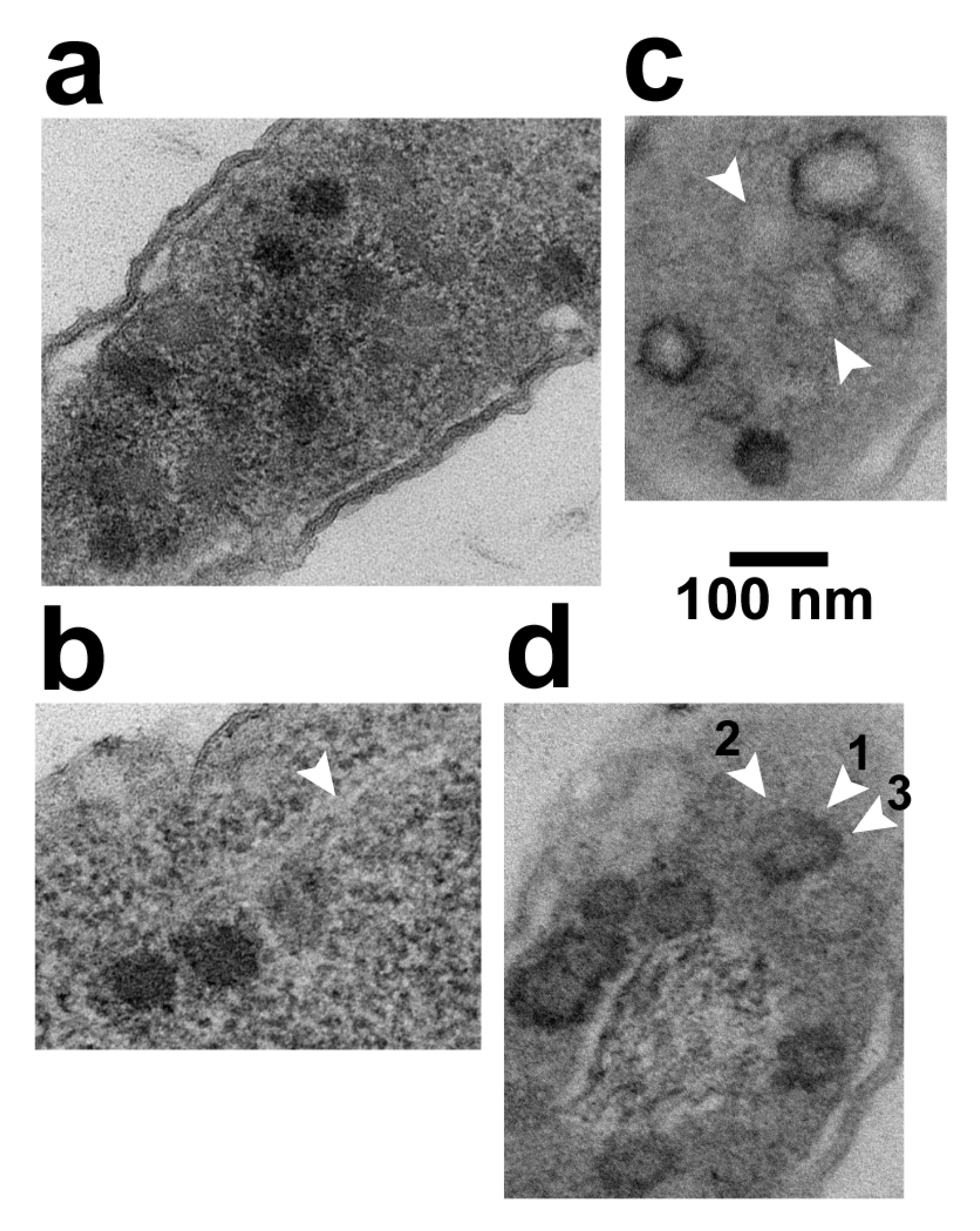
2.4. Strategy 1: LBP* Shells in Specimens of the Previous Section
2.5. Strategy#2: A Proflavine-Induced Increase in N (LBP*) (Visualization of Single DNA Duplexes)
2.6. Strategy #2: Alternative Staining
2.7. Results for Liquid Culture
3. Discussion
3.1. Intracellular Hyper-Expanded Capsids: Proposed Conformation for gp10
3.2. Phage DNA Packaging
3.3. Potential Relationship to Neurodegenerative Disease Causation
4. Materials and Methods
4.1. Propagation of Phages: Termination, Fixation and Agarose-Embedding of Liquid Cultures
4.2. Thin Sectioning and Electron Microscopy (EM-TS)
4.3. Interpretation of Electron Micrographs
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Fujisawa, H.; Morita, M. Phage DNA packaging. Genes Cells 1997, 2, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Casjens, S.R. The DNA-packaging nanomotor of tailed bacteriophages. Nat. Rev. Microbiol. 2011, 9, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.B.; Feiss, M. The bacteriophage DNA packaging motor. Annu. Rev. Genet. 2008, 42, 647–681. [Google Scholar] [CrossRef] [PubMed]
- Serwer, P.; Jiang, W. Dualities in the analysis of phage DNA packaging motors. Bacteriophage 2012, 2, 239–255. [Google Scholar] [CrossRef] [Green Version]
- Sharp, K.A.; Lu, X.J.; Cingolani, G.; Harvey, S.C. DNA conformational changes play a force-generating role during bacteriophage genome packaging. Biophys. J. 2019, 116, 2172–2180. [Google Scholar] [CrossRef]
- Pajunen, M.I.; Elizondo, M.R.; Skurnik, M.; Kieleczawa, J.; Molineux, I.J. Complete nucleotide sequence and likely recombinatorial origin of bacteriophage T3. J. Mol. Biol. 2002, 319, 1115–1132. [Google Scholar] [CrossRef]
- Baker, M.L.; Jiang, W.; Rixon, F.J.; Chiu, W. Common ancestry of herpesviruses and tailed DNA bacteriophages. J. Virol. 2005, 79, 14967–14970. [Google Scholar] [CrossRef] [Green Version]
- Cardone, G.; Winkler, D.C.; Trus, B.L.; Cheng, N.; Heuser, J.E.; Newcomb, W.W.; Brown, J.C.; Steven, A.C. Visualization of the herpes simplex virus portal in situ by cryo-electron tomography. Virology 2007, 361, 426–434. [Google Scholar] [CrossRef] [Green Version]
- McElwee, M.; Vijayakrishnan, S.; Rixon, F.; Bhella, D. Structure of the herpes simplex virus portal-vertex. PLoS Biol. 2018, 16, e2006191. [Google Scholar] [CrossRef] [Green Version]
- Thaljeh, L.F.; Rothschild, J.A.; Naderi, M.; Coghill, L.M.; Brown, J.M.; Brylinski, M. Hinge region in DNA packaging terminase pUL15 of herpes simplex birus: A potential allosteric target for antiviral drugs. Biomolecules 2019, 9, 603. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Yang, Q.; Wang, M.; Jia, R.; Chen, S.; Zhu, D.; Liu, M.; Wu, Y.; Zhao, X.; Zhang, S.; et al. Terminase large subunit provides a new drug target for herpesvirus treatment. Viruses 2019, 11, 219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serwer, P.; Wright, E.T. ATP-Driven contraction of phage T3 capsids with DNA incompletely packaged in vivo. Viruses 2017, 9, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serwer, P.; Wright, E.T. Nanomedicine and phage capsids. Viruses 2018, 10, 307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harper, D.R.; Parracho, H.M.R.T.; Walker, J.; Sharp, R.; Hughes, G.; Werthén, M.; Lehman, S.; Morales, S. Bacteriophages and biofilms. Antibiotics (Basel) 2014, 3, 270–284. [Google Scholar] [CrossRef]
- Simmons, M.; Drescher, K.; Nadell, C.D.; Bucci, V. Phage mobility is a core determinant of phage-bacteria coexistence in biofilms. ISME J. 2018, 12, 531–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serwer, P.; Hunter, B.; Wright, E.T. Cell-gel interactions of in-gel propagating bacteria. BMC Res. Notes 2018, 11, 699. [Google Scholar] [CrossRef] [Green Version]
- Lickfield, K.G.; Menge, B.; Hohn, B.; Hohn, T. Morphogenesis of bacteriophage lambda: Electron microscopy of thin sections. J. Mol. Biol. 1976, 103, 299–318. [Google Scholar] [CrossRef]
- Kellenberger, E.; Eiserling, F.A.; Boy de la Tour, E. Studies on the morphopoiesis of the head of phage T-even. 3. The cores of head-related structures. J. Ultrastruct. Res. 1967, 21, 335–360. [Google Scholar] [CrossRef]
- Kuhn, A.; Moncany, M.L.; Kellenberger, E.; Hausmann, R. Involvement of the bacterial groM gene product in bacteriophage T7 reproduction. I. Arrest at the level of DNA packaging. J. Virol. 1982, 41, 657–673. [Google Scholar] [CrossRef] [Green Version]
- Karska-Wysocki, B.; Zollinger, M.; Mamet-Bradley, M.D. Characterization of morphogenetic intermediates and progeny of normal and alkylated bacteriophage T7. Virology 1987, 157, 285–297. [Google Scholar] [CrossRef]
- Lickfeld, K.G.; Menge, B.; Wunderli, H.; van den Broek, J.; Kellenberger, E. The interpretation and quantitation of sliced intracellular bacteriophages and phage-related particles. J. Ultrastruct. Res. 1977, 60, 148–168. [Google Scholar] [CrossRef]
- Guo, F.; Liu, Z.; Fang, P.A.; Zhang, Q.; Wright, E.T.; Wu, W.; Zhang, C.; Vago, F.; Ren, Y.; Jakana, J.; et al. Capsid expansion mechanism of bacteriophage T7 revealed by multistate atomic models derived from cryo-EM reconstructions. Proc. Natl. Acad. Sci. USA 2014, 111, E4606–E4614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serwer, P. Flattening and shrinkage of bacteriophage T7 after preparation for electron microscopy by negative staining. J. Ultrastruct. Res. 1977, 58, 235–243. [Google Scholar] [CrossRef]
- Serwer, P.; Hayes, S.J.; Watson, R.H. Conformation of DNA packaged in bacteriophage T7. Analysis by use of ultraviolet light-induced DNA-capsid cross-linking. J. Mol. Biol. 1992, 223, 999–1011. [Google Scholar] [CrossRef]
- Meyers, C.G.; Pettitt, B.M. Phage-like packing structures with mean field sequence dependence. J. Comput. Chem. 2017, 38, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Serwer, P.; Watson, R.H.; Hayes, S.J. Heterogeneity of the procapsid of bacteriophage T3. J. Virol. 1985, 55, 232–237. [Google Scholar] [CrossRef] [Green Version]
- Serwer, P.; Watson, R.H.; Hayes, S.J. Formation of the right before the left mature DNA end during packaging-cleavage of bacteriophage T7 DNA concatemers. J. Mol. Biol. 1992, 226, 311–317. [Google Scholar] [CrossRef]
- Silverman, L.; Glick, D. The reactivity and staining of tissue proteins with phosphotungstic acid. J. Cell Biol. 1969, 40, 761–767. [Google Scholar] [CrossRef] [Green Version]
- Pauling, L.; Corey, R.B. The pleated sheet, a new layer configuration of polypeptide chains. Proc. Natl. Acad. Sci. USA 1951, 37, 251–256. [Google Scholar] [CrossRef] [Green Version]
- Milner-White, E.J.; Russell, M.J. Predicting the conformations of peptides and proteins in early evolution. A review article submitted to Biology. Direct. Biol. Direct 2008, 3, 3. [Google Scholar]
- Daggett, V. Alpha-sheet: The toxic conformer in amyloid diseases? Acc. Chem. Res. 2006, 39, 594–602. [Google Scholar] [CrossRef]
- Serwer, P.; Wright, E.T. Testing a proposed paradigm shift in analysis of phage DNA packaging. Bacteriophage 2017, 6, e1268664. [Google Scholar] [CrossRef] [Green Version]
- Rao, V.B.; Black, L.W. Evidence that a T4 DNA packaging enzyme is a processed form of the major capsid gene product. Cell 1985, 42, 967–977. [Google Scholar] [CrossRef]
- Hamada, K.; Fujisawa, H.; Minagawa, T. Characterization of ATPase activity of a defined in vitro system for packaging of bacteriophage T3 DNA. Virology 1987, 159, 244–249. [Google Scholar] [CrossRef]
- Blobel, G. Christian de Duve (1917–2013). Nature 2013, 498, 300. [Google Scholar] [CrossRef] [Green Version]
- Zorca, S.M.; Zorca, C.E. The legacy of a founding father of modern cell biology: George Emil Palade (1912–2008). Yale J. Biol. Med. 2011, 84, 113–116. [Google Scholar]
- Parry, B.R.; Surovtsev, I.V.; Cabeen, M.T.; O’Hern, C.S.; Dufresne, E.R.; Jacobs-Wagner, C. The bacterial cytoplasm has glass-like properties and is fluidized by metabolic activity. Cell 2014, 156, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Sheats, J.; Cicuta, P.; Sclavi, B.; Cosentino Lagomarsino, M.; Dorfman, K.D. Subdiffusion of loci and cytoplasmic particles are different in compressed Escherichia coli cells. Commun. Biol. 2018, 1, 176. [Google Scholar] [CrossRef] [Green Version]
- Koch, A.L. Shrinkage of growing Escherichia coli cells by osmotic challenge. J. Bacteriol. 1984, 159, 919–924. [Google Scholar] [CrossRef] [Green Version]
- Scheie, P. Osmotic pressure in Escherichia coli as rendered detectable by lysozyme attack. J. Bact. 1973, 114, 549–555. [Google Scholar] [CrossRef] [Green Version]
- Itzhaki, R.F. Corroboration of a major role for herpes simplex virus type 1 in Alzheimer’s disease. Front. Aging Neurosci. 2018, 10, 324. [Google Scholar] [CrossRef] [Green Version]
- Eimer, W.A.; Vijaya Kumar, D.K.; Navalpur Shanmugam, N.K.; Rodriguez, A.S.; Mitchell, T.; Washicosky, K.J.; György, B.; Breakefield, X.O.; Tanzi, R.E.; Moir, R.D. Alzheimer’s disease-associated β-amyloid is rapidly seeded by herpesviridae to protect against brain infection. Neuron 2018, 99, 56–63. [Google Scholar] [CrossRef] [Green Version]
- Watts, J.C.; Prusiner, S.B. Aβ prions and the pathobiology of Alzheimer’s disease. Cold Spring Harb. Perspect. Med. 2018, 8, a023507. [Google Scholar] [CrossRef] [Green Version]
- Jucker, M.; Walker, L.C. Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1341–1349. [Google Scholar] [CrossRef]
- Shea, D.; Hsu, C.-C.; Bi, T.M.; Paranjapye, N.; Childers, M.C.; Cochran, J.; Colson, P.; Tomberlin, C.P.; Wang, L.; Paris, D.; et al. α-Sheet secondary structure in amyloid β-peptide drives aggregation and toxicity in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2019, 116, 8895–8900. [Google Scholar] [CrossRef] [Green Version]
- Bandea, C.I. Aβ, tau, α-synuclein, huntingtin, TDP-43, PrP and AA are members of the innate immune system: A unifying hypothesis on the etiology of AD, PD, HD, ALS, CJD and RSA as innate immunity disorders. bioRχiv 2013. Available online: http://biorxiv.org/content/early/2013/11/18/000604 (accessed on 23 September 2019). [CrossRef] [Green Version]
- Serwer, P. Hypothesis for the cause and therapy of neurodegenerative diseases. Med. Hypotheses 2018, 110, 60–63. [Google Scholar] [CrossRef]
- Itzhaki, R.F.; Lin, W.R.; Shang, D.; Wilcock, G.K.; Faragher, B.; Jamieson, G.A. Herpes simplex virus type 1 in brain and risk of Alzheimer’s disease. Lancet 1997, 349, 241–244. [Google Scholar] [CrossRef]
- Salloway, S.; Gur, T.; Berzin, T.; Tavares, R.; Zipser, B.; Correia, S.; Hovanesian, V.; Fallon, J.; Kuo-Leblanc, V.; Glass, D.; et al. Effect of APOE genotype on microvascular basement membrane in Alzheimer’s disease. J. Neurol. Sci. 2001, 203–204, 183–187. [Google Scholar] [CrossRef]
- Adams, M.H. Bacteriophages; Interscience Publishers, Inc.: New York, NY, USA, 1959. [Google Scholar]
- Glauert, A. Fixation, Dehydration and Embedding of Biological Specimens; North-Holland Publishing Co.: New York, NY, USA, 1975. [Google Scholar]
- Peachey, L.D. Thin sections. I. A study of section thickness and physical distortion produced during microtomy. J. Biophys. Biochem. Cytol. 1958, 4, 233–242. [Google Scholar] [CrossRef]
- Reynolds, E.S. The use of lead citrate at high pH as an electron-opaque stain in electron microscopy. J. Cell Biol. 1963, 17, 208–212. [Google Scholar] [CrossRef] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serwer, P.; Hunter, B.; Wright, E.T. Electron Microscopy of In-Plaque Phage T3 Assembly: Proposed Analogs of Neurodegenerative Disease Triggers. Pharmaceuticals 2020, 13, 18. https://doi.org/10.3390/ph13010018
Serwer P, Hunter B, Wright ET. Electron Microscopy of In-Plaque Phage T3 Assembly: Proposed Analogs of Neurodegenerative Disease Triggers. Pharmaceuticals. 2020; 13(1):18. https://doi.org/10.3390/ph13010018
Chicago/Turabian StyleSerwer, Philip, Barbara Hunter, and Elena T. Wright. 2020. "Electron Microscopy of In-Plaque Phage T3 Assembly: Proposed Analogs of Neurodegenerative Disease Triggers" Pharmaceuticals 13, no. 1: 18. https://doi.org/10.3390/ph13010018
APA StyleSerwer, P., Hunter, B., & Wright, E. T. (2020). Electron Microscopy of In-Plaque Phage T3 Assembly: Proposed Analogs of Neurodegenerative Disease Triggers. Pharmaceuticals, 13(1), 18. https://doi.org/10.3390/ph13010018