Bornyl Derivatives of p-(Benzyloxy)Phenylpropionic Acid: In Vivo Evaluation of Antidiabetic Activity

 , ,
, ,  , ,
, ,
Abstract
:
1. Introduction
2. Results
2.1. Compound Selection
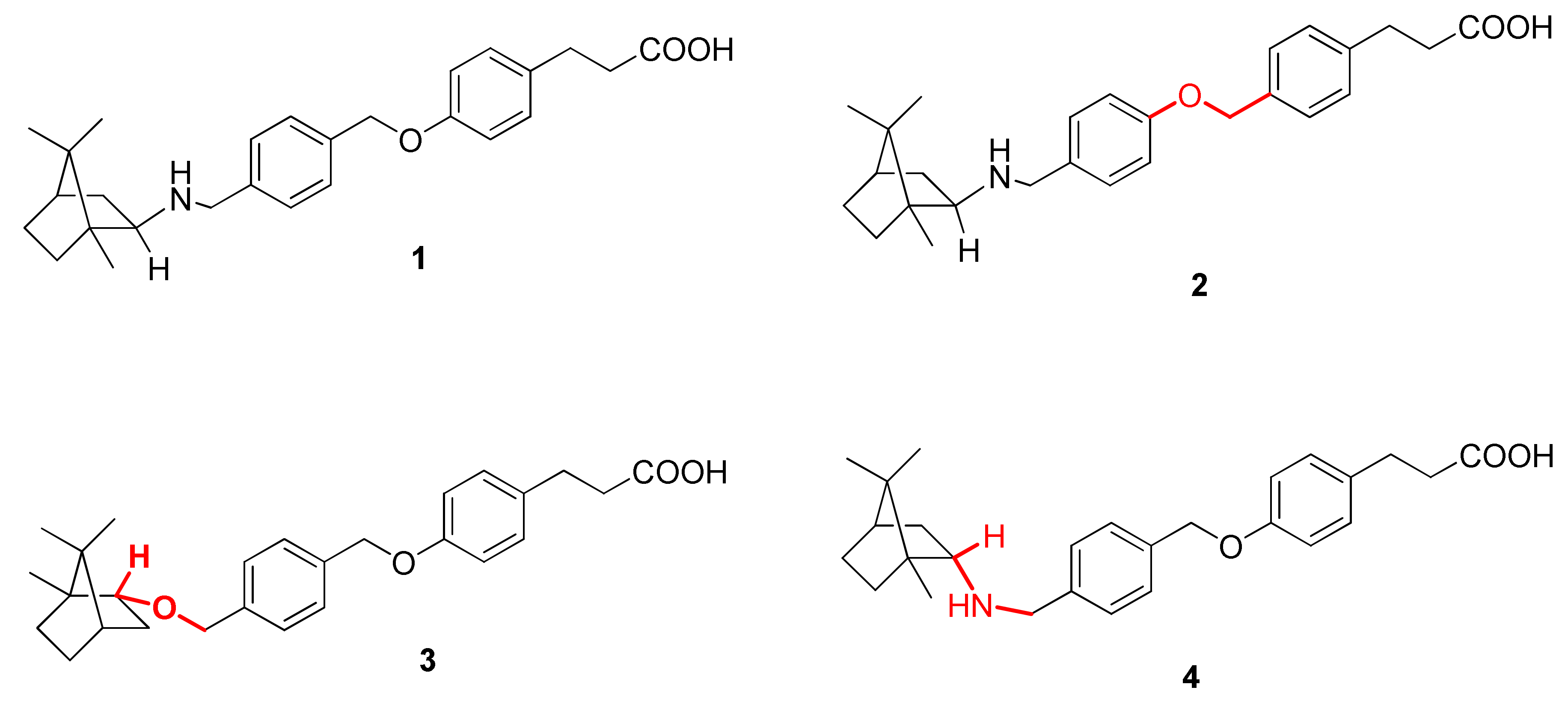
2.2. Synthesis
2.3. In Silico Analysis
- 1 if Indki > mean (Indki), 0 otherwise;
- 1 if ΔEki < mean (ΔEki), 0 otherwise.
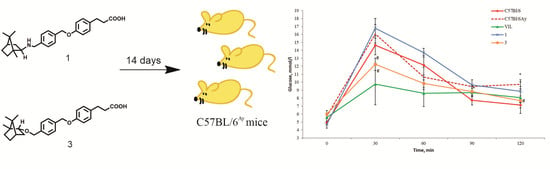
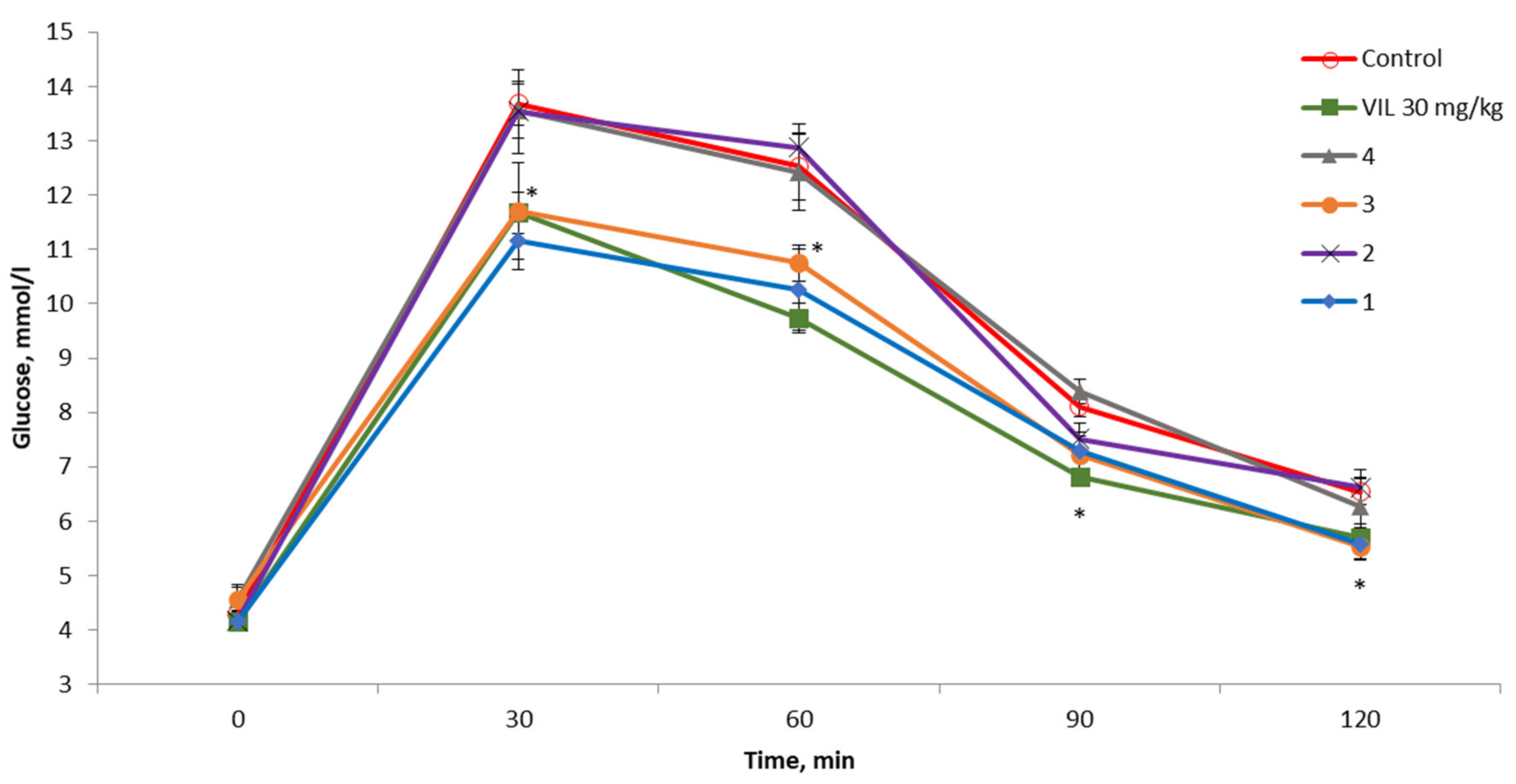
2.4. The OGTT in C57BL/6 Mice
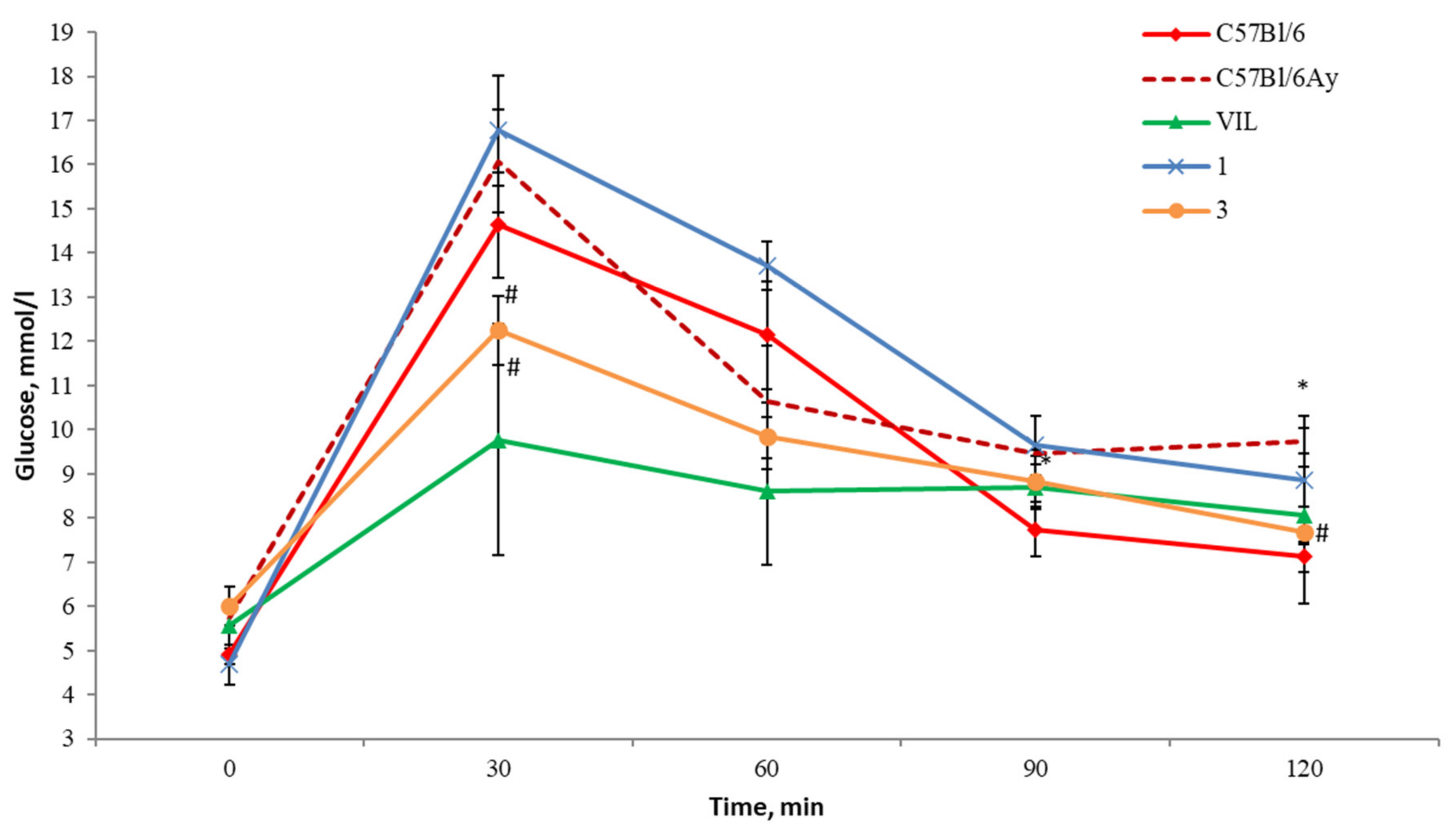
2.5. The OGTT in AY Mice
2.6. A Biochemical Blood Assay in Treated AY Mice
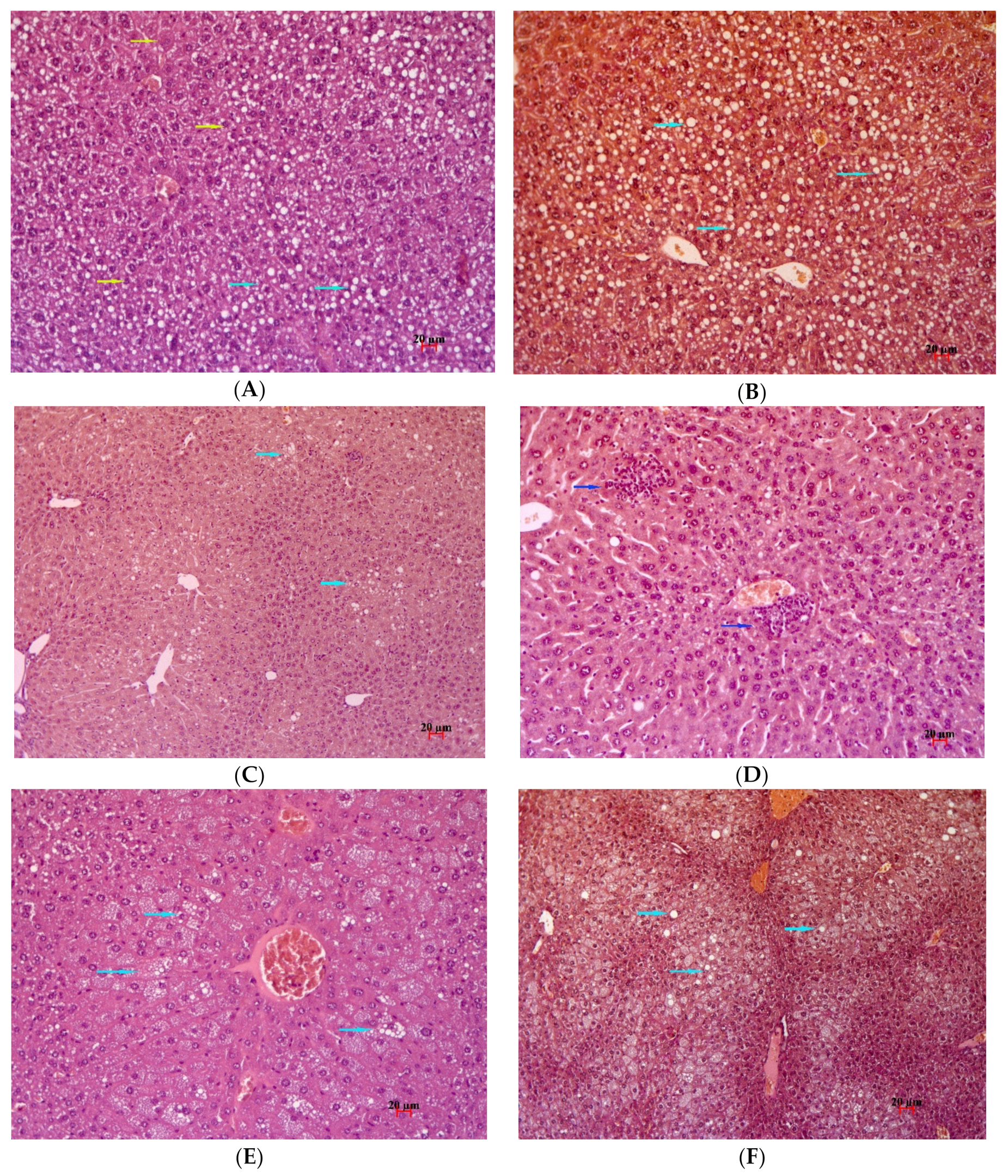
2.7. Histological Examination
3. Discussion
- –
- For PPARD, the highest Ind value corresponds to the highest docking energy ΔE;
- –
- For FFAR1 also, the highest value of the Ind estimate corresponds to the highest docking energy ΔE;
- –
- For DPP4, the lowest value of the Ind estimate corresponds to the minimum docking energy ΔE.
4. Materials and Methods
4.1. Chemistry
4.1.1. General Procedure for Hydrolysis
4.1.2. 3-(4-((4-(((1R,2R,4R)-1,7,7-Trimethylbicyclo[2.2.1]heptan-2-ylamino)methyl)- phenoxy)methyl)phenyl)propanoic acid hydrochloride 2
4.1.3. 3-(4-(4-(((1S,2R,4R)-1,7,7-Trimethylbicyclo[2.2.1]heptan-2-yloxy)methyl)- benzyloxy)phenyl)propanoic acid 3
4.1.4. 3-(4-(4-(((1R,2S,4R)-1,7,7-Trimethylbicyclo[2.2.1]heptan-2-ylamino)methyl)- benzyloxy)phenyl)propanoic acid hydrochloride 4
4.1.5. Methyl 3-(4-(4-(((1R,2S,4R)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-ylamino)- methyl)phenethyl)phenyl)propanoate 7
4.1.6. Methyl 3-(4-(4-(((1S,2R,4R)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-yloxy)methyl)- benzyloxy)phenyl)Propanoate 9
4.1.7. 4-(4-(Bromomethyl)benzyloxy)benzaldehyde 12
4.1.8. Diethyl 2-(4-((4-Formylphenoxy)methyl)benzyl)malonate 13
4.1.9. Ethyl 3-(4-((4-Formylphenoxy)methyl)phenyl)propanoate 14
4.1.10. Ethyl 3-(4-((4-(((1R,2R,4R)-1,7,7-Trimethylbicyclo[2.2.1]heptan-2-ylamino)methyl)- phenoxy)methyl)phenyl)propanoate 15
4.2. Biological Experiments
4.2.1. Animals
4.2.2. The OGTT
4.2.3. The Design of the Experiment on AY Mice
4.2.4. Biochemical Assays
4.2.5. Histological Examination
4.3. Computational Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Roglic, G. WHO Global report on diabetes: A summary. Int. J. Non-Commun. Dis. 2016, 1, 3–8. [Google Scholar] [CrossRef]
- Sonksen, P.; Sonksen, J. Insulin: Understanding its action in health and disease. Br. J. Anaesth. 2000, 85, 69–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabish, S.A. Is Diabetes Becoming the Biggest Epidemic of the Twenty-first Century? Int. J. Health Sci. 2007, 1, V–VIII. [Google Scholar]
- Riddle, M.C.; Herman, W.H. The Cost of Diabetes Care—An Elephant in the Room. Diabetes Care 2018, 45, 929–932. [Google Scholar] [CrossRef] [Green Version]
- Carbone, S.; Buono, M.G.D.; Ozemek, C.; Lavie, C.J. Obesity, risk of diabetes and role of physical activity, exercise training and cardiorespiratory fitness. Prog. Cardiovasc. Dis. 2019, 62, 327–333. [Google Scholar] [CrossRef]
- American Diabetes Association. 9. Pharmacologic approaches to glycemic treatment: Standards of medical care in diabetes-2019. Diabetes Care 2019, 42, 90–102. [Google Scholar] [CrossRef] [Green Version]
- Vieira, R.; Souto, S.B.; Sánchez-López, E.; Machado, A.L.; Severino, P.; Jose, S.; Santini, A.; Fortuna, A.; García, M.L.; Silva, A.M.; et al. Sugar-Lowering Drugs for Type 2 Diabetes Mellitus and Metabolic Syndrome—Review of Classical and New Compounds: Part-I. Pharmaceuticals 2019, 12, 152. [Google Scholar] [CrossRef] [Green Version]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Habtemariam, S. Antidiabetic Potential of Monoterpenes: A Case of Small Molecules Punching above Their Weight. Int. J. Mol. Sci. 2017, 19, 4. [Google Scholar] [CrossRef] [Green Version]
- Kodikonda, M.; Naik, P.R. Ameliorative effect of borneol, a natural bycyclic monoterpene against hyperglycemia, hyperlipidemia and oxidative stress in streptozotocin-induced diabetic Wistar rats. Biomed. Pharmacother. 2017, 96, 336–347. [Google Scholar]
- Rogachev, A.D.; Kuranov, S.O.; Salakhutdinov, N.F. Chemical approach to the design of effective antidiabetic agents. Russ. Chem. Rev. 2016, 85, 1313–1337. [Google Scholar] [CrossRef]
- Sparks, S.M.; Aquino, C.; Banker, P.; Collins, J.L.; Cowan, D.; Diaz, C.; Dock, S.T.; Hertzog, D.L.; Liang, X.; Swiger, E.D.; et al. Exploration of phenylpropanoic acids as agonists of the free fatty acid receptor 4 (FFA4): Identification of an orally efficacious FFA4 agonist. Bioorg. Med. Chem. Lett. 2017, 27, 1278–1283. [Google Scholar] [CrossRef] [PubMed]
- Hiroyuki, M.; Hashimoto, Y. Structural development studies of subtype-selective ligands for peroxisome proliferator-activated receptors (PPARs) based on the 3,4-disubstituted phenylpropanoic acid scaffold as a versatile template. PPAR Res. 2008, 689859. [Google Scholar]
- Tang, Y.-B.; Liu, J.-Z.; Zhang, S.-E.; Du, X.; Nie, F.; Tian, J.-Y.; Ye, F.; Huang, K.; Hu, J.-P.; Li, Y.; et al. 3-Phenylpropanoic acid-based phosphotyrosine (pTyr) mimetics: Hit evolution to a novel orally active protein tyrosine phosphatase 1B (PTP1B) inhibitor. ChemMedChem 2014, 9, 918–921. [Google Scholar] [CrossRef]
- Kuranov, S.O.; Luzina, O.A.; Onopchenko, O.; Pishel, I.; Zozulya, S.; Gureev, M.; Salakhutdinov, N.F.; Krasavin, M. Exploring bulky natural and natural-like periphery in the design of p-(benzyloxy)phenylpropionic acid agonists of free fatty acid receptor 1 (GPR40). Bioorg. Chem. 2020, 99, 103830. [Google Scholar] [CrossRef] [PubMed]
- Levien, T.L.; Baker, D.E. New Drugs in Development for the Treatment of Diabetes. Diabetes Spectrum 2009, 22, 92–106. [Google Scholar] [CrossRef] [Green Version]
- Croasdell, G. American Diabetes association—70th scientific sessions—Research on novel therapeutics. Drugs 2010, 13, 588. [Google Scholar]
- Tyurenkov, I.N.; Kurkin, D.V.; Volotova, E.V.; Bakulin, D.A.; Lomkina, E.M. Drug discovery for type 2 diabetes mellitus and metabolic syndrome: Ten novel biological targets. Diabetes Mellit. 2015, 18, 101–109. [Google Scholar] [CrossRef] [Green Version]
- Bateman, A. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, 506–515. [Google Scholar]
- Vassiliev, P.M.; Spasov, A.A.; Kosolapov, V.A.; Kucheryavenko, A.F.; Gurova, N.A.; Anisimova, V.A. Consensus Drug Design Using IT Microcosm. In Application of Computational Techniques in Pharmacy and Medicine; Gorb, L., Kuzmin, V., Muratov, E., Eds.; Springer: Dordrecht, The Netherlands, 2014; Volume 17, pp. 369–431. [Google Scholar]
- Vasilyev, P.M.; Luzina, O.A.; Babkov, D.A.; Appazova, D.T.; Salakhutdinov, N.F.; Spasov, A.A. Studying dependences between the chemotype structure of some natural compounds and the spectrum of their targeted activities correlated with the hypoglycemic effect. J. Struct. Chem. 2019, 60, 1827–1832. [Google Scholar] [CrossRef]
- Kubinyi, H. Similarity and Dissimilarity: A Medicinal Chemist’s View. In 3D QSAR in Drug Design. Three-Dimensional Quantitative Structure Activity Relationships; Kubinyi, H., Folkers, G., Martin, Y.C., Eds.; Springer: Dordrecht, The Netherlands, 2002; Volume 2. [Google Scholar]
- MarvinSketch, ChemAxon Kft. Available online: http://www.chemaxon.com/products/marvin/marvinsketch/ (accessed on 17 November 2020).
- MOPAC. Stewart Computational Chemistry. Available online: http://openmopac.net (accessed on 17 November 2020).
- Vassiliev, P.M.; Spasov, A.A.; Yanaliyeva, L.R.; Kochetkov, A.N.; Vorfolomeyeva, V.V.; Klochkov, V.G.; Appazova, D.T. Neural network modeling of multitarget RAGE inhibitory activity. Biochem. Suppl. Ser. B Biomed. Chem. 2019, 13, 256–263. [Google Scholar]
- RCSB PDB, Protein Data Bank. Available online: https://www.rcsb.org/ (accessed on 17 November 2020).
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comp. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piskunova, Y.V.; Kazantceva, A.Y.; Baklanov, A.V.; Bazhan, N.M. Mutation yellow in agouti loci prevents age-related increase of skeletal muscle genes regulating free fatty acids oxidation. Vavilov J. Genet. Breed. 2018, 22, 265–272. [Google Scholar] [CrossRef]
- Oh, D.Y.; Olefsky, J.M. G protein-coupled receptors as targets for anti-diabetic therapeutics. Nat. Rev. Drug Discov. 2016, 15, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Raab, S.; Wang, H.; Uhles, S.; Cole, N.; Alvarez-Sanchez, R.; Künnecke, B.; Ullmer, C.; Matile, H.; Bedoucha, M.; Norcross, R.D.; et al. Incretin-like effects of small molecule trace amine-associated receptor 1 agonists. Mol. Metab. 2015, 5, 47–56. [Google Scholar] [CrossRef]
- Ayala, J.E.; Samuel, V.T.; Morton, G.J.; Obici, S.; Croniger, C.M.; Shulman, G.I.; Wasserman, D.H.; McGuinness, O.P.; NIH Mouse Metabolic Phenotyping Center Consortium. Standard operating procedures for describing and performing metabolic tests of glucose homeostasis in mice. Dis. Model. Mech. 2010, 3, 525–534. [Google Scholar] [CrossRef] [Green Version]
- Lambeir, A.M.; Durinx, C.; Scharpé, S.; Meester, I.D. Dipeptidyl-Peptidase IV from Bench to Bedside: An Update on Structural Properties, Functions, and Clinical Aspects of the Enzyme DPP IV. Crit. Rev. Clin. Lab. Sci. 2003, 40, 209–294. [Google Scholar] [CrossRef]
- Burkey, B.F.; Li, X.; Bolognese, L.; Balkan, B.; Mone, M.; Russell, M.; Hughes, T.E.; Wang, P.R. Acute and Chronic Effects of the Incretin Enhancer Vildagliptin in Insulin-Resistant Rats. J. Pharmacol. Exp. Ther. 2005, 315, 688–695. [Google Scholar] [CrossRef] [Green Version]
- Carman, R.; Greenfield, K. The endo—And exo 1,7,7-Trimethylbicyclo[2.2.1]heptan- 2-amines (Bornan-2-amines) and their acetamides. Aust. J. Chem. 1984, 37, 1785–1790. [Google Scholar] [CrossRef]
- Tai, M.M. A Mathematical Model for the Determination of Total Area under Glucose Tolerance and Other Metabolic Curves. Diabetes Care 1994, 17, 152–154. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Name 2 | Protein Symbol 3 | UniProt 4 | Ligand 5 |
|---|---|---|---|
| Cleavage of poly- and oligosaccharides | |||
| Pancreatic alpha-amylase | AMY2A | P04746 | inhibitor |
| Maltase-glucoamylase, intestinal | MGAM | O43451 | inhibitor |
| Insulinogens | |||
| Sulfonylurea receptor 1 | SUR1 | Q09428 | inhibitor |
| Incretin mimetics | |||
| Dipeptidyl peptidase 4 | DPP4 | P27487 | inhibitor |
| Gastric inhibitory polypeptide receptor | GIPR | P48546 | agonist |
| Glucagon-like peptide receptor 1 | GLP1R | P43220 | agonist |
| Trace amine-associated receptor 1 | TAAR1 | Q96RJ0 | agonist |
| Insulin resistance | |||
| Free fatty acid receptor 1 (GPR40) | FFAR1 | O14842 | agonist |
| Free fatty acid receptor 2 (GPR43) | FFAR2 | O15552 | agonist |
| Free fatty acid receptor 3 (GPR41) | FFAR3 | O14843 | agonist |
| Free fatty acid receptor 4 (GPR120) | FFAR4 | Q5NUL3 | agonist |
| Peroxisome proliferator-activated receptor alpha | PPARA | Q07869 | agonist |
| Peroxisome proliferator-activated receptor delta | PPARD | Q03181 | agonist |
| Peroxisome proliferator-activated receptor gamma | PPARG | P37231 | agonist |
| Protein-tyrosine phosphatase 1B (PTP1B) | PTPN1 | P18031 | inhibitor |
| Gluconeogenesis and glycogenolysis | |||
| Glycogen phosphorylase, liver form | PYGL | P06737 | inhibitor |
| Glycogen phosphorylase, muscle form | PYGM | P11217 | inhibitor |
| Glucagon | GCG | P01275 | inhibitor |
| Glucagon receptor | GCGR | P47871 | inhibitor |
| Phosphoenolpyruvate carboxykinase, cytosolic | PCK1 | P35558 | inhibitor |
| Fructose-1,6-bisphosphatase 1 | FBP1 | P09467 | inhibitor |
| Glycolysis and glycogen synthesis | |||
| Pyruvate dehydrogenase kinase 4 | PDK4 | Q16654 | activator |
| 6-phosphofructokinase, muscle type | PFKM | P08237 | activator |
| Glucose transport | |||
| Sodium/glucose cotransporter 2 | SGLT2 | P31639 | inhibitor |
| Target Name | Protein Symbol | IndMean 1 | TMean 2 |
|---|---|---|---|
| Peroxisome proliferator-activated receptor delta | PPARD | 2.93 | 0.624 |
| Free fatty acid receptor 1 (GPR40) | FFAR1 | 1.87 | 0.684 |
| Dipeptidyl peptidase 4 | DPP4 | 1.86 | 0.478 |
| Glycogen phosphorylase, liver form | PYGL | 1.70 | 0.460 |
| Protein-tyrosine phosphatase 1B (PTP1B) | PTPN1 | 1.59 | 0.418 |
| Free fatty acid receptor 4 (GPR120) | FFAR4 | 1.58 | 0.651 |
| Pancreatic alpha-amylase | AMY2A | 1.57 | 0.195 |
| 6-phosphofructokinase, muscle type | PFKM | 1.00 | 0.230 |
| Gastric inhibitory polypeptide receptor | GIPR | 0.35 | 0.426 |
| Peroxisome proliferator-activated receptor alpha | PPARA | 0.33 | 0.627 |
| Glucagon-like peptide receptor 1 | GLP1R | 0.00 | 0.181 |
| Pyruvate dehydrogenase kinase 4 | PDK4 | 0.00 | 0.145 |
| Trace amine-associated receptor 1 | TAAR1 | −0.18 | 0.441 |
| Maltase-glucoamylase, intestinal | MGAM | −0.25 | 0.118 |
| Peroxisome proliferator-activated receptor gamma | PPARG | −0.28 | 0.586 |
| Glycogen phosphorylase, muscle form | PYGM | −0.33 | 0.381 |
| Sulfonylurea receptor 1 | SUR1 | −0.50 | 0.479 |
| Free fatty acid receptor 3 (GPR41) | FFAR3 | −0.75 | 0.194 |
| Glucagon receptor | GCGR | −0.76 | 0.430 |
| Sodium/glucose cotransporter 2 | SGLT2 | −1.31 | 0.491 |
| Fructose-1,6-bisphosphatase 1 | FBP1 | −1.45 | 0.384 |
| Free fatty acid receptor 2 (GPR43) | FFAR2 | −1.45 | 0.459 |
| Glucagon | GCG | −1.65 | 0.486 |
| Phosphoenolpyruvate carboxykinase, cytosolic | PCK1 | −2.50 | 0.228 |
| Target Name | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Peroxisome proliferator-activated receptor delta | 2.70 | 2.50 | 3.80 | 2.70 |
| Free fatty acid receptor 1 (GPR40) | 1.40 | 1.60 | 3.08 | 1.40 |
| Dipeptidyl peptidase 4 | 2.00 | 2.54 | 0.89 | 2.00 |
| Glycogen phosphorylase, liver form | 1.70 | 1.70 | 1.70 | 1.70 |
| Protein-tyrosine phosphatase 1B (PTP1B) | 1.79 | 1.71 | 1.08 | 1.79 |
| Free fatty acid receptor 4 (GPR120) | 1.70 | 1.90 | 1.00 | 1.70 |
| Pancreatic alpha-amylase | 1.40 | 1.90 | 1.58 | 1.40 |
| 6-phosphofructokinase, muscle type | 1.00 | 1.00 | 1.00 | 1.00 |
| Gastric inhibitory polypeptide receptor | 0.32 | 0.51 | 0.27 | 0.32 |
| Peroxisome proliferator-activated receptor alpha | 0.20 | −1.10 | 2.00 | 0.20 |
| Glucagon-like peptide receptor 1 | 0.00 | 0.00 | 0.00 | 0.00 |
| Pyruvate dehydrogenase kinase 4 | 0.00 | 0.00 | 0.00 | 0.00 |
| Trace amine-associated receptor 1 | −0.90 | −1.10 | 2.20 | −0.90 |
| Maltase-glucoamylase, intestinal | −0.20 | −0.20 | −0.40 | −0.20 |
| Peroxisome proliferator-activated receptor gamma | −0.90 | −0.90 | 1.60 | −0.90 |
| Glycogen phosphorylase, muscle form | −1.10 | −0.80 | 1.70 | −1.10 |
| Sulfonylurea receptor 1 | −0.50 | −0.50 | −0.50 | −0.50 |
| Free fatty acid receptor 3 (GPR41) | −0.75 | −0.75 | −0.75 | −0.75 |
| Glucagon receptor | −0.83 | −0.66 | −0.70 | −0.83 |
| Sodium/glucose cotransporter 2 | −1.90 | −2.10 | 0.65 | −1.90 |
| Fructose-1,6-bisphosphatase 1 | −1.60 | −0.70 | −1.90 | −1.60 |
| Free fatty acid receptor 2 (GPR43) | −2.70 | −2.20 | 1.80 | −2.70 |
| Glucagon | −1.73 | −1.73 | −1.40 | −1.73 |
| Phosphoenolpyruvate carboxykinase, cytosolic | −2.50 | −2.50 | −2.50 | −2.50 |
| Total sum of indices of activity levels | −1.40 | 0.12 | 16.21 | −1.40 |
| Sum of significant indices of activity levels | 14.21 | 15.36 | 24.36 | 14.21 |
| Target | Compound | Ind 1 | ΔE 2 | BinInd 4 | BinΔE 5 |
|---|---|---|---|---|---|
| Peroxisome proliferator- activated receptor delta | 1 | 2.70 | −9.30 | 0 | 0 |
| 2 | 2.50 | −9.40 | 0 | 0 | |
| 3 | 3.80 | −9.80 | 1 | 1 | |
| 4 | 2.70 | −9.70 | 0 | 1 | |
| Mean 3 | 2.93 | −9.55 | |||
| Free fatty acid receptor 1 (GPR40) | 1 | 1.40 | −10.00 | 0 | 0 |
| 2 | 1.60 | −9.80 | 0 | 0 | |
| 3 | 3.08 | −10.20 | 1 | 1 | |
| 4 | 1.40 | −10.10 | 0 | 1 | |
| Mean | 1.87 | −10.03 | |||
| Dipeptidyl peptidase 4 | 1 | 2.00 | −9.10 | 1 | 1 |
| 2 | 2.54 | −9.10 | 1 | 1 | |
| 3 | 0.89 | −8.80 | 0 | 0 | |
| 4 | 2.00 | −9.10 | 1 | 1 | |
| Mean | 1.86 | −9.03 | |||
| Spearman test RSp | 0.7143 | ||||
| p | 9.06 × 10−3 | ||||
| Group | TC, mmol/L | TGs, mmol/L | HDL-C, mg/dL | LDL-C, mg/dL | Glu, mmol/L |
|---|---|---|---|---|---|
| C57BL/6 mice | 1.59 ± 0.14 | 0.54 ± 0.04 | 82.66 ± 5.29 | 15.13 ± 2.02 | 5.55 ± 0.38 |
| AY mice | 2.59 ± 0.27 * | 0.64 ± 0.03 | 99.33 ± 6.38 | 10.36 ± 1.49 | 7.15 ± 0.69 * |
| VIL, 30 mg/kg | 3.26 ± 0.10 * | 0.46 ± 0.04 | 109.67 ± 5.23 * | 13.13 ± 0.95 | 8.41 ± 0.43 * |
| 1, 30 mg/kg | 1.79 ± 0.21 # | 0.58 ± 0.02 | 78.30 ± 5.73 # | 9.38 ± 1.25 | 5.16 ± 0.52 # |
| 3, 30 mg/kg | 2.80 ± 0.25 * | 0.64 ± 0.06 | 90.48 ± 8.6 | 10.78 ± 1.38 | 6.58 ± 1.06 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuranov, S.; Luzina, O.; Khvostov, M.; Baev, D.; Kuznetsova, D.; Zhukova, N.; Vassiliev, P.; Kochetkov, A.; Tolstikova, T.; Salakhutdinov, N. Bornyl Derivatives of p-(Benzyloxy)Phenylpropionic Acid: In Vivo Evaluation of Antidiabetic Activity. Pharmaceuticals 2020, 13, 404. https://doi.org/10.3390/ph13110404
Kuranov S, Luzina O, Khvostov M, Baev D, Kuznetsova D, Zhukova N, Vassiliev P, Kochetkov A, Tolstikova T, Salakhutdinov N. Bornyl Derivatives of p-(Benzyloxy)Phenylpropionic Acid: In Vivo Evaluation of Antidiabetic Activity. Pharmaceuticals. 2020; 13(11):404. https://doi.org/10.3390/ph13110404
Chicago/Turabian StyleKuranov, Sergey, Olga Luzina, Mikhail Khvostov, Dmitriy Baev, Darya Kuznetsova, Nataliya Zhukova, Pavel Vassiliev, Andrey Kochetkov, Tatyana Tolstikova, and Nariman Salakhutdinov. 2020. "Bornyl Derivatives of p-(Benzyloxy)Phenylpropionic Acid: In Vivo Evaluation of Antidiabetic Activity" Pharmaceuticals 13, no. 11: 404. https://doi.org/10.3390/ph13110404
APA StyleKuranov, S., Luzina, O., Khvostov, M., Baev, D., Kuznetsova, D., Zhukova, N., Vassiliev, P., Kochetkov, A., Tolstikova, T., & Salakhutdinov, N. (2020). Bornyl Derivatives of p-(Benzyloxy)Phenylpropionic Acid: In Vivo Evaluation of Antidiabetic Activity. Pharmaceuticals, 13(11), 404. https://doi.org/10.3390/ph13110404