Molecular Mechanisms of the Teratogenic Effects of Thalidomide


Abstract
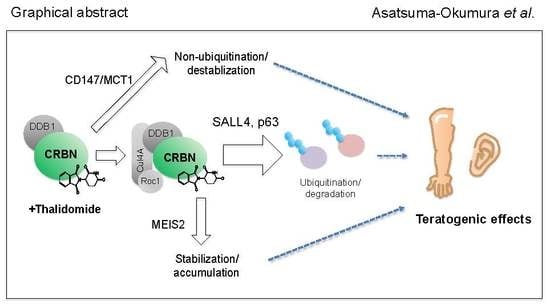
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Teratogenic Activity of Thalidomide
3. The Direct Target of Thalidomide
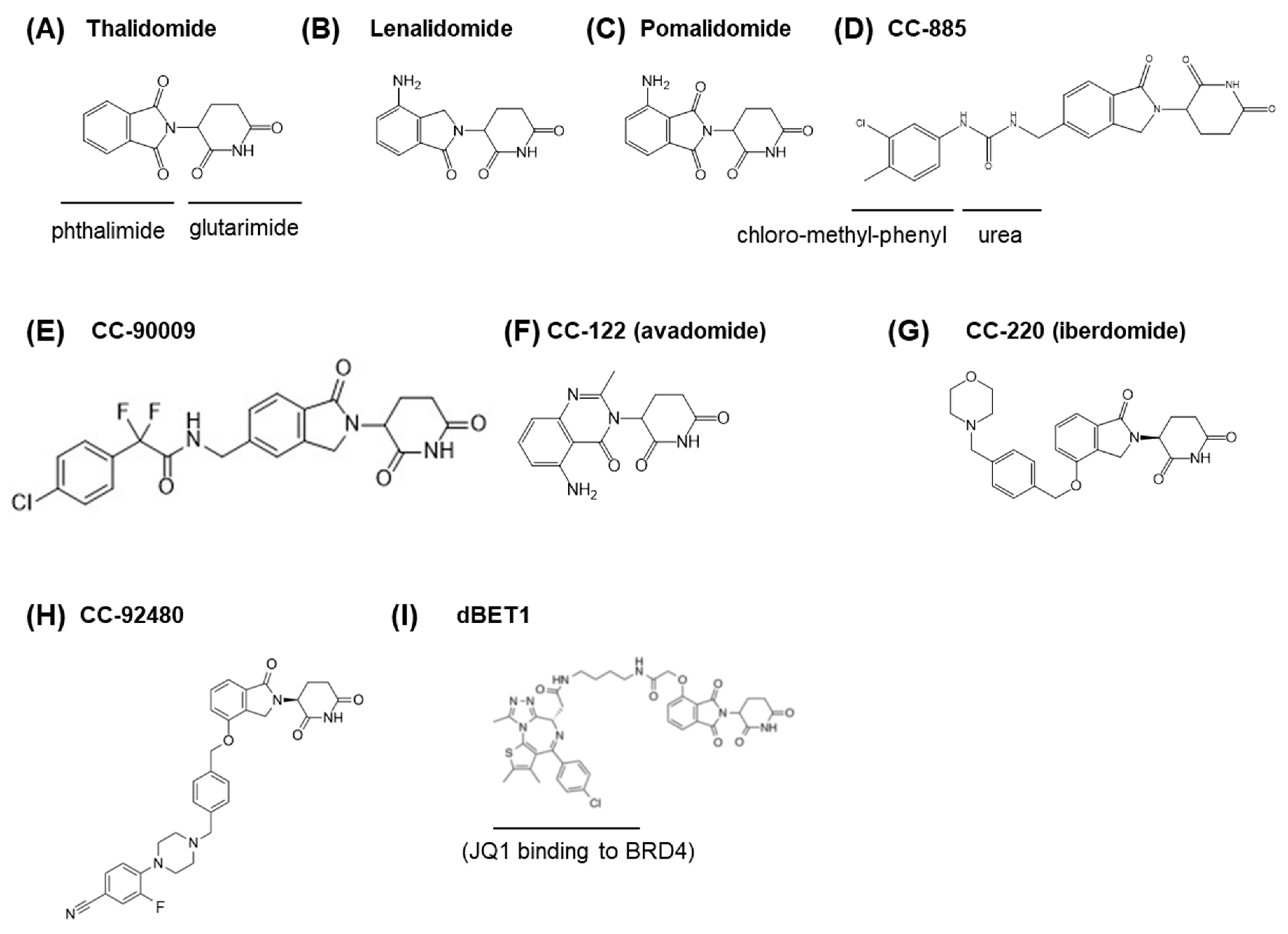
4. CRBN as a Therapeutic Target of Thalidomide and Its Derivatives
5. Ligand-Dependent Substrate Recognition of CRL4CRBN
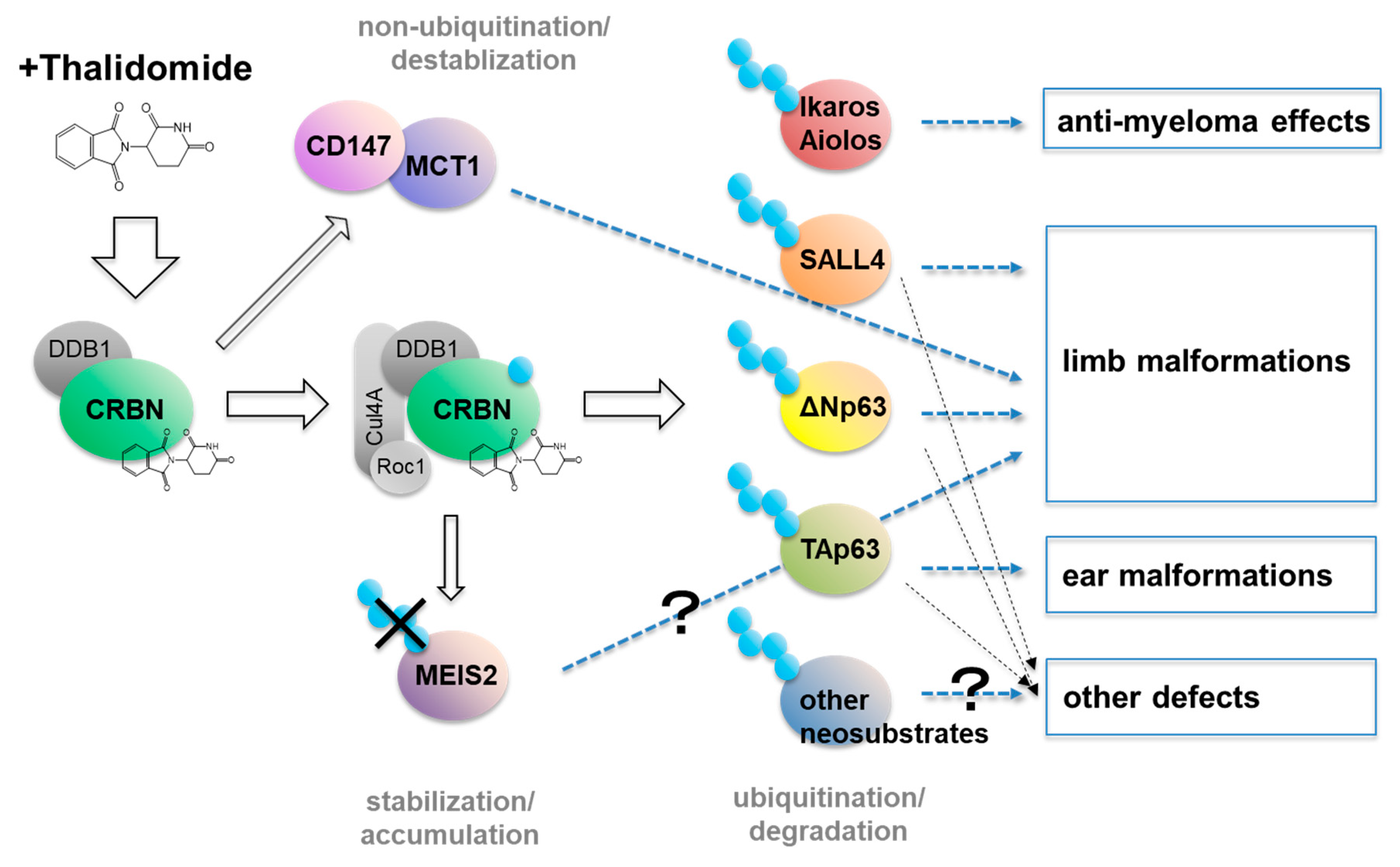
5.1. Ikaros and Aiolos
5.2. CK1α
5.3. GSPT1
5.4. ZFP91 and Other Zinc Finger Proteins
6. Structure of the CRBN–Drug–Neosubstrate Complex
7. Teratogenic Mechanisms Associated With CRBN
7.1. MEIS2
7.2. CD147
7.3. SALL4
7.4. p63
8. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- McBride, W.G. Thalidomide and congenital malformations. Lancet 1961, 1, 358. [Google Scholar]
- Lenz, W. Thalidomide and congenital abnormalities. Lancet 1962, 1, 271–272. [Google Scholar]
- McBride, W.G. Thalidomide embryopathy. Teratology 1977, 16, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Lenz, W. A short history of thalidomide embryopathy. Teratology 1988, 38, 203–215. [Google Scholar] [CrossRef]
- Sheskin, J. Thalidomide in the Treatment of Lepra Reactions. Clin. Pharmacol. Ther. 1965, 6, 303–306. [Google Scholar] [CrossRef]
- Gutierrez-Rodriguez, O. Thalidomide. A promising new treatment for rheumatoid arthritis. Arthritis Rheum. 1984, 27, 1118–1121. [Google Scholar] [CrossRef]
- Hamza, M.H. Treatment of Behcet’s disease with thalidomide. Clin. Rheumatol. 1986, 5, 365–371. [Google Scholar] [CrossRef]
- Vogelsang, G.B.; Hess, A.D.; Santos, G.W. Thalidomide for treatment of graft-versus-host disease. Bone Marrow Transplant 1988, 3, 393–398. [Google Scholar] [CrossRef]
- Atra, E.; Sato, E.I. Treatment of the cutaneous lesions of systemic lupus erythematosus with thalidomide. Clin. Exp. Rheumatol. 1993, 11, 487–493. [Google Scholar]
- Sampaio, E.P.; Sarno, E.N.; Galilly, R.; Cohn, Z.A.; Kaplan, G. Thalidomide selectively inhibits tumor necrosis factor alpha production by stimulated human monocytes. J. Exp. Med. 1991, 173, 699–703. [Google Scholar] [CrossRef] [Green Version]
- Moreira, A.L.; Sampaio, E.P.; Zmuidzinas, A.; Frindt, P.; Smith, K.A.; Kaplan, G. Thalidomide exerts its inhibitory action on tumor necrosis factor alpha by enhancing mRNA degradation. J. Exp. Med. 1993, 177, 1675–1680. [Google Scholar] [CrossRef] [PubMed]
- Makonkawkeyoon, S.; Limson-Pobre, R.N.; Moreira, A.L.; Schauf, V.; Kaplan, G. Thalidomide inhibits the replication of human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. USA 1993, 90, 5974–5978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Amato, R.J.; Loughnan, M.S.; Flynn, E.; Folkman, J. Thalidomide is an inhibitor of angiogenesis. Proc. Natl. Acad. Sci. USA 1994, 91, 4082–4085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singhal, S.; Mehta, J.; Desikan, R.; Ayers, D.; Roberson, P.; Eddlemon, P.; Munshi, N.; Anaissie, E.; Wilson, C.; Dhodapkar, M.; et al. Antitumor activity of thalidomide in refractory multiple myeloma. N. Engl. J. Med. 1999, 341, 1565–1571. [Google Scholar] [CrossRef] [Green Version]
- Bartlett, J.B.; Dredge, K.; Dalgleish, A.G. The evolution of thalidomide and its IMiD derivatives as anticancer agents. Nat. Rev. Cancer 2004, 4, 314–322. [Google Scholar] [CrossRef]
- Melchert, M.; List, A. The thalidomide saga. Int. J. Biochem. Cell Boil. 2007, 39, 1489–1499. [Google Scholar] [CrossRef]
- Zeldis, J.B.; Williams, B.A.; Thomas, S.D.; Elsayed, M.E. STEPS™: A comprehensive program for controlling and monitoring access to thalidomide. Clin. Ther. 1999, 21, 319–330. [Google Scholar] [CrossRef]
- Brandenburg, N.A.; Bwire, R.; Freeman, J.; Houn, F.; Sheehan, P.; Zeldis, J.B. Effectiveness of Risk Evaluation and Mitigation Strategies (REMS) for Lenalidomide and Thalidomide: Patient Comprehension and Knowledge Retention. Drug Saf. 2017, 40, 333–341. [Google Scholar] [CrossRef] [Green Version]
- Castilla, E.E.; Ashton-Prolla, P.; Barreda-Mejia, E.; Brunoni, D.; Cavalcanti, D.P.; Correa-Neto, J.; Delgadillo, J.L.; Dutra, M.G.; Felix, T.; Giraldo, A.; et al. Thalidomide, a current teratogen in South America. Teratology 1996, 54, 273–277. [Google Scholar] [CrossRef]
- Schuler-Faccini, L.; Soares, R.C.; de Sousa, A.C.; Maximino, C.; Luna, E.; Schwartz, I.V.; Waldman, C.; Castilla, E.E. New cases of thalidomide embryopathy in Brazil. Birth Defects Res. Part A Clin. Mol. Teratol. 2007, 79, 671–672. [Google Scholar] [CrossRef]
- Vianna, F.S.; de Oliveira, M.Z.; Sanseverino, M.T.; Morelo, E.F.; de Lyra Rabello Neto, D.; Lopez-Camelo, J.; Camey, S.A.; Schuler-Faccini, L. Pharmacoepidemiology and thalidomide embryopathy surveillance in Brazil. Reprod Toxicol. 2015, 53, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Parman, T.; Wiley, M.J.; Wells, P.G. Free radical-mediated oxidative DNA damage in the mechanism of thalidomide teratogenicity. Nat. Med. 1999, 5, 582–585. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.M.; Harris, K.K.; Philbert, M.A.; Harris, C. Thalidomide modulates nuclear redox status and preferentially depletes glutathione in rabbit limb versus rat limb. J. Pharmacol. Exp. Ther. 2002, 300, 768–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knobloch, J.; Shaughnessy, J.D., Jr.; Ruther, U. Thalidomide induces limb deformities by perturbing the Bmp/Dkk1/Wnt signaling pathway. FASEB J. 2007, 21, 1410–1421. [Google Scholar] [CrossRef] [PubMed]
- Therapontos, C.; Erskine, L.; Gardner, E.R.; Figg, W.D.; Vargesson, N. Thalidomide induces limb defects by preventing angiogenic outgrowth during early limb formation. Proc. Natl. Acad. Sci. USA 2009, 106, 8573–8578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a primary target of thalidomide teratogenicity. Science 2010, 327, 1345–1350. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Handa, H. Myeloid disease: Another action of a thalidomide derivative. Nature 2015, 523, 167–168. [Google Scholar] [CrossRef]
- Asatsuma-Okumura, T.; Ito, T.; Handa, H. Molecular mechanisms of cereblon-based drugs. Pharmacol. Ther. 2019, 202, 132–139. [Google Scholar] [CrossRef]
- Vargesson, N. Thalidomide-induced teratogenesis: history and mechanisms. Birth Defects Res. Part C Embryo Today Rev. 2015, 105, 140–156. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Ando, H.; Handa, H. Teratogenic effects of thalidomide: molecular mechanisms. Cell. Mol. Life Sci. 2011, 68, 1569–1579. [Google Scholar] [CrossRef]
- Miller, M.T.; Stromland, K. Teratogen update: thalidomide: a review, with a focus on ocular findings and new potential uses. Teratology 1999, 60, 306–321. [Google Scholar] [CrossRef]
- Spouge, D.; Baird, P.A. Imperforate anus in 700,000 consecutive liveborn infants. Am. J. Med Genet. Suppl. 1986, 2, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Knobloch, J.; Ruther, U. Shedding light on an old mystery: thalidomide suppresses survival pathways to induce limb defects. Cell Cycle 2008, 7, 1121–1127. [Google Scholar] [CrossRef] [PubMed]
- Newman, C.G. Clinical observations on the thalidomide syndrome. Proc. R. Soc. Med. 1977, 70, 225–227. [Google Scholar] [CrossRef]
- Smithells, R.W.; Newman, C.G. Recognition of thalidomide defects. J. Med Genet. 1992, 29, 716–723. [Google Scholar] [CrossRef] [Green Version]
- Vianna, F.S.; Schuler-Faccini, L.; Leite, J.C.; de Sousa, S.H.; da Costa, L.M.; Dias, M.F.; Morelo, E.F.; Doriqui, M.J.; Maximino, C.M.; Sanseverino, M.T. Recognition of the phenotype of thalidomide embryopathy in countries endemic for leprosy: new cases and review of the main dysmorphological findings. Clin. Dysmorphol. 2013, 22, 59–63. [Google Scholar] [CrossRef]
- Smithells, R.W. Thalidomide and malformations in Liverpool. Lancet 1962, 279, 1270–1273. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.T.; Stromland, K.K. What can we learn from the thalidomide experience: An ophthalmologic perspective. Curr. Opin. Ophthalmol. 2011, 22, 356–364. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.T.; Stromland, K.; Ventura, L.; Johansson, M.; Bandim, J.M.; Gillberg, C. Autism associated with conditions characterized by developmental errors in early embryogenesis: A mini review. Int. J. Dev. Neurosci. 2005, 23, 201–219. [Google Scholar] [CrossRef]
- Asatsuma-Okumura, T.; Ando, H.; De Simone, M.; Yamamoto, J.; Sato, T.; Shimizu, N.; Asakawa, K.; Yamaguchi, Y.; Ito, T.; Guerrini, L.; et al. P63 is a cereblon substrate involved in thalidomide teratogenicity. Nat. Chem. Biol. 2019, 15, 1077–1084. [Google Scholar] [CrossRef]
- Siamwala, J.H.; Veeriah, V.; Priya, M.K.; Rajendran, S.; Saran, U.; Sinha, S.; Nagarajan, S.; Pradeep, T.; Chatterjee, S. Nitric oxide rescues thalidomide mediated teratogenicity. Sci. Rep. 2012, 2, 679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brent, R.L. Drug Testing in Animals for Teratogenic Effects. Thalidomide in the Pregnant Rat. J. Pediatr. 1964, 64, 762–770. [Google Scholar] [CrossRef]
- Lewandoski, M.; Sun, X.; Martin, G.R. Fgf8 signalling from the AER is essential for normal limb development. Nat. Genet. 2000, 26, 460–463. [Google Scholar] [CrossRef] [PubMed]
- Moon, A.M.; Capecchi, M.R. Fgf8 is required for outgrowth and patterning of the limbs. Nat. Genet. 2000, 26, 455–459. [Google Scholar] [CrossRef]
- Hansen, J.M.; Gong, S.G.; Philbert, M.; Harris, C. Misregulation of gene expression in the redox-sensitive NF-kappab-dependent limb outgrowth pathway by thalidomide. Dev. Dyn. 2002, 225, 186–194. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, N.; Sugimoto, K.; Tang, J.; Nishi, T.; Sato, I.; Hiramoto, M.; Aizawa, S.; Hatakeyama, M.; Ohba, R.; Hatori, H.; et al. High-performance affinity beads for identifying drug receptors. Nat. Biotechnol. 2000, 18, 877–881. [Google Scholar] [CrossRef]
- Nishio, K.; Masaike, Y.; Ikeda, M.; Narimatsu, H.; Gokon, N.; Tsubouchi, S.; Hatakeyama, M.; Sakamoto, S.; Hanyu, N.; Sandhu, A.; et al. Development of novel magnetic nano-carriers for high-performance affinity purification. Colloids Surf. B Biointerfaces 2008, 64, 162–169. [Google Scholar] [CrossRef]
- Sakamoto, S.; Hatakeyama, M.; Ito, T.; Handa, H. Tools and methodologies capable of isolating and identifying a target molecule for a bioactive compound. Bioorganic Med Chem 2012, 20, 1990–2001. [Google Scholar] [CrossRef]
- Higgins, J.J.; Pucilowska, J.; Lombardi, R.Q.; Rooney, J.P. A mutation in a novel ATP-dependent Lon protease gene in a kindred with mild mental retardation. Neurology 2004, 63, 1927–1931. [Google Scholar] [CrossRef]
- Groisman, R.; Polanowska, J.; Kuraoka, I.; Sawada, J.; Saijo, M.; Drapkin, R.; Kisselev, A.F.; Tanaka, K.; Nakatani, Y. The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell 2003, 113, 357–367. [Google Scholar] [CrossRef] [Green Version]
- Petroski, M.D.; Deshaies, R.J. Function and regulation of cullin-RING ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2005, 6, 9–20. [Google Scholar] [CrossRef] [Green Version]
- Angers, S.; Li, T.; Yi, X.; MacCoss, M.J.; Moon, R.T.; Zheng, N. Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature 2006, 443, 590–593. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Zhou, P. DCAFs, the missing link of the CUL4-DDB1 ubiquitin ligase. Mol. Cell 2007, 26, 775–780. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.; Behnke, M.; Chen, S.; Cruickshank, A.A.; Dick, L.R.; Grenier, L.; Klunder, J.M.; Ma, Y.T.; Plamondon, L.; Stein, R.L. Potent and selective inhibitors of the proteasome: Dipeptidyl boronic acids. Bioorganic Med. Chem. Lett. 1998, 8, 333–338. [Google Scholar] [CrossRef]
- Zhu, Y.X.; Braggio, E.; Shi, C.X.; Bruins, L.A.; Schmidt, J.E.; Van Wier, S.; Chang, X.B.; Bjorklund, C.C.; Fonseca, R.; Bergsagel, P.L.; et al. Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide. Blood 2011, 118, 4771–4779. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Girona, A.; Mendy, D.; Ito, T.; Miller, K.; Gandhi, A.K.; Kang, J.; Karasawa, S.; Carmel, G.; Jackson, P.; Abbasian, M.; et al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia 2012, 26, 2326–2335. [Google Scholar] [CrossRef] [PubMed]
- Kronke, J.; Hurst, S.N.; Ebert, B.L. Lenalidomide induces degradation of IKZF1 and IKZF3. Oncoimmunology 2014, 3, e941742. [Google Scholar] [CrossRef] [Green Version]
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.K.; Bradner, J.E.; Kaelin, W.G., Jr. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 2014, 343, 305–309. [Google Scholar] [CrossRef] [Green Version]
- Heizmann, B.; Kastner, P.; Chan, S. The Ikaros family in lymphocyte development. Curr. Opin. Immunol. 2018, 51, 14–23. [Google Scholar] [CrossRef]
- Gandhi, A.K.; Kang, J.; Havens, C.G.; Conklin, T.; Ning, Y.; Wu, L.; Ito, T.; Ando, H.; Waldman, M.F.; Thakurta, A.; et al. Immunomodulatory agents lenalidomide and pomalidomide co-stimulate T cells by inducing degradation of T cell repressors Ikaros and Aiolos via modulation of the E3 ubiquitin ligase complex CRL4(CRBN.). Br. J. Haematol. 2014, 164, 811–821. [Google Scholar] [CrossRef] [Green Version]
- List, A.; Kurtin, S.; Roe, D.J.; Buresh, A.; Mahadevan, D.; Fuchs, D.; Rimsza, L.; Heaton, R.; Knight, R.; Zeldis, J.B. Efficacy of lenalidomide in myelodysplastic syndromes. N. Engl. J. Med. 2005, 352, 549–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- List, A.; Dewald, G.; Bennett, J.; Giagounidis, A.; Raza, A.; Feldman, E.; Powell, B.; Greenberg, P.; Thomas, D.; Stone, R.; et al. Lenalidomide in the myelodysplastic syndrome with chromosome 5q deletion. N. Engl. J. Med. 2006, 355, 1456–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kronke, J.; Fink, E.C.; Hollenbach, P.W.; MacBeth, K.J.; Hurst, S.N.; Udeshi, N.D.; Chamberlain, P.P.; Mani, D.R.; Man, H.W.; Gandhi, A.K.; et al. Lenalidomide induces ubiquitination and degradation of CK1alpha in del(5q) MDS. Nature 2015, 523, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Matyskiela, M.E.; Lu, G.; Ito, T.; Pagarigan, B.; Lu, C.C.; Miller, K.; Fang, W.; Wang, N.Y.; Nguyen, D.; Houston, J.; et al. A novel cereblon modulator recruits GSPT1 to the CRL4(CRBN) ubiquitin ligase. Nature 2016, 535, 252–257. [Google Scholar] [CrossRef]
- Hansen, J.D.; Condroski, K.; Correa, M.; Muller, G.; Man, H.W.; Ruchelman, A.; Zhang, W.; Vocanson, F.; Crea, T.; Liu, W.; et al. Protein Degradation via CRL4(CRBN) Ubiquitin Ligase: Discovery and Structure-Activity Relationships of Novel Glutarimide Analogs That Promote Degradation of Aiolos and/or GSPT1. J. Med. Chem. 2018, 61, 492–503. [Google Scholar] [CrossRef]
- Chamberlain, P.P.; Hamann, L.G. Development of targeted protein degradation therapeutics. Nat. Chem. Biol. 2019, 15, 937–944. [Google Scholar] [CrossRef]
- An, J.; Ponthier, C.M.; Sack, R.; Seebacher, J.; Stadler, M.B.; Donovan, K.A.; Fischer, E.S. pSILAC mass spectrometry reveals ZFP91 as IMiD-dependent substrate of the CRL4(CRBN) ubiquitin ligase. Nat. Commun. 2017, 8, 15398. [Google Scholar] [CrossRef]
- Sievers, Q.L.; Petzold, G.; Bunker, R.D.; Renneville, A.; Slabicki, M.; Liddicoat, B.J.; Abdulrahman, W.; Mikkelsen, T.; Ebert, B.L.; Thoma, N.H. Defining the human C2H2 zinc finger degrome targeted by thalidomide analogs through CRBN. Science 2018, 362. [Google Scholar] [CrossRef] [Green Version]
- Fischer, E.S.; Bohm, K.; Lydeard, J.R.; Yang, H.; Stadler, M.B.; Cavadini, S.; Nagel, J.; Serluca, F.; Acker, V.; Lingaraju, G.M.; et al. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 2014, 512, 49–53. [Google Scholar] [CrossRef] [Green Version]
- Chamberlain, P.P.; Lopez-Girona, A.; Miller, K.; Carmel, G.; Pagarigan, B.; Chie-Leon, B.; Rychak, E.; Corral, L.G.; Ren, Y.J.; Wang, M.; et al. Structure of the human Cereblon-DDB1-lenalidomide complex reveals basis for responsiveness to thalidomide analogs. Nat. Struct. Mol. Biol. 2014, 21, 803–809. [Google Scholar] [CrossRef]
- Sheereen, A.; Alaamery, M.; Bawazeer, S.; Al Yafee, Y.; Massadeh, S.; Eyaid, W. A missense mutation in the CRBN gene that segregates with intellectual disability and self-mutilating behaviour in a consanguineous Saudi family. J. Med. Genet. 2017, 54, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Petzold, G.; Fischer, E.S.; Thoma, N.H. Structural basis of lenalidomide-induced CK1alpha degradation by the CRL4(CRBN) ubiquitin ligase. Nature 2016, 532, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Blaschke, G.; Kraft, H.P.; Fickentscher, K.; Kohler, F. [Chromatographic separation of racemic thalidomide and teratogenic activity of its enantiomers (author’s transl)]. Arzneimittelforschung 1979, 29, 1640–1642. [Google Scholar] [PubMed]
- Nishimura, K.; Hashimoto, Y.; Iwasaki, S. (S)-form of alpha-methyl-N(alpha)-phthalimidoglutarimide, but not its (R)-form, enhanced phorbol ester-induced tumor necrosis factor-alpha production by human leukemia cell HL-60: implication of optical resolution of thalidomidal effects. Chem. Pharm. Bull. (Tokyo) 1994, 42, 1157–1159. [Google Scholar] [CrossRef] [Green Version]
- Mori, T.; Ito, T.; Liu, S.; Ando, H.; Sakamoto, S.; Yamaguchi, Y.; Tokunaga, E.; Shibata, N.; Handa, H.; Hakoshima, T. Structural basis of thalidomide enantiomer binding to cereblon. Sci. Rep. 2018, 8, 1294. [Google Scholar] [CrossRef]
- Fratta, I.D.; Sigg, E.B.; Maiorana, K. Teratogenic Effects of Thalidomide in Rabbits, Rats, Hamsters, and Mice. Toxicol. Appl. Pharmacol. 1965, 7, 268–286. [Google Scholar] [CrossRef]
- Chesi, M.; Matthews, G.M.; Garbitt, V.M.; Palmer, S.E.; Shortt, J.; Lefebure, M.; Stewart, A.K.; Johnstone, R.W.; Bergsagel, P.L. Drug response in a genetically engineered mouse model of multiple myeloma is predictive of clinical efficacy. Blood 2012, 120, 376–385. [Google Scholar] [CrossRef] [Green Version]
- Gemechu, Y.; Millrine, D.; Hashimoto, S.; Prakash, J.; Sanchenkova, K.; Metwally, H.; Gyanu, P.; Kang, S.; Kishimoto, T. Humanized cereblon mice revealed two distinct therapeutic pathways of immunomodulatory drugs. Proc. Natl. Acad. Sci. USA 2018, 115, 11802–11807. [Google Scholar] [CrossRef] [Green Version]
- Fink, E.C.; McConkey, M.; Adams, D.N.; Haldar, S.D.; Kennedy, J.A.; Guirguis, A.A.; Udeshi, N.D.; Mani, D.R.; Chen, M.; Liddicoat, B.; et al. Crbn (I391V) is sufficient to confer in vivo sensitivity to thalidomide and its derivatives in mice. Blood 2018, 132, 1535–1544. [Google Scholar] [CrossRef] [Green Version]
- Capdevila, J.; Tsukui, T.; Rodriquez Esteban, C.; Zappavigna, V.; Izpisua Belmonte, J.C. Control of vertebrate limb outgrowth by the proximal factor Meis2 and distal antagonism of BMPs by Gremlin. Mol. Cell 1999, 4, 839–849. [Google Scholar] [CrossRef]
- Eichner, R.; Heider, M.; Fernandez-Saiz, V.; van Bebber, F.; Garz, A.K.; Lemeer, S.; Rudelius, M.; Targosz, B.S.; Jacobs, L.; Knorn, A.M.; et al. Immunomodulatory drugs disrupt the cereblon-CD147-MCT1 axis to exert antitumor activity and teratogenicity. Nat. Med. 2016, 22, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Wang, Z.; Zhao, J.J.; Calimeri, T.; Meng, J.; Hideshima, T.; Fulciniti, M.; Kang, Y.; Ficarro, S.B.; Tai, Y.T.; et al. The Cyclophilin A-CD147 complex promotes the proliferation and homing of multiple myeloma cells. Nat. Med. 2015, 21, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Donovan, K.A.; An, J.; Nowak, R.P.; Yuan, J.C.; Fink, E.C.; Berry, B.C.; Ebert, B.L.; Fischer, E.S. Thalidomide promotes degradation of SALL4, a transcription factor implicated in Duane Radial Ray syndrome. Elife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Al-Baradie, R.; Yamada, K.; St Hilaire, C.; Chan, W.M.; Andrews, C.; McIntosh, N.; Nakano, M.; Martonyi, E.J.; Raymond, W.R.; Okumura, S.; et al. Duane radial ray syndrome (Okihiro syndrome) maps to 20q13 and results from mutations in SALL4, a new member of the SAL family. Am. J. Hum. Genet. 2002, 71, 1195–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohlhase, J.; Heinrich, M.; Schubert, L.; Liebers, M.; Kispert, A.; Laccone, F.; Turnpenny, P.; Winter, R.M.; Reardon, W. Okihiro syndrome is caused by SALL4 mutations. Hum. Mol. Genet. 2002, 11, 2979–2987. [Google Scholar] [CrossRef] [Green Version]
- Kohlhase, J.; Schubert, L.; Liebers, M.; Rauch, A.; Becker, K.; Mohammed, S.N.; Newbury-Ecob, R.; Reardon, W. Mutations at the SALL4 locus on chromosome 20 result in a range of clinically overlapping phenotypes, including Okihiro syndrome, Holt-Oram syndrome, acro-renal-ocular syndrome, and patients previously reported to represent thalidomide embryopathy. J. Med. Genet. 2003, 40, 473–478. [Google Scholar] [CrossRef] [Green Version]
- Kohlhase, J.; Holmes, L.B. Mutations in SALL4 in malformed father and daughter postulated previously due to reflect mutagenesis by thalidomide. Birth Defects Res. Part A Clin. Mol. Teratol. 2004, 70, 550–551. [Google Scholar] [CrossRef]
- Matyskiela, M.E.; Couto, S.; Zheng, X.; Lu, G.; Hui, J.; Stamp, K.; Drew, C.; Ren, Y.; Wang, M.; Carpenter, A.; et al. SALL4 mediates teratogenicity as a thalidomide-dependent cereblon substrate. Nat. Chem. Biol. 2018, 14, 981–987. [Google Scholar] [CrossRef]
- Belair, D.G.; Lu, G.; Waller, L.E.; Gustin, J.A.; Collins, N.D.; Kolaja, K.L. Thalidomide Inhibits Human iPSC Mesendoderm Differentiation by Modulating CRBN-dependent Degradation of SALL4. Sci. Rep. 2020, 10, 2864. [Google Scholar] [CrossRef] [Green Version]
- Sakaki-Yumoto, M.; Kobayashi, C.; Sato, A.; Fujimura, S.; Matsumoto, Y.; Takasato, M.; Kodama, T.; Aburatani, H.; Asashima, M.; Yoshida, N.; et al. The murine homolog of SALL4, a causative gene in Okihiro syndrome, is essential for embryonic stem cell proliferation, and cooperates with Sall1 in anorectal, heart, brain and kidney development. Development 2006, 133, 3005–3013. [Google Scholar] [CrossRef] [Green Version]
- Asakawa, K.; Kawakami, K. Protocadherin-Mediated Cell Repulsion Controls the Central Topography and Efferent Projections of the Abducens Nucleus. Cell Rep. 2018, 24, 1562–1572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerrini, L.; Costanzo, A.; Merlo, G.R. A symphony of regulations centered on p63 to control development of ectoderm-derived structures. J. Biomed. Biotechnol. 2011, 2011, 864904. [Google Scholar] [CrossRef] [PubMed]
- Restelli, M.; Lopardo, T.; Lo Iacono, N.; Garaffo, G.; Conte, D.; Rustighi, A.; Napoli, M.; Del Sal, G.; Perez-Morga, D.; Costanzo, A.; et al. DLX5, FGF8 and the Pin1 isomerase control DeltaNp63alpha protein stability during limb development: a regulatory loop at the basis of the SHFM and EEC congenital malformations. Hum. Mol. Genet. 2014, 23, 3830–3842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinne, T.; Hamel, B.; van Bokhoven, H.; Brunner, H.G. Pattern of p63 mutations and their phenotypes--update. Am. J. Med. Genet. A 2006, 140, 1396–1406. [Google Scholar] [CrossRef]
- Mills, A.A.; Zheng, B.; Wang, X.J.; Vogel, H.; Roop, D.R.; Bradley, A. P63 is a p53 homologue required for limb and epidermal morphogenesis. Nature 1999, 398, 708–713. [Google Scholar] [CrossRef]
- Yang, A.; Schweitzer, R.; Sun, D.; Kaghad, M.; Walker, N.; Bronson, R.T.; Tabin, C.; Sharpe, A.; Caput, D.; Crum, C.; et al. P63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 1999, 398, 714–718. [Google Scholar] [CrossRef]
- Chen, H.; Beasley, A.; Hu, Y.; Chen, X. A Zebrafish Model for Studies on Esophageal Epithelial Biology. PLoS ONE 2015, 10, e0143878. [Google Scholar] [CrossRef]
- Yang, A.; Kaghad, M.; Caput, D.; McKeon, F. On the shoulders of giants: p63, p73 and the rise of p53. Trends Genet. 2002, 18, 90–95. [Google Scholar] [CrossRef]
- Rouleau, M.; Medawar, A.; Hamon, L.; Shivtiel, S.; Wolchinsky, Z.; Zhou, H.; De Rosa, L.; Candi, E.; de la Forest Divonne, S.; Mikkola, M.L.; et al. TAp63 is important for cardiac differentiation of embryonic stem cells and heart development. Stem Cells 2011, 29, 1672–1683. [Google Scholar] [CrossRef]
- Terrinoni, A.; Serra, V.; Bruno, E.; Strasser, A.; Valente, E.; Flores, E.R.; van Bokhoven, H.; Lu, X.; Knight, R.A.; Melino, G. Role of p63 and the Notch pathway in cochlea development and sensorineural deafness. Proc. Natl. Acad. Sci. USA 2013, 110, 7300–7305. [Google Scholar] [CrossRef] [Green Version]
- Latina, A.; Viticchie, G.; Lena, A.M.; Piro, M.C.; Annicchiarico-Petruzzelli, M.; Melino, G.; Candi, E. DeltaNp63 targets cytoglobin to inhibit oxidative stress-induced apoptosis in keratinocytes and lung cancer. Oncogene 2016, 35, 1493–1503. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.X.; Tu, H.C.; Dong, Y.; Skanderup, A.J.; Wang, Y.; Takeda, S.; Ganesan, Y.T.; Han, S.; Liu, H.; Hsieh, J.J.; et al. DeltaNp63 Inhibits Oxidative Stress-Induced Cell Death, Including Ferroptosis, and Cooperates with the BCL-2 Family to Promote Clonogenic Survival. Cell Rep. 2017, 21, 2926–2939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chamberlain, P.P.; Cathers, B.E. Cereblon modulators: Low molecular weight inducers of protein degradation. Drug Discov. Today Technol. 2019, 31, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Hagner, P.R.; Man, H.W.; Fontanillo, C.; Wang, M.; Couto, S.; Breider, M.; Bjorklund, C.; Havens, C.G.; Lu, G.; Rychak, E.; et al. CC-122, a pleiotropic pathway modifier, mimics an interferon response and has antitumor activity in DLBCL. Blood 2015, 126, 779–789. [Google Scholar] [CrossRef]
- Rasco, D.W.; Papadopoulos, K.P.; Pourdehnad, M.; Gandhi, A.K.; Hagner, P.R.; Li, Y.; Wei, X.; Chopra, R.; Hege, K.; DiMartino, J.; et al. A First-in-Human Study of Novel Cereblon Modulator Avadomide (CC-122) in Advanced Malignancies. Clin. Cancer Res. 2019, 25, 90–98. [Google Scholar] [CrossRef] [Green Version]
- Matyskiela, M.E.; Zhang, W.; Man, H.W.; Muller, G.; Khambatta, G.; Baculi, F.; Hickman, M.; LeBrun, L.; Pagarigan, B.; Carmel, G.; et al. A Cereblon Modulator (CC-220) with Improved Degradation of Ikaros and Aiolos. J. Med. Chem. 2018, 61, 535–542. [Google Scholar] [CrossRef]
- Schafer, P.H.; Ye, Y.; Wu, L.; Kosek, J.; Ringheim, G.; Yang, Z.; Liu, L.; Thomas, M.; Palmisano, M.; Chopra, R. Cereblon modulator iberdomide induces degradation of the transcription factors Ikaros and Aiolos: immunomodulation in healthy volunteers and relevance to systemic lupus erythematosus. Ann. Rheum. Dis. 2018, 77, 1516–1523. [Google Scholar] [CrossRef] [Green Version]
- Bjorklund, C.C.; Kang, J.; Amatangelo, M.; Polonskaia, A.; Katz, M.; Chiu, H.; Couto, S.; Wang, M.; Ren, Y.; Ortiz, M.; et al. Iberdomide (CC-220) is a potent cereblon E3 ligase modulator with antitumor and immunostimulatory activities in lenalidomide- and pomalidomide-resistant multiple myeloma cells with dysregulated CRBN. Leukemia 2019, 34, 1197–1201. [Google Scholar] [CrossRef] [Green Version]
- Hansen, J.D.; Correa, M.; Nagy, M.A.; Alexander, M.; Plantevin, V.; Grant, V.; Whitefield, B.; Huang, D.; Kercher, T.; Harris, R.; et al. Discovery of CRBN E3 Ligase Modulator CC-92480 for the Treatment of Relapsed and Refractory Multiple Myeloma. J. Med. Chem. 2020. [Google Scholar] [CrossRef]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef] [Green Version]
- Lai, A.C.; Toure, M.; Hellerschmied, D.; Salami, J.; Jaime-Figueroa, S.; Ko, E.; Hines, J.; Crews, C.M. Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew. Chem. Int. Ed. Engl. 2016, 55, 807–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burslem, G.M.; Crews, C.M. Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell 2020, 181, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Winter, G.E.; Buckley, D.L.; Paulk, J.; Roberts, J.M.; Souza, A.; Dhe-Paganon, S.; Bradner, J.E. DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. [Google Scholar] [CrossRef] [Green Version]
- Silva, M.C.; Ferguson, F.M.; Cai, Q.; Donovan, K.A.; Nandi, G.; Patnaik, D.; Zhang, T.; Huang, H.T.; Lucente, D.E.; Dickerson, B.C.; et al. Targeted degradation of aberrant tau in frontotemporal dementia patient-derived neuronal cell models. Elife 2019, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.; Xiong, Y.; Safaee, N.; Nowak, R.P.; Donovan, K.A.; Yuan, C.J.; Nabet, B.; Gero, T.W.; Feru, F.; Li, L.; et al. Exploring Targeted Degradation Strategy for Oncogenic KRAS(G12C). Cell Chem. Biol. 2020, 27, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [Green Version]
- Matyskiela, M.E.; Clayton, T.; Zheng, X.; Mayne, C.; Tran, E.; Carpenter, A.; Pagarigan, B.; McDonald, J.; Rolfe, M.; Hamann, L.G.; et al. Crystal structure of the SALL4-pomalidomide-cereblon-DDB1 complex. Nat. Struct. Mol. Biol. 2020, 27, 319–322. [Google Scholar] [CrossRef]
- Jo, S.; Lee, K.H.; Song, S.; Jung, Y.K.; Park, C.S. Identification and functional characterization of cereblon as a binding protein for large-conductance calcium-activated potassium channel in rat brain. J. Neurochem. 2005, 94, 1212–1224. [Google Scholar] [CrossRef]
- Liu, J.; Ye, J.; Zou, X.; Xu, Z.; Feng, Y.; Zou, X.; Chen, Z.; Li, Y.; Cang, Y. CRL4A(CRBN) E3 ubiquitin ligase restricts BK channel activity and prevents epileptogenesis. Nat. Commun. 2014, 5, 3924. [Google Scholar] [CrossRef]
- Rajadhyaksha, A.M.; Ra, S.; Kishinevsky, S.; Lee, A.S.; Romanienko, P.; DuBoff, M.; Yang, C.; Zupan, B.; Byrne, M.; Daruwalla, Z.R.; et al. Behavioral characterization of cereblon forebrain-specific conditional null mice: a model for human non-syndromic intellectual disability. Behav. Brain Res. 2012, 226, 428–434. [Google Scholar] [CrossRef] [Green Version]
- Ando, H.; Sato, T.; Ito, T.; Yamamoto, J.; Sakamoto, S.; Nitta, N.; Asatsuma-Okumura, T.; Shimizu, N.; Mizushima, R.; Aoki, I.; et al. Cereblon Control of Zebrafish Brain Size by Regulation of Neural Stem Cell Proliferation. iScience 2019, 15, 95–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, M.D.; Boichenko, I.; Coles, M.; Zanini, F.; Lupas, A.N.; Hernandez Alvarez, B. Thalidomide mimics uridine binding to an aromatic cage in cereblon. J. Struct. Biol. 2014, 188, 225–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boichenko, I.; Bar, K.; Deiss, S.; Heim, C.; Albrecht, R.; Lupas, A.N.; Hernandez Alvarez, B.; Hartmann, M.D. Chemical Ligand Space of Cereblon. ACS Omega 2018, 3, 11163–11171. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asatsuma-Okumura, T.; Ito, T.; Handa, H. Molecular Mechanisms of the Teratogenic Effects of Thalidomide. Pharmaceuticals 2020, 13, 95. https://doi.org/10.3390/ph13050095
Asatsuma-Okumura T, Ito T, Handa H. Molecular Mechanisms of the Teratogenic Effects of Thalidomide. Pharmaceuticals. 2020; 13(5):95. https://doi.org/10.3390/ph13050095
Chicago/Turabian StyleAsatsuma-Okumura, Tomoko, Takumi Ito, and Hiroshi Handa. 2020. "Molecular Mechanisms of the Teratogenic Effects of Thalidomide" Pharmaceuticals 13, no. 5: 95. https://doi.org/10.3390/ph13050095
APA StyleAsatsuma-Okumura, T., Ito, T., & Handa, H. (2020). Molecular Mechanisms of the Teratogenic Effects of Thalidomide. Pharmaceuticals, 13(5), 95. https://doi.org/10.3390/ph13050095