Novel Nonsymmetrical 1,4-Dihydropyridines as Inhibitors of Nonsymmetrical MRP-Efflux Pumps for Anticancer Therapy

Abstract
:1. Introduction
2. Results and Discussion
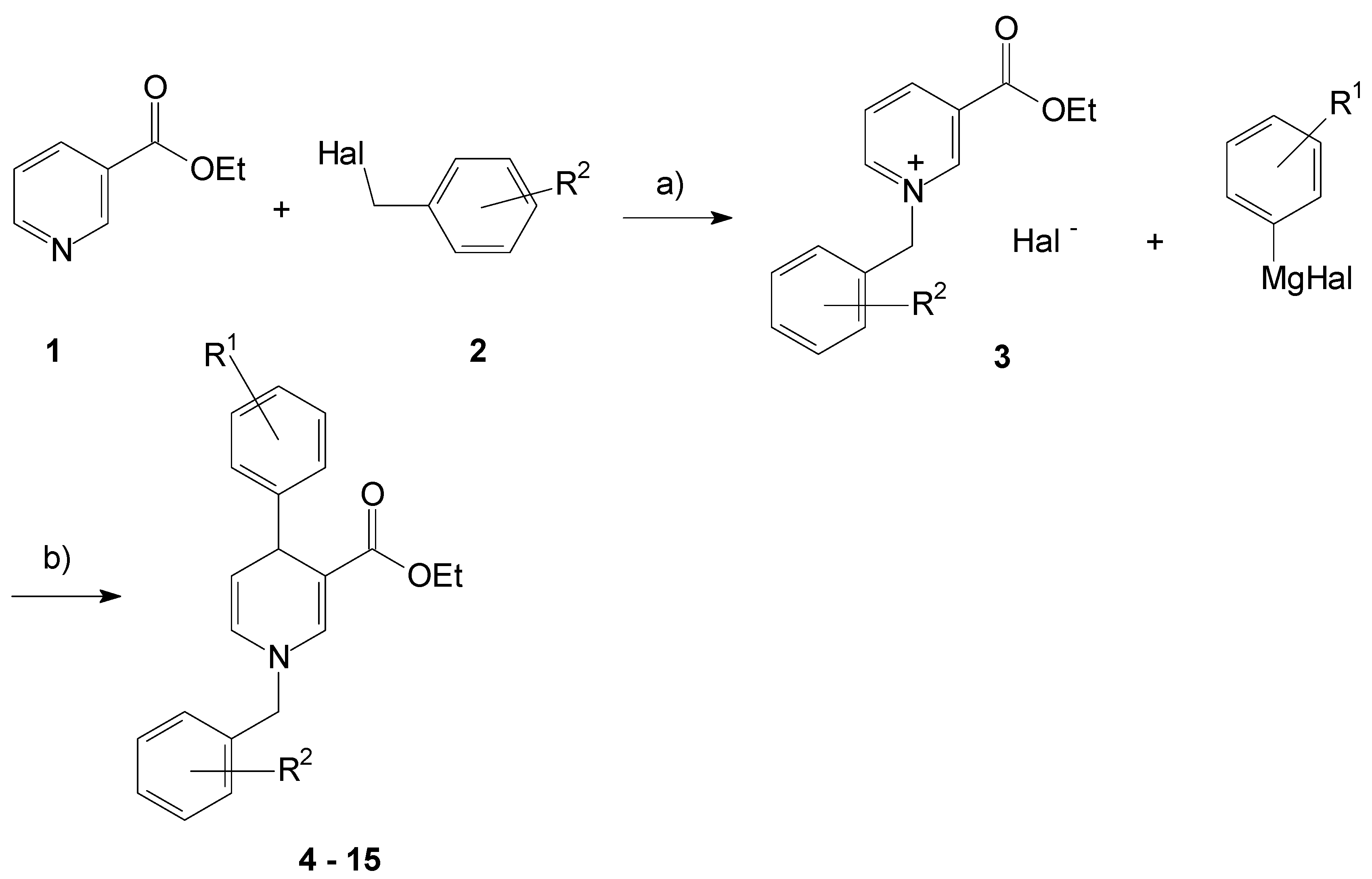
2.1. Synthesis of the 1,4-Dihydropyridines
2.2. MRP Efflux Pump Inhibition of the 1,4-Dihydropyridines
2.3. In Vitro MRP Resistance Studies of Drug Reversal
3. Materials and Methods
3.1. Chemical Reagents and Instruments
3.2. General Procedure for the Synthesis of Compound 3
3.3. General Procedure for the Synthesis of Compounds 4–15
3.4. MRP Inhibition Assay
3.5. MRP Reversal Assay
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Meegan, M.J.; O’ Boyle, N.M. Special issue “anticancer drugs”. Pharmaceuticals 2019, 12, 134. [Google Scholar] [CrossRef] [Green Version]
- The International Agency for Research on Cancer. Available online: https://www.wh.int/cancer/PRGlobocanFinal.pdf (accessed on 24 May 2020).
- Sampson, A.; Peterson, B.G.; Tan, K.W.; Iram, S.H. Doxorubicin as a fluorescent reporter identifies novel MRP1 (ABCC1) inhibitors missed by the calcein-based high content screening anticancer agents. Biomed. Pharmacother. 2019, 118, 109289. [Google Scholar] [CrossRef]
- Krug, M.; Hilgeroth, A. Recent advances in the development of multi-kinase inhibitors. Mini Rev. Med. Chem. 2008, 8, 1312–1327. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, H.; Assaref, Y.G.; Zhao, K.; Xu, X.; Xie, J.; Yang, D.H.; Chen, Z.S. Overcoming ABC transporter-mediated multidrug resistance: Molecular mechanisms and novel therapeutic drug strategies. Drug Resist. Update 2016, 27, 14–29. [Google Scholar] [CrossRef]
- Lu, J.F.; Pokharel, D.; Bebawy, M. MRP1 and its role in anticancer drug resistance. Drug Metab. Rev. 2015, 2532, 1–14. [Google Scholar]
- Li, F.; Zhou, X.; Zhou, H.; Jia, J.; Li, I.; Zhai, S.; Yan, B. Reducing both pgp overexpression and drug-efflux with Anti-Cancer gold-paclitaxel nanoconjugates. PLoS ONE 2016, 11, e0160042. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-K.; Wang, Y.-J.; Gupta, P.; Chen, Z.-S. Multidrug resistance proteins (MRPs) and cancer therapy. AAPS J. 2015, 17, 802–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majidinia, M.; Mirza-Aghazadeh-Attari, M.; Rahimi, M.; Mihanfar, A.; Karimian, A.; Safa, A.; Yousefi, B. Overcoming multidrug resistance in cancer: Recent progress in nanotechnology and new horizons. IUMB Life 2020, 72, 855–871. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-S.; Coi, I.; Choi, D.-H. Effects of nifedipine on the pharmakokinetics of repaglidine in rats: Possible role of CYP3A4 and P-glycoprotein inhibition by nifedipine. Pharmacol. Rep. 2013, 65, 1411–1430. [Google Scholar] [CrossRef]
- Sharma, M.G.; Rajani, D.P.; Patel, H.M. Green approach for synthesis of bioactive Hantzsch 1,4-dihydropyridine derivatives based on thiophene moiety via multicomponent reaction. R. Soc. Open Sci. 2017, 4, 170006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hantzsch, A.; Justus, I. On the synthesis of pyridine-like compounds from aceto acetic ether and aldehyde ammoni. Ann. Chem. 1882, 215, 1–82. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Gong, H.; Quan, Z.; Li, L.; Ye, H. One-pot, three component synthesis of 1,4-dihydropyridines in PEG-400. Synth. Commun. 2011, 41, 3251–3258. [Google Scholar] [CrossRef]
- Kuckländer, U.; Hilgeroth, A. Versuche zur darstellung N-substituierter dihydropyridine nach hantzsch. Arch. Pharm. Pharm. Med. Chem. 1994, 327, 287–294. [Google Scholar] [CrossRef]
- Triverdi, A.R.; Dodiya, D.K.; Dholariya, B.H.; Kataria, V.B.; Bhuva, V.R.; Shah, V.H. Synthesis and biological evaluation of some novel N-aryl-1,4-dihydropyridines. Bioorg. Med. Chem. Lett. 2011, 21, 5181–5183. [Google Scholar] [CrossRef] [PubMed]
- Cataldi, M.; Bruno, F. 1,4-diyhdropyridines: The multiple personalities of a blockbuster family. Transl. Med. 2012, 4, 12–26. [Google Scholar]
- Shahraki, O.; Zargan, F.; Edraki, N.; Khoshneviszadeh, M.; Firuzi, O.; Miri, R. Molecular dynamics simulation and molecular docking studies of 1,4-Dihydropyridines as P-glycoprotein’s allosteric inhibitors. J. Biomol. Struc. Dyn. 2018, 36, 112–125. [Google Scholar] [CrossRef]
- Zhou, S.F. Structure, function and regulation of P-glycoprotein and its clinical relevance in drug disposition. Xenobiotica 2008, 38, 802–832. [Google Scholar] [CrossRef] [PubMed]
- Sharom, F.J. Complex interplay between the P-glycoprotein multidrug efflux pump and the membrane: Its role in modulating protein function. Front. Oncol. 2014, 4, 41. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, R.J.; Ferreira, M.-J.U.; dos Santos, D.J.V.A. Insight on P-glycoprotein’s efflux mechanism obtained by molecular dynamics simulation. J. Chem. Theory Comput. 2012, 8, 1853–1864. [Google Scholar] [CrossRef]
- Cakil, Y.D.; Khunweeraphong, N.; Parveen, Z.; Schmid, D.; Artaker, M.; Ecker, G.F.; Sitte, H.H.; Pusch, O.; Stockner, T.; Chiba, P. Pore-exposed tyrosine residues of P-glycoprotein are important hydrogen-bonding partners for drugs. Mol. Pharmacol. 2014, 85, 420–428. [Google Scholar] [CrossRef]
- Dastvan, R.; Mishra, S.; Peskova, Y.B.; Nakamoto, R.K.; Mchaourab, H.S. Mechanism of allosteric modulation of P-glycoprotein by transport substrates and inhibitors. Science 2019, 364, 689–692. [Google Scholar] [CrossRef]
- Rosenberg, M.F.; Mao, Q.; Holzenburg, A.; Ford, R.C.; Deeley, R.G.; Cole, S.P. The structure of multidrug resistance protein 1 (MRP1/ABCC1) crystallization and single-particle analysis. J. Biol. Chem. 2001, 276, 16076–16082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, S.M.; Li, R.; Kanwar, J.R.; Zhou, S.F. Structural and functional properties of human multidrug resistance protein 1 (MRP1/ABCC1). Curr. Med. Chem. 2011, 18, 439–481. [Google Scholar] [CrossRef]
- Iram, S.H.; Cole, S.P. Expression and function of human MRP1 (ABCC1) is dependent on amino acids in cytoplasmic loop 5 and its interface with nucleotide binding domain 2. J. Biol. Chem. 2011, 286, 7202–7213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, S.; Kawabe, T.; Nada, S.; Yamaguchi, A. Identification of basic residues involved in drug export function of human multidrug resistance-associated protein 2. J. Biol. Chem. 2000, 275, 39617–39624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eva, A.; Robbins, K.C.; Andersen, P.R.; Srinivasan, A.; Tronick, S.R.; Reddy, E.P.; Ellmore, N.W.; Galen, A.T.; Lautenberg, J.A.; Papas, T.S.; et al. Cellular genes analogous to retroviral onco genes are transcribed in human tumor cells. Nature 1982, 295, 116–119. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Patel, A.; Ma, S.L.; Li, X.J.; Zhang, Y.K.; Yang, P.-Q. In vitro, in vivo and ex-vivo characterization of ibrutinib: A potent inhibitor of MRP1 efflux function. Br. J. Pharmacol. 2014, 12889. [Google Scholar]


{kind=link}
| FAR Value a | |||||
|---|---|---|---|---|---|
| Cpd. | R1 | R2 | MRP1 | MRP2 | Ratio MRP2/MRP1 |
| 4 | 4-OMe | 3-OMe | 1.07 ± 0.17 | 2.01 ± 0.23 | 1.88 |
| 5 | 4-OMe | 4-OMe | 0.81 ± 0.15 | 1.40 ± 0.26 | 1.75 |
| 6 | 4-OMe | 3-Me | 1.16 ± 0.18 | 1.32 ± 0.32 | 1.13 |
| 7 | 4-OMe | 4-Me | 1.04 ± 0.11 | 1.55 ± 0.34 | 1.49 |
| 8 | 3-OMe | 3-OMe | 1.35 ± 0.04 | 1.33 ± 0.32 | 0.99 |
| 9 | 3-OMe | 4-OMe | 1.08 ± 0.13 | 1.59 ± 0.31 | 1.47 |
| 10 | 3-OMe | 3-Me | 1.12 ± 0.12 | 1.46 ± 0.34 | 1.30 |
| 11 | 3-OMe | 4-Me | 1.50 ± 0.19 | 1.57 ± 0.31 | 1.05 |
| 12 | 3,4-OMe | 3-OMe | 1.32 ± 0.13 | 1.48 ± 0.37 | 1.17 |
| 13 | 3,4-OMe | 4-OMe | 1.31 ± 0.17 | 1.13 ± 0.21 | 0.86 |
| 14 | 3,4-OMe | 3-Me | 1.17 ± 0.19 | 1.78 ± 0.43 | 1.52 |
| 15 | 3,4-OMe | 4-Me | 1.00 ± 0.09 | 1.39 ± 0.32 | 1.39 |
| Probenecid | 1.23 ± 0.11 | 1.00 ± 0.21 | 0.86 | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kreutzer, D.; Ritter, C.A.; Hilgeroth, A. Novel Nonsymmetrical 1,4-Dihydropyridines as Inhibitors of Nonsymmetrical MRP-Efflux Pumps for Anticancer Therapy. Pharmaceuticals 2020, 13, 146. https://doi.org/10.3390/ph13070146
Kreutzer D, Ritter CA, Hilgeroth A. Novel Nonsymmetrical 1,4-Dihydropyridines as Inhibitors of Nonsymmetrical MRP-Efflux Pumps for Anticancer Therapy. Pharmaceuticals. 2020; 13(7):146. https://doi.org/10.3390/ph13070146
Chicago/Turabian StyleKreutzer, David, Christoph A. Ritter, and Andreas Hilgeroth. 2020. "Novel Nonsymmetrical 1,4-Dihydropyridines as Inhibitors of Nonsymmetrical MRP-Efflux Pumps for Anticancer Therapy" Pharmaceuticals 13, no. 7: 146. https://doi.org/10.3390/ph13070146
APA StyleKreutzer, D., Ritter, C. A., & Hilgeroth, A. (2020). Novel Nonsymmetrical 1,4-Dihydropyridines as Inhibitors of Nonsymmetrical MRP-Efflux Pumps for Anticancer Therapy. Pharmaceuticals, 13(7), 146. https://doi.org/10.3390/ph13070146