
Identification of Novel Anthracycline Resistance Genes and Their Inhibitors




Abstract
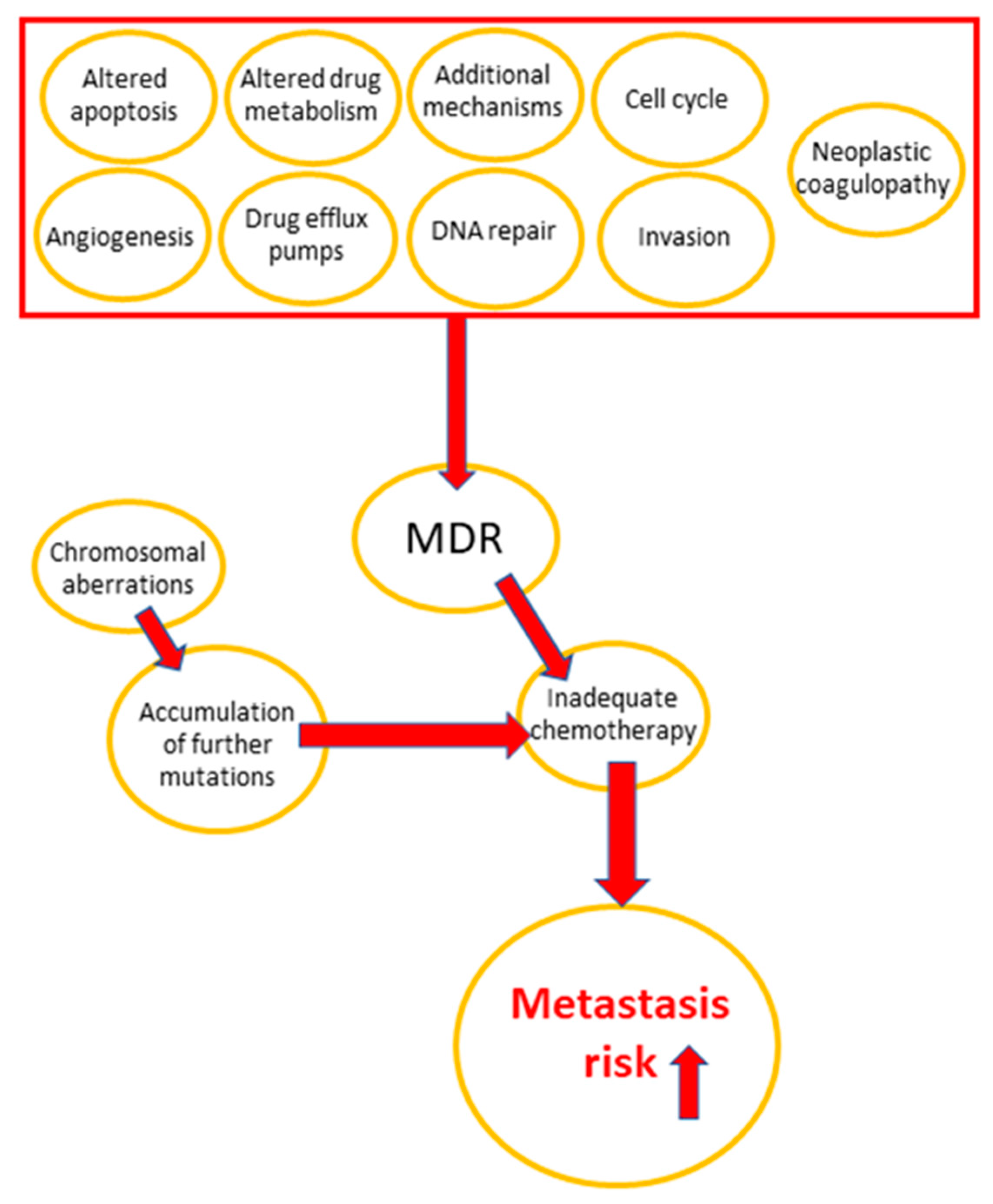
:1. Introduction
2. Results
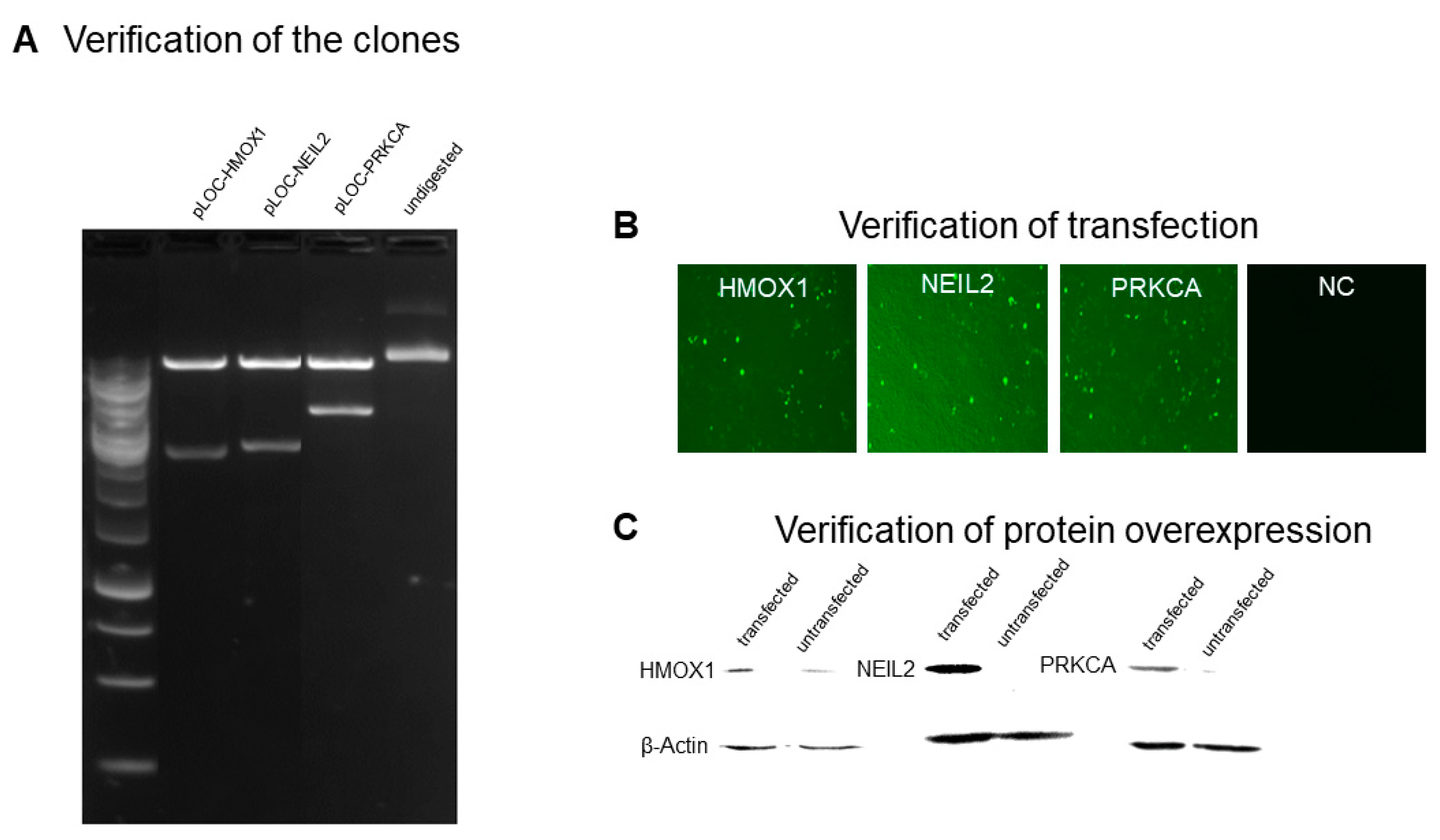
2.1. Generation of Transfectant Cell Lines
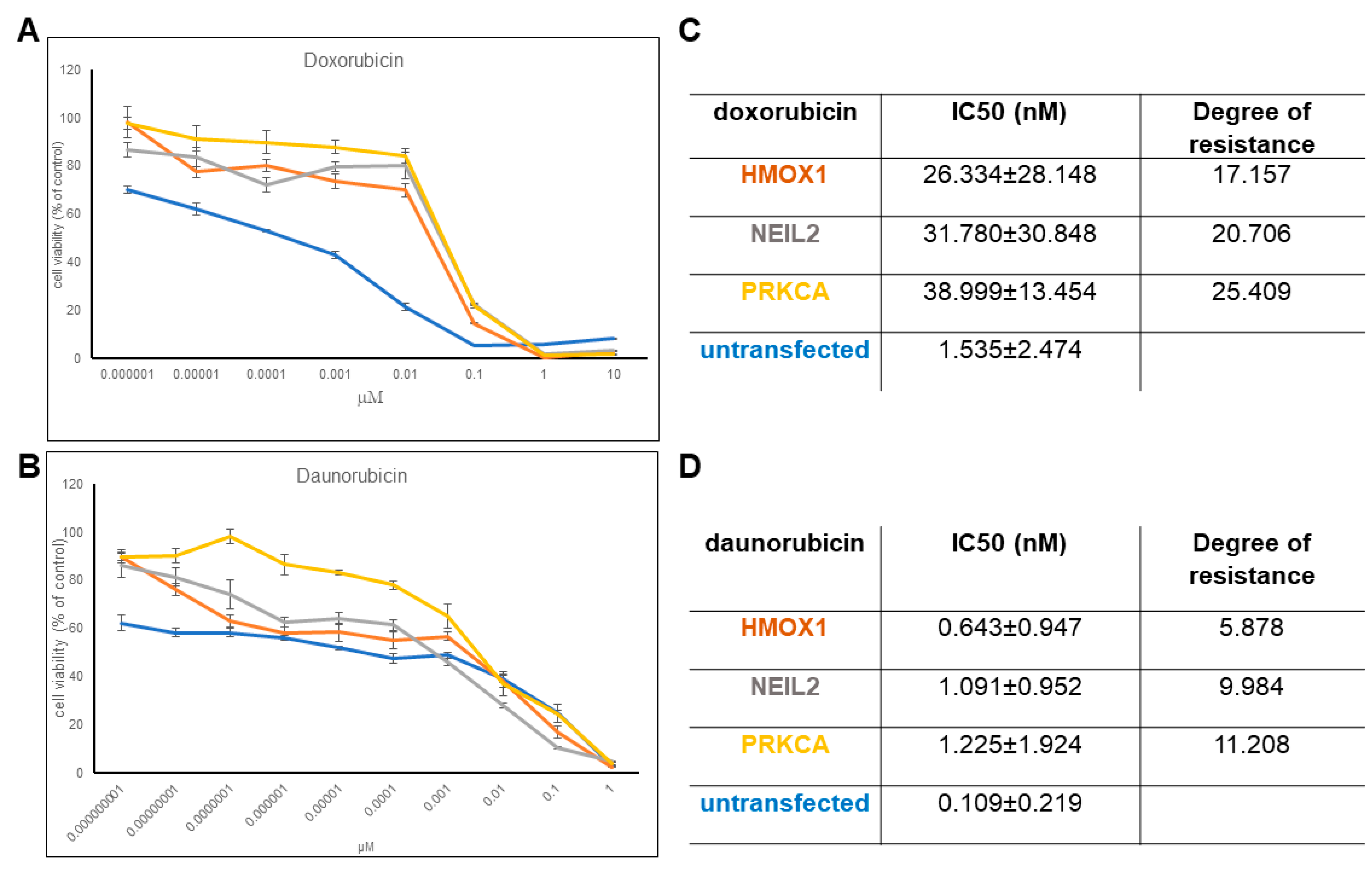
2.2. Resistance of Transfectant Cell Lines toward Anthracyclines
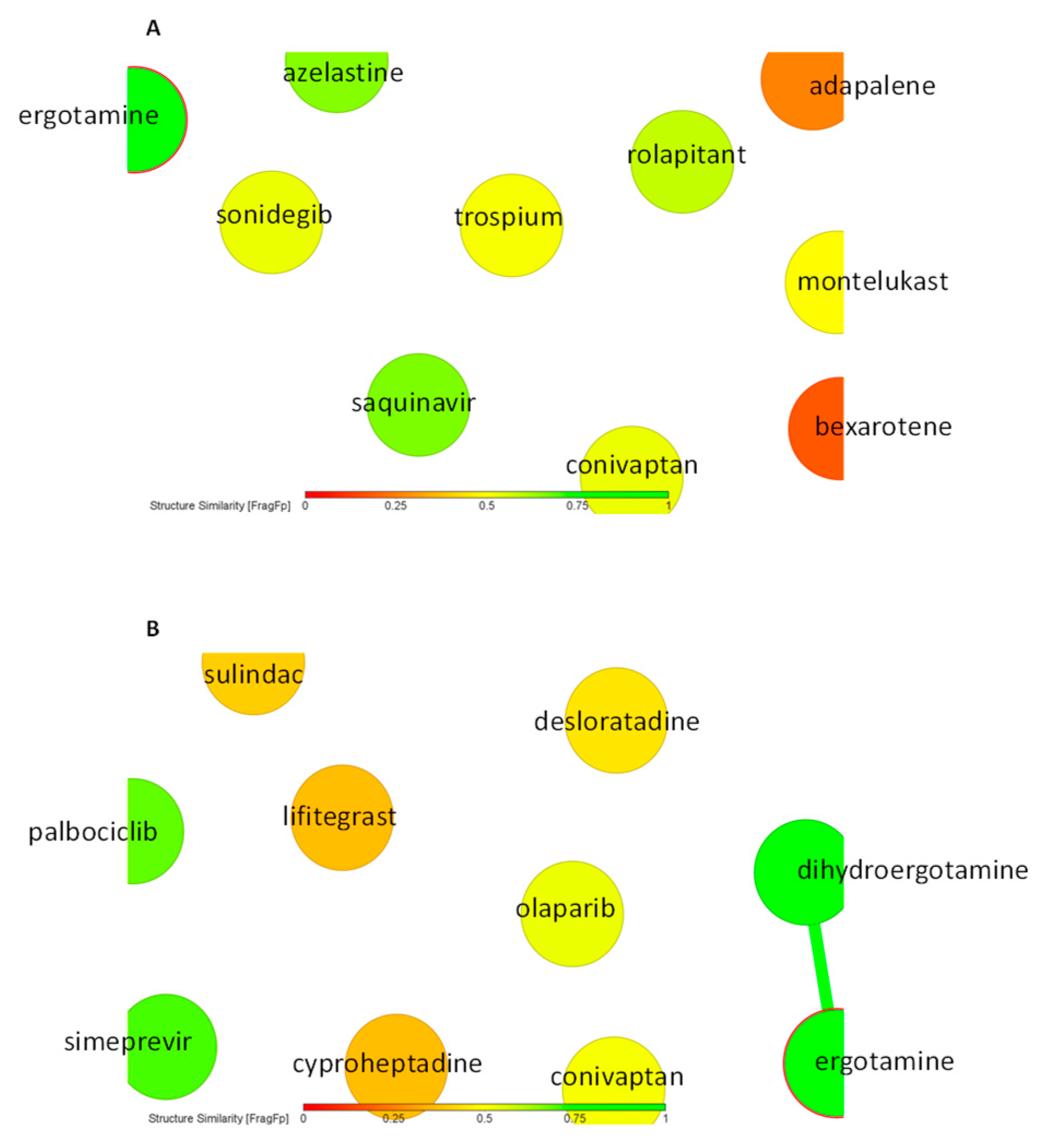
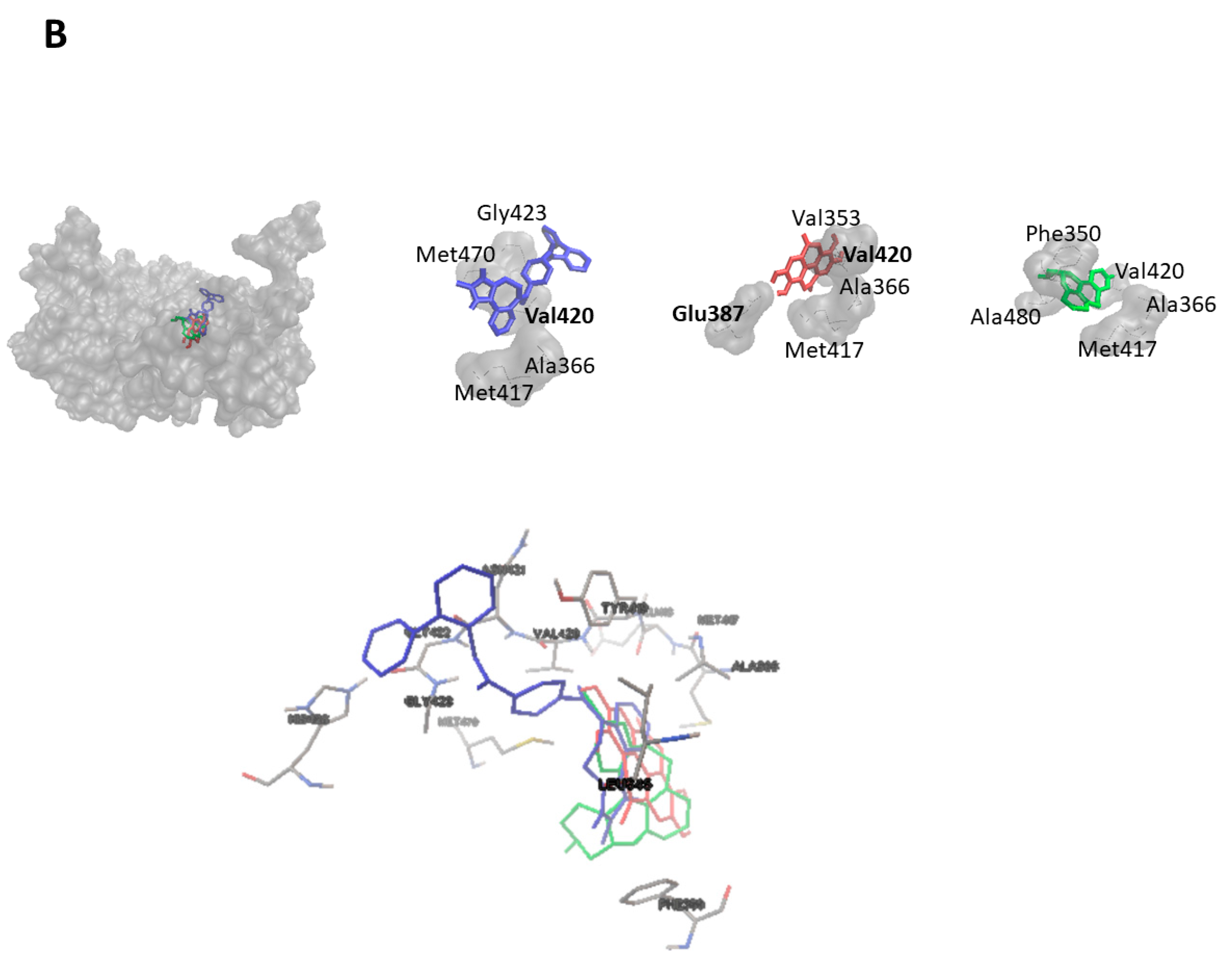
2.3. Virtual Screening for HMOX1 and PRKCA Inhibitors
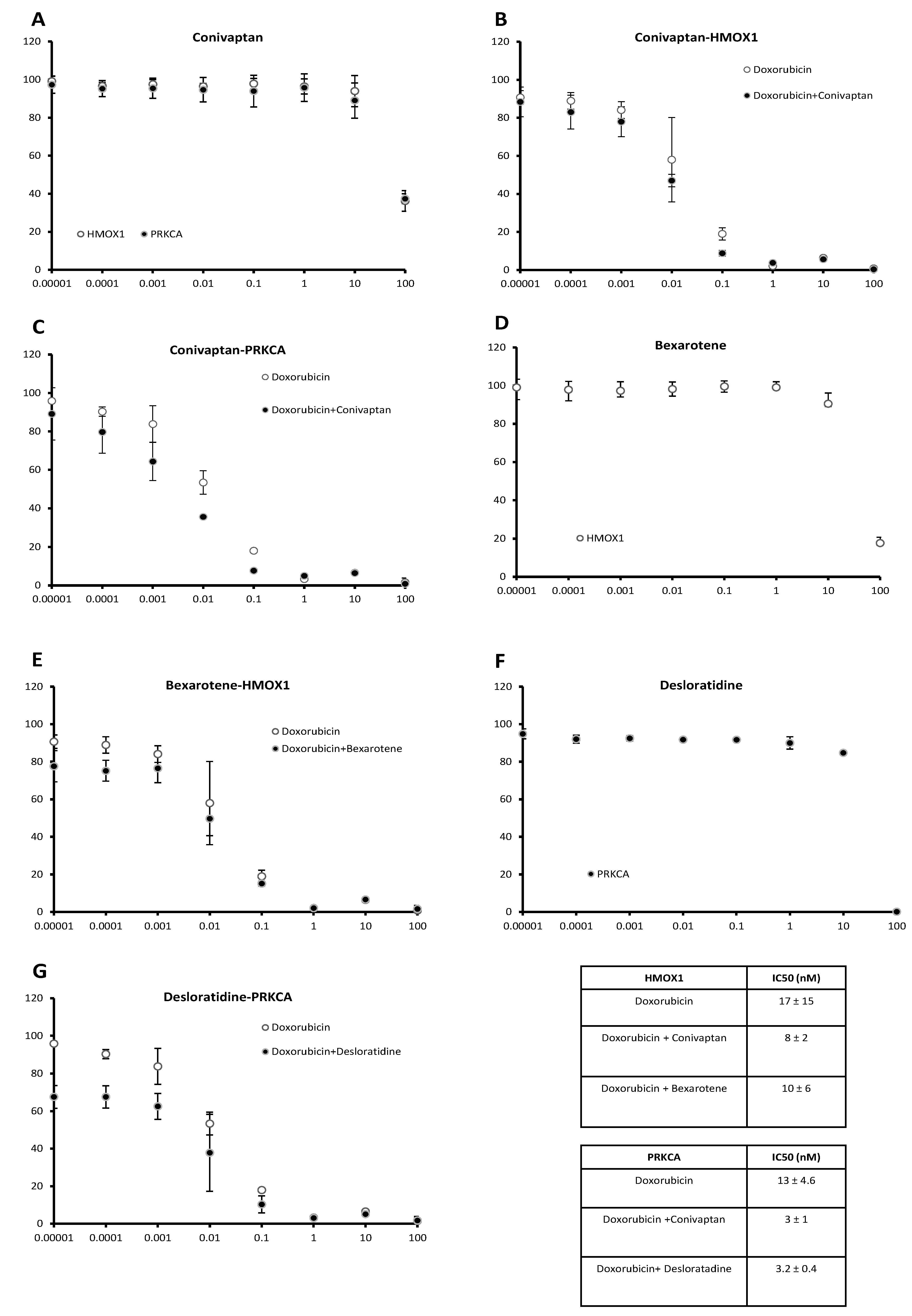
2.4. Doxorubicin-Sensitizing Effects of HMOX1 and PRKCA Inhibitors
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Establishment of Stably Transfected Cell Lines
4.3. Cytotoxicity Assay
4.4. In Silico Screening and Molecular Docking
4.5. Similarity Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Wang, J.; Seebacher, N.; Shi, H.; Kan, Q.; Duan, Z. Novel strategies to prevent the development of multidrug resistance (MDR) in cancer. Oncotarget 2017, 8, 84559–84571. [Google Scholar] [CrossRef] [Green Version]
- Kadioglu, O.; Cao, J.; Kosyakova, N.; Mrasek, K.; Liehr, T.; Efferth, T. Genomic and transcriptomic profiling of resistant CEM/ADR-5000 and sensitive CCRF-CEM leukaemia cells for unravelling the full complexity of multi-factorial multidrug resistance. Sci. Rep. 2016, 6, 36754. [Google Scholar] [CrossRef] [Green Version]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Ling, V. The molecular basis of multidrug resistance in cancer: The early years of P-glycoprotein research. FEBS Lett. 2006, 580, 998–1009. [Google Scholar] [CrossRef] [Green Version]
- Gottesman, M.M. Mechanisms of cancer drug resistance. Annu. Rev. Med. 2002, 53, 615–627. [Google Scholar] [CrossRef] [Green Version]
- Jabbour, E.; Cortes, J.E.; Kantarjian, H. Second-line therapy and beyond resistance for the treatment of patients with chronic myeloid leukemia post imatinib failure. Clin. Lymphoma Myeloma 2009, 9 (Suppl. 3), 272–279. [Google Scholar] [CrossRef]
- Eghtedar, A.; Kantarjian, H.; Jabbour, E.; O’Brien, S.; Burton, E.; Garcia-Manero, G.; Verstovsek, S.; Ravandi, F.; Borthakur, G.; Konopleva, M.; et al. Outcome after failure of second generation tyrosine kinase inhibitors treatment as first-line therapy for patients with chronic myeloid leukemia. Clin. Lymphoma Myeloma Leuk. 2013, 13, 477–484. [Google Scholar] [CrossRef] [Green Version]
- Pollyea, D.A.; Kohrt, H.E.; Medeiros, B.C. Acute myeloid leukaemia in the elderly: A review. Br. J. Haematol. 2011, 152, 524–542. [Google Scholar] [CrossRef]
- Efferth, T.; Volm, M. Multiple resistance to carcinogens and xenobiotics: P-glycoproteins as universal detoxifiers. Arch. Toxicol. 2017, 91, 2515–2538. [Google Scholar] [CrossRef]
- Efferth, T.; Konkimalla, V.B.; Wang, Y.F.; Sauerbrey, A.; Meinhardt, S.; Zintl, F.; Mattern, J.; Volm, M. Prediction of broad spectrum resistance of tumors towards anticancer drugs. Clin. Cancer Res. 2008, 14, 2405–2412. [Google Scholar] [CrossRef] [Green Version]
- Holohan, C.; van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Erin, N.; Grahovac, J.; Brozovic, A.; Efferth, T. Tumor microenvironment and epithelial mesenchymal transition as targets to overcome tumor multidrug resistance. Drug Resist. Updates 2020, 53, 100715. [Google Scholar] [CrossRef]
- Boulos, J.C.; Yousof Idres, M.R.; Efferth, T. Investigation of cancer drug resistance mechanisms by phosphoproteomics. Pharmacol. Res. 2020, 160, 105091. [Google Scholar] [CrossRef] [PubMed]
- Yan, G.E.; Efferth, T. Broad-spectrum cross-resistance to anticancer drugs mediated by epidermal growth factor receptor. Anticancer Res. 2019, 39, 3585–3593. [Google Scholar] [CrossRef] [PubMed]
- Hientz, K.; Mohr, A.; Bhakta-Guha, D.; Efferth, T. The role of p53 in cancer drug resistance and targeted chemotherapy. Oncotarget 2017, 8, 8921–8946. [Google Scholar] [CrossRef] [Green Version]
- Wright, P.K. Targeting vesicle trafficking: An important approach to cancer chemotherapy. Recent Pat. Anti-Cancer Drug Discov. 2008, 3, 137–147. [Google Scholar] [CrossRef]
- Chiang, S.K.; Chen, S.E.; Chang, L.C. A Dual Role of Heme Oxygenase-1 in Cancer Cells. Int. J. Mol. Sci. 2018, 20, 39. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.F.; Li, W.; Ma, J.Y.; Shao, N.; Zhang, Y.J.; Liu, R.M.; Wu, W.B.; Lin, Y.; Wang, S.M. Knockdown of heme oxygenase-1 promotes apoptosis and autophagy and enhances the cytotoxicity of doxorubicin in breast cancer cells. Oncol. Lett. 2015, 10, 2974–2980. [Google Scholar] [CrossRef] [Green Version]
- Tan, Q.; Wang, H.; Hu, Y.; Hu, M.; Li, X.; Ma, Y.; Wei, C.; Song, L. Src/STAT3-dependent heme oxygenase-1 induction mediates chemoresistance of breast cancer cells to doxorubicin by promoting autophagy. Cancer Sci. 2015, 106, 1023–1032. [Google Scholar] [CrossRef]
- Ma, D.; Fang, Q.; Wang, P.; Gao, R.; Wu, W.; Lu, T.; Cao, L.; Hu, X.; Wang, J. Induction of heme oxygenase-1 by Na+-H+ exchanger 1 protein plays a crucial role in imatinib-resistant chronic myeloid leukemia cells. J. Biol. Chem. 2015, 290, 12558–12571. [Google Scholar] [CrossRef] [Green Version]
- Isakov, N. Protein kinase C (PKC) isoforms in cancer, tumor promotion and tumor suppression. Semin. Cancer Biol. 2018, 48, 36–52. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.J.; Xu, H.; Qu, J.W.; Zhao, W.Z.; Zhao, Y.B.; Wang, J.H. Modulation of drug resistance in ovarian cancer cells by inhibition of protein kinase C-alpha (PKC-alpha) with small interference RNA (siRNA) agents. Asian Pac. J. Cancer Prev. 2012, 13, 3631–3636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masanek, U.; Stammler, G.; Volm, M. Modulation of multidrug resistance in human ovarian cancer cell lines by inhibition of P-glycoprotein 170 and PKC isoenzymes with antisense oligonucleotides. J. Exp. Ther. Oncol. 2002, 2, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.K.; Shehzad, A.; Jung, J.C.; Sonn, J.K.; Lee, J.T.; Park, J.W.; Lee, Y.S. Protein kinase Calpha protects against multidrug resistance in human colon cancer cells. Mol. Cells 2012, 34, 61–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Yu, G.Z.; Yu, D.H.; Zhu, M.H. PKC alpha-induced drug resistance in pancreatic cancer cells is associated with transforming growth factor-beta 1. J. Exp. Clin. Cancer Res. 2010, 29, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awasthi, S.; Singhal, S.S.; Awasthi, Y.C.; Martin, B.; Woo, J.H.; Cunningham, C.C.; Frankel, A.E. RLIP76 and cancer. Clin. Cancer Res. 2008, 14, 4372–4377. [Google Scholar] [CrossRef] [Green Version]
- Singhal, S.S.; Yadav, S.; Singhal, J.; Drake, K.; Awasthi, Y.C.; Awasthi, S. The role of PKCalpha and RLIP76 in transport-mediated doxorubicin-resistance in lung cancer. FEBS Lett. 2005, 579, 4635–4641. [Google Scholar] [CrossRef] [Green Version]
- Dey, S.; Maiti, A.K.; Hegde, M.L.; Hegde, P.M.; Boldogh, I.; Sarkar, P.S.; Abdel-Rahman, S.Z.; Sarker, A.H.; Hang, B.; Xie, J.; et al. Increased risk of lung cancer associated with a functionally impaired polymorphic variant of the human DNA glycosylase NEIL2. DNA Repair 2012, 11, 570–578. [Google Scholar] [CrossRef] [Green Version]
- Das, A.; Wiederhold, L.; Leppard, J.B.; Kedar, P.; Prasad, R.; Wang, H.; Boldogh, I.; Karimi-Busheri, F.; Weinfeld, M.; Tomkinson, A.E.; et al. NEIL2-initiated, APE-independent repair of oxidized bases in DNA: Evidence for a repair complex in human cells. DNA Repair 2006, 5, 1439–1448. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, M.; Hernandez, M.; Cheungpasitporn, W.; Kashani, K.B.; Riaz, I.; Rangaswami, J.; Herzog, E.; Guglin, M.; Krittanawong, C. Hyponatremia in heart failure: Pathogenesis and management. Curr. Cardiol. Rev. 2019, 15, 252–261. [Google Scholar] [CrossRef]
- Buckley, M.S.; Patel, S.A.; Hattrup, A.E.; Kazem, N.H.; Jacobs, S.C.; Culver, M.A. Conivaptan for treatment of hyponatremia in neurologic and neurosurgical adults. Ann. Pharmacother. 2013, 47, 1194–1200. [Google Scholar] [CrossRef] [PubMed]
- Rianthavorn, P.; Cain, J.P.; Turman, M.A. Use of conivaptan to allow aggressive hydration to prevent tumor lysis syndrome in a pediatric patient with large-cell lymphoma and SIADH. Pediatric Nephrol. 2008, 23, 1367–1370. [Google Scholar] [CrossRef]
- Raftopoulos, H. Diagnosis and management of hyponatremia in cancer patients. Support Care Cancer 2007, 15, 1341–1347. [Google Scholar] [CrossRef] [PubMed]
- Kamijo, H.; Miyagaki, T. Mycosis fungoides and Sezary syndrome: Updates and review of current therapy. Curr. Treat. Options Oncol. 2021, 22, 10. [Google Scholar] [CrossRef]
- Farol, L.T.; Hymes, K.B. Bexarotene: A clinical review. Expert Rev. Anticancer Ther. 2004, 4, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Kassira, S.; Korta, D.Z.; Chapman, L.W.; Dann, F. Review of treatment for alopecia totalis and alopecia universalis. Int. J. Dermatol. 2017, 56, 801–810. [Google Scholar] [CrossRef]
- Iriarte Sotes, P.; Armisen, M.; Usero-Barcena, T.; Rodriguez Fernandez, A.; Otero Rivas, M.M.; Gonzalez, M.T.; Meijide Calderon, A.; Veleiro, B.; Urtigal, the Galician group of interest in urticaria. Efficacy and safety of up-dosing antihistamines in chronic spontaneous urticaria: A systematic review of the literature. J. Investig. Allergol. Clin. Immunol. 2021, 31, 282–291. [Google Scholar] [CrossRef]
- DuBuske, L.M. Review of desloratadine for the treatment of allergic rhinitis, chronic idiopathic urticaria and allergic inflammatory disorders. Expert Opin. Pharmacother. 2005, 6, 2511–2523. [Google Scholar] [CrossRef]
- Fritz, I.; Wagner, P.; Olsson, H. Improved survival in several cancers with use of H1-antihistamines desloratadine and loratadine. Transl. Oncol. 2021, 14, 101029. [Google Scholar] [CrossRef]
- O’Brien, J.; Wilson, I.; Orton, T.; Pognan, F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur. J. Biochem. 2000, 267, 5421–5426. [Google Scholar] [CrossRef]
- Abdelfatah, S.; Bockers, M.; Asensio, M.; Kadioglu, O.; Klinger, A.; Fleischer, E.; Efferth, T. Isopetasin and S-isopetasin as novel P-glycoprotein inhibitors against multidrug-resistant cancer cells. Phytomedicine 2021, 86, 153196. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.N.; Vlahakis, J.Z.; Szarek, W.A.; Nakatsu, K.; Jia, Z. X-ray crystal structure of human heme oxygenase-1 in complex with 1-(adamantan-1-yl)-2-(1H-imidazol-1-yl)ethanone: A common binding mode for imidazole-based heme oxygenase-1 inhibitors. J. Med. Chem. 2008, 51, 5943–5952. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ren, F.; Li, B.; Song, Z.; Chen, P.; Ouyang, L. Ellagic acid exerts antitumor effects via the PI3K signaling pathway in endometrial cancer. J. Cancer 2019, 10, 3303–3314. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | AutoDock 4.2.6 LBE (kcal/mol) | Interacting Amino Acid Residues |
|---|---|---|
| Adapalene | −12.383 ± 0.025 | Lys18, His25, Ala28, Glu29, Thr135, Leu138, Gly139, Ser142, Leu147, Lys179, Arg183, Phe207, Asn210 |
| Montelukast | −12.307 ± 0.671 | Lys18, Thr21, Lys22, His25, Tyr134, Arg136, Gly139, Ser142, Gly143, Leu147, Lys179, Arg183, Phe207 |
| Bexarotene | −11.527 ± 0.051 | Lys18, His25, Tyr134, Thr135, Arg136, Leu138, Gly139, Ser142, Leu147, Lys179, Arg183, Phe207 |
| Conivaptan | −10.837 ± 0.427 | His25, Val50, Leu54, Ile57, Tyr134, Arg136, Leu138, Gly139, Asp140, Ser142, Gly143, Leu147, Phe166, Phe167, Phe207, Asn210, Leu213, Phe214 |
| Sonidegib | −10.420 ± 0.226 | Val59, Glu62, Glu63, Ile65, Glu66, Val77, Tyr78, Phe79, Pro80, Leu83, His84, Lys86, Tyr137 |
| Trospium | −10.010 ± 0.044 | His25, Met34, Phe37, Phe47, Val50, Leu54, Thr135, Arg136, Asp140, Leu147, Phe167, Phe207, Asn210 |
| Azelastine | −9.950 ± <0.001 | His25, Ala28, Glu29, Met34, Gln38, Val50, Thr135, Arg136, Leu147, Phe207, Asn210, Phe214 |
| Ergotamine | −9.877 ± 0.035 | Tyr55, His56, Val59, Tyr107, Gln112, Arg113, Val115, Lys116, His119 |
| Saquinavir | −8.180 ± 0.370 | Leu49, Tyr97, Gln102, Glu103, Val104, Ile105, Pro106, Tyr107, Thr108, Pro109, Gln112, Leu220 |
| Rolapitant | −7.727 ± 0.156 | Glu62, Glu63, Ile65, Glu66, Tyr78, Phe79, Pro80, Leu83, His84, Lys86, Tyr137 |
| 1-(Adamantan-1-yl)-2-(1H-imidazol-1-yl)ethenone | −7.333 ± 0.032 | Glu62, Ile65, Glu66, Tyr78, Phe79, Pro80, Leu83, His84, Tyr137 |
| Compounds | AutoDock 4.2.6 LBE (kcal/mol) | Interacting Amino Acid Residues |
|---|---|---|
| Lifitegrast | −13.080 ± 0.123 | His455, Lys456, Met489, Asp491, Gly492, Tyr515, Gly516, Lys517, Ser518, Pro577, Gly587, Glu588 |
| Conivaptan | −12.207 ± 0.015 | Leu345, Phe350, Ala366, Met417, Glu418, Tyr419, Val420, Asn421, Gly422, Gly423, His428, Met470 |
| Dihydroergotamine | −11.187 ± 0.061 | Leu345, Phe350, Val353, Ala366, Lys368, Glu387, Met417, Tyr419, Val420, Asn421, Met470, Ala480, Asp481 |
| Olaparib | −10.763 ± 0.042 | Phe350, Val353, Ala366, Lys368, Glu387, Thr401, Met417, Tyr419, Asp467, Asn468, Met470, Ala480, Asp481 |
| Simeprevir | −10.663 ± 0.086 | Leu345, Val353, Ala366, Tyr419, Val420, Asn421, Gly422, Gly423, Asp424, Tyr427, His428, Met470, Lys617 |
| Ergotamine | −10.663 ± 0.291 | Leu393, Leu394, Asp395, Lys396, Pro397, Pro398, Gln402, Leu403, Ile449, Phe453, Glu606, Asn607 |
| Desloratadine | −10.450 ± <0.001 | Phe350, Ala366, Lys368, Met417, Tyr419, Val420, Asn468, Ala480, Asp481 |
| Palbociclib | −9.810 ± 0.0529 | Phe350, Ala366, Lys368, Glu387, Leu391, Thr401, Met417, Glu418, Val420, Asp424, Asp467, Met470, Ala480, Asp481 |
| Cyproheptadine | −9.760 ± <0.001 | Gly346, Phe350, Val353, Ala366, Lys368, Glu387, Thr401, Met417, Glu418, Val420, Asp481 |
| Sulindac | −9.207 ± 0.183 | Lys347, Gly351, Lys352, Ile369, Leu370, Lys371, Val374, Val375, Asp378 |
| Ellagic acid | −7.550 ± <0.001 | Val353, Ala366, Lys368, Glu387, Thr401, Met417, Val420 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kadioglu, O.; Elbadawi, M.; Fleischer, E.; Efferth, T. Identification of Novel Anthracycline Resistance Genes and Their Inhibitors. Pharmaceuticals 2021, 14, 1051. https://doi.org/10.3390/ph14101051
Kadioglu O, Elbadawi M, Fleischer E, Efferth T. Identification of Novel Anthracycline Resistance Genes and Their Inhibitors. Pharmaceuticals. 2021; 14(10):1051. https://doi.org/10.3390/ph14101051
Chicago/Turabian StyleKadioglu, Onat, Mohamed Elbadawi, Edmond Fleischer, and Thomas Efferth. 2021. "Identification of Novel Anthracycline Resistance Genes and Their Inhibitors" Pharmaceuticals 14, no. 10: 1051. https://doi.org/10.3390/ph14101051
APA StyleKadioglu, O., Elbadawi, M., Fleischer, E., & Efferth, T. (2021). Identification of Novel Anthracycline Resistance Genes and Their Inhibitors. Pharmaceuticals, 14(10), 1051. https://doi.org/10.3390/ph14101051