3.1. Chemical Synthesis
3.1.1. General Methods
Commercially available reagents and solvents were used without further purification unless stated otherwise. Preparative normal phase chromatography was performed on a CombiFlash Rf 150 (Teledyne Isco) with pre-packed RediSep Rf silica gel cartridges. Thin-layer chromatography was performed with aluminum-backed sheets with silica gel 60 F254 (Merck, ref 1.05554), and spots were visualized with UV light and 1% aqueous solution of KMnO
4. Melting points were determined in open capillary tubes with an MFB 595010M Gallenkamp. Next, 400 MHz 1H and 100.6 MHz 13C NMR spectra were recorded on a Varian Mercury 400 or on a Bruker 400 Avance III spectrometers. The chemical shifts are reported in ppm (δ scale) relative to internal tetramethylsilane, and coupling constants are reported in Hertz (Hz). Assignments given for the NMR spectra of selected new compounds have been carried out on the basis of DEPT, COSY
1H/
1H (standard procedures), and COSY
1H/
13C (gHSQC and gHMBC sequences) experiments. IR spectra were run on Perkin-Elmer Spectrum RX I, Perkin-Elmer Spectrum TWO or Nicolet Avatar 320 FT-IR spectrophotometers. Absorption values are expressed as wavenumbers (cm
−1); only significant absorption bands are given. High-resolution mass spectrometry (HRMS) analyses were performed with an LC/MSD TOF Agilent Technologies spectrometer. The elemental analyses were carried out in a Flash 1112 series Thermofinnigan elemental microanalyzer (A5) to determine C, H and N. The structure of all new compounds was confirmed by elemental analysis and/or accurate mass measurement, IR,
1H NMR, and
13C NMR (check them in
Supplementary Materials). The analytical samples of all the new compounds, which were subjected to pharmacological evaluation, possessed purity ≥95% as evidenced by their elemental analyses.
3.1.2. Synthesis of t-Butyl 4-(2-((9-Methyl-5,6,8,9,10,11-hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-yl)amino)-2-oxoethyl)piperidine-1-carboxylate, 4a
To a suspension of 9-methyl-5,6,8,9,10,11-hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-amine hydrochloride (500 mg, 1.89 mmol), 2a, in EtOAc (5 mL), 2-(1-(t-butoxycarbonyl)piperidin-4-yl)acetic acid (461 mg, 1.89 mmol), 3, HOBt (384 mg, 2.84 mmol), EDC·HCl (440 mg, 2.84 mmol), and Et3N (767 mg, 7.58 mmol) were added. The mixture was stirred at room temperature for 24 h. Water (10 mL) and DCM (20 mL) were added to the resulting suspension and the two phases were separated. The organic phase was washed with sat. NaHCO3 aqueous solution (10 mL), brine (10 mL), 2N HCl solution (10 mL) and 2N NaOH (10 mL), dried over anh. Na2SO4, filtered, and concentrated under vacuum to give 4a as a yellow solid (515 mg, 60% yield). 1H-NMR (400 MHz, CDCl3) δ: 0.92 (s, 3 H), 1.11 (dq, J = 4.4 Hz, J’ = 11.6 Hz, 2 H), 1.4 (s, 9 H), 1.54 (d, J = 13.6 Hz, 2 H), 1.63–1.68 (complex signal, 4 H), 1.84 (s, 2 H), 1.91 (m, 1 H), 1.97 (s, 2 H), 2.0 (d, J = 12.8 Hz, 2 H), 2.14–2.18 (complex signal, 2 H), 2.69 (t, J = 13.2 Hz, 2 H), 3.06 (t, J = 6 Hz, 2 H), 4.06 (broad signal, 2 H), 5.14 (s, 1 H), 7.02–7.08 (complex signal, 4 H).
3.1.3. Synthesis of N-(9-Methyl-5,6,8,9,10,11-hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-yl)-2-(piperidin-4-yl)acetamide, 5a
To a solution of 4a (250 mg, 0.55 mmol) in DCM (4 mL) was added 4M HCl in 1,4-dioxane (0.5 mL). The reaction mixture was stirred at room temperature for 3 days. Then, the solvent was evaporated under vacuum and the residue was dissolved in DCM (10 mL) and washed with 5N NaOH solution, dried over anh. Na2SO4, filtered, and concentrated under vacuum to give 5a as a yellow solid (189 mg, 97% yield). 1H-NMR (400 MHz, CDCl3) δ: 0.91 (s, 3 H), 1.12 (dq, J = 4 Hz, J’ = 12.0 Hz, 2 H), 1.53 (d, J = 13.2 Hz, 2 H), 1.62–1.71 (complex signal, 4 H), 1.84 (s, 2 H), 1.88 (m, 1 H), 1.95–2.01 (complex signal, 4 H), 2.14–2.19 (complex signal, 2 H), 2.6 (dt, J = 2.8 Hz, J’ = 12.0 Hz, 2 H), 3.00–3.07 (complex signal, 4 H), 5.15 (s, 1 H), 7.02–7.09 (complex signal, 4 H).
3.1.4. Synthesis of N-(5,6,8,9,10,11-Hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-yl)-2-(piperidin-4-yl)acetamide, 5b
To a suspension of 5,6,8,9,10,11-hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-amine (90 mg, 0.42 mmol) 2b, in DMF (2 mL), 2-(1-(t-butoxycarbonyl)piperidin-4-yl)acetic acid (123 mg, 0.51 mmol), 3, HATU (239 mg, 0.63 mmol), and DIPEA (162 mg, 1.26 mmol) were added. The mixture was stirred at room temperature for 24 h. Solvent was concentrated in vacuo and EtOAc (10 mL) was added. The mixture was washed with NaHCO3 sat. (2 × 30 mL). The organic phase was dried over anh. Na2SO4, filtered, and concentrated under vacuum to give carbamate 4b (185 mg) as a yellow gum that was used as such without further purification. HCl 4 M in dioxane (2 mL) and dioxane (2 mL) were added to the previous gum of 4b and the mixture was stirred at room temperature for 2 h. Na2CO3 sat. was added until pH = 12 followed by EtOAc (15 mL) and the mixture was partitioned. The aqueous layer was extracted with EtOAc/MeOH 9/1 (2 × 10 mL). All organic phases were joined, dried over anh. Na2SO4, filtered, and concentrated under vacuum. The resulting crude was purified by column chromatography in silica gel (SiO2, DCM/Methanol mixtures) and gave 5b as an orangish solid (108 mg, 72% overall yield), mp 185–186 °C. IR (ATR) v: 3301, 2907, 2855, 2349, 1653, 1556, 1450, 1356, 1320, 1280, 1137, 1046, 951, 929, 794, 765, 737, 660, 631, 616 cm−1. 1H-NMR (400 MHz, CD3OD) δ: 1.20 [m, 2 H, 3′′(5′′)-Hax], 1.63–1.77 [complex signal, 4 H, 10′(13′)-Hax, 3′′(5′′)-Heq], 1.83 (m, 1 H, 4′′-H), 1.98 [m, 2 H, 10′(13′)-Heq], 2.02 (d, J = 7.2 Hz, 2 H, 8′-H), 2.06 (m, 2 H, 2-H), 2.12–2.26 [complex signal, 4 H, 6′(12′)-H], 2.32 (m, 1 H, 9′-H), 2.59 [m, 2 H, 2′′(6′′)-Hax], 2.98–3.08 [complex signal, 4 H, 5′(11′)-H, 2′′(6′′)-Heq], 7.03 [s, 4 H, 1′(4′)-H, 2′(3′)-H]. 13C-NMR (100.6 MHz, CD3OD) δ: 32.6 (CH, C9′), 33.1 [CH2, C3′′(5′′)], 35.0 (CH, C4′′), 35.7 [CH2, C10′(13′)], 40.4 [CH2, C6′(12′)], 41.5 (CH2, C2), 42.6 [CH, C5′(11′)], 45.2 (CH2, C8′), 46.7 [CH2, 2′′(6′′)], 53.8 (C, C7′), 127.3 [CH, C2′(3′)], 129.0 [CH, C1′(4′)], 148.0 [C, C4a’(11a’)], 173.9 (C, CO). HRMS: Calcd for [C22H30N2O + H]+: 339.2438, found: 339.2431.
3.1.5. Synthesis of N-(5,6,8,9,10,11-Hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-yl-9-d)-2-(piperidin-4-yl)acetamide, 5c
To a suspension of 5,6,8,9,10,11-hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-9-d-7-amine (90 mg, 0.42 mmol), 2c, in DMF (2 mL), 2-(1-(t-butoxycarbonyl)piperidin-4-yl)acetic acid (123 mg, 0.51 mmol), 3, HATU (239 mg, 0.63 mmol), and DIPEA (162 mg, 1.26 mmol) were added. The mixture was stirred at room temperature for 24 h. Solvent was concentrated in vacuo and EtOAc (10 mL) was added. The mixture was washed with NaHCO3 sat. (2 x 30 mL). The organic phase was dried over anh. Na2SO4, filtered, and concentrated under vacuum to give 4c (185 mg) as a yellow gum that was used as such without further purification. HCl 4 M in dioxane (2 mL) and dioxane (2 mL) were added to the aforementioned gum of 4c (185 mg, 0.42 mmol), and the mixture was stirred at room temperature for 2 h. Na2CO3 sat. was added until pH = 12 followed by EtOAc (15 mL) and the mixture was partitioned. The aqueous layer was extracted with EtOAc/MeOH 9/1 (2 × 10 mL). All organic phases were joined, dried over anh. Na2SO4, filtered, and concentrated under vacuum. The resulting crude was purified by column chromatography in silica gel (SiO2, DCM/Methanol mixtures) and gave 5c as an orangish solid (129 mg, 90% yield), mp: 188–189 °C. IR (ATR) v: 3300, 2909, 2852, 2351, 1654, 1555, 1492, 1449, 1356, 1297, 1280, 1046, 928, 840, 794, 764, 737, 655, 616 cm−1. 1H-NMR (400 MHz, CD3OD) δ: 1.24 [m, 2 H, 3′′(5′′)-Hax], 1.69–1.76 [complex signal, 4 H, 10′(13′)-Hax, 3′′(5′′)-Heq], 1.86 (m, 1 H, 4′′-H), 1.99 [m, 2 H, 10′(13′)-Heq], 2.02–2.07 (complex signal, 4 H, 2-H, 8′-H), 2.19 [m, 4 H, 6′(12′)-H], 2.66 (dt, J = 12.6 Hz, J’ = 2.8 Hz, 2 H, 2′′(6′′)-Hax), 3.04 [m, 2 H, 5′(11′)-H], 3.09 [dt, J = 12.6 Hz, J’ = 2.8 Hz, 2 H, 2′′(6′′)-Heq], 7.03 [s, 4 H, 1′(4′)-H, 2′(3′)-H]. 13C-NMR (100.6 MHz, CD3OD) δ: 32.4 [CH2, C3′′(5′′)], 32.5 (m, CD, C9′), 34.5 (CH, C4′′), 35.6 [CH2, C10′(13′)], 40.4 [CH2, C6′(12′)], 41.4 (CH2, C2), 42.6 [CH, C5′(11′)], 44.9 (CH2, C8′), 46.4 [CH2, 2′′(6′′)], 53.8 (C, C7′), 127.3 [CH, C2′(3′)], 129.0 [CH, C1′(4′)], 148.0 [C, C4a’(11a’)], 173.7 (C, CO). HRMS: Calcd for [C22H29DN2O + H]+: 340.2494, found: 340.2500.
3.1.6. Synthesis of N-(9-Fluoro-5,6,8,9,10,11-hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-yl)-2-(piperidin-4-yl)acetamide, 5d
To a suspension of 9-fluoro-5,6,8,9,10,11-hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-amine hydrochloride (200 mg, 0.75 mmol), 2d, in DMF (3 mL), 2-(1-(t-butoxycarbonyl)piperidin-4-yl)acetic acid (218 mg, 0.90 mmol), HATU (430 mg, 1.13 mmol), and DIPEA (386 mg, 3.00 mmol) were added. The mixture was stirred at room temperature for 24 h. Solvent was concentrated in vacuo and EtOAc (15 mL) was added. The mixture was washed with NaHCO3 sat. (2 × 30 mL). The organic phase was dried over anh. Na2SO4, filtered, and concentrated under vacuum. The resulting crude of 4d was used as such without further purification. 1H-NMR (400 MHz, CDCl3) δ: 1.10 (m, 2 H), 1.44 (s, 9 H), 1.47 (s, 2 H), 1.66 (d, J = 13.5 Hz, 2 H), 1.87–2.05 (complex signal, 6 H), 2.09–2.24 (complex signal, 5 H), 2.27 (d, J = 6.2 Hz, 2 H), 2.68 (m, 2 H), 3.23 (m, 2 H), 5.23 (s, 1 H), 7.05–7.15 (complex signal, 4 H). HCl 4 M in dioxane (2 mL), and dioxane (2 mL) were added to 4d (341 mg, 0.75 mmol) and the mixture was stirred at room temperature for 4 h. Na2CO3 sat. was added until pH = 12 followed by EtOAc (20 mL) and the mixture was partitioned. The aqueous layer was extracted with EtOAc/MeOH 9/1 (2 × 15 mL). All organic phases were joined, dried over anh. Na2SO4, filtered, and concentrated under vacuum. The resulting crude was purified by column chromatography in silica gel (SiO2, DCM/Methanol/NH3 mixtures) and gave 5d as a yellowish solid (186 mg, 70% yield). 1H NMR (400 MHz, DMSO-d6) δ: 0.98 (m, 2 H), 1.50 (m, 2 H), 1.72 (m, 2 H), 1.90 (d, J = 7.2 Hz, 2 H), 1.98–2.02 (complex signal, 4 H), 2.06–2.13 (complex signal, 4 H), 2.39 (dm, J = 12.0 Hz, 2 H), 2.86 (dm, J = 12.0 Hz, 2 H), 3.21 (s, 2 H), 7.11 (s, 4 H), 7.61 (s, 1 H).
3.1.7. Synthesis of N-(9-Chloro-5,6,8,9,10,11-hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-yl)-2-(piperidin-4-yl)acetamide Hydrochloride, 5e
To a suspension of 9-chloro-5,6,8,9,10,11-hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-amine hydrochloride (178 mg, 0.63 mmol), 2e, in DMF (3 mL), 2-(1-(t-butoxycarbonyl)piperidin-4-yl)acetic acid (183 mg, 0.75 mmol), HATU (357 mg, 0.94 mmol), and DIPEA (326 mg, 2.52 mmol) were added. The mixture was stirred at room temperature for 6 h. Solvent was concentrated in vacuo and EtOAc (15 mL) was added. The mixture was washed with NaHCO3 sat. (2 × 30 mL). The organic phase was dried over anh. Na2SO4, filtered, and concentrated under vacuum. The resulting crude of 4e was used as such without further purification. 1H-NMR (400 MHz, CDCl3) δ: 1.44 (s, 9 H), 1.69 (m, 2 H), 1.91 (m, 1 H), 1.95–2.01 (complex signal, 4 H), 2.04 (s, 1 H), 2.12–2.32 (complex signal, 6 H), 2.38 (m, 2 H), 2.53 (s, 1 H), 2.70 (d, J = 12.1 Hz, 2 H), 3.17 (t, J = 6.5 Hz, 2 H), 4.07 (m, 2 H), 5.21 (s, 1 H), 7.04–7.15 (complex signal, 4 H). HCl 4 M in dioxane (2 mL) and dioxane (2 mL) were added to 4e (296 mg, 0.63 mmol) and the mixture was stirred at room temperature overnight. Saturated aqueous NaHCO3 solution (20 mL) was added followed by EtOAc (15 mL) and the mixture was partitioned. The aqueous phase was further extracted with EtOAc/MeOH 9/1 (2 × 15 mL). All organic phases were joined, dried over anh. Na2SO4, filtered, and solvents were concentrated under vacuum. The resulting crude was purified by column chromatography in silica gel (SiO2, DCM/MeOH/NH3 mixtures). Fractions containing the desired product were collected and concentrated in vacuo to afford a reddish solid. HCl 2 M in Et2O (5 mL) was added to form its hydrochloride, followed by filtration of the solid to afford 5e as an orange solid (84 mg, 33% yield), mp: > 300 °C. IR (ATR) v: 3300, 2937, 1655, 1557, 1493, 1450, 1357, 1298, 1280, 1206, 1089, 930, 793, 765, 632, 614 cm−1. 1H-NMR (400 MHz, CD3OD) δ: 1.47 [m, 2 H, 3′′(5′′)-Hax], 1.90 [d, J = 14.1 Hz, 2 H, 3′′(5′′)-Heq], 1.99–2.09 [complex signal, 5 H, 6′(12′)-Hax, 10′(13′)-Hax, 4′′-H], 2.14 (d, J = 7.2 Hz, 2 H, 2-H), 2.22 [dd, J = 13.2 Hz, J’ = 6.0 Hz, 2 H, 6′(12′)-Heq], 2.38 [dd, J = 13.2 Hz, J’ = 6.0 Hz, 2 H, 10′(13′)-Heq], 2.49 (s, 2 H, 8′-H), 2.97 [t, J = 12.5 Hz, 2 H, 2′′(6′′)-Hax], 3.18 [broad t, J = 6.2 Hz, 2 H, 5′(11′)-H], 3.36 [d, J = 12.4 Hz, 2 H, 2′′(6′′)-Heq], 7.03–7.14 [complex signal, 4 H, 1′(4′)-H, 2′(3′)-H]. 13C-NMR (100.6 MHz, CD3OD) δ: 29.5 [CH2, C3′′(5′′)], 32.7 (CH, C4′′), 38.9 [CH2, C6′(12′)], 42.5 [CH, C5′(11′)], 43.7 (CH2, C2), 45.1 [CH2, 2′′(6′′)], 45.9 [CH2, C10′(13′)], 50.9 (CH2, C8′), 57.6 (C, C7′), 70.2 (C, C9′), 127.9 [CH, C2′(3′)], 129.1 [CH, C1′(4′)], 146.1 [C, C4a’(11a’)], 173.1 (C, CO). Anal. Calcd for C22H29ClN2O · 1 HCl: C 64.54, H 7.39, N 6.84. Calcd for C22H29ClN2O · 2 HCl: C 59.27, H 7.01, N 6.28. Found: C 59.30, H 7.00, N 6.37. HRMS: Calcd for [C22H29ClN2O + H]+: 373.2041, found: 373.2047.
3.1.8. Synthesis of 2-(1-Acetylpiperidin-4-yl)-N-(9-methyl-5,6,8,9,10,11-hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-yl)acetamide, 6a
To a solution of 5a (200 mg, 0.57 mmol) in anh. DCM (5 mL) under argon atmosphere was added anh. Et3N (69 mg, 0.68 mmol). The mixture was cooled down to 0°C and acetyl chloride (45 mg, 0.57 mmol) was added dropwise. Then, the reaction mixture was stirred at room temperature overnight and quenched by addition of 2N HCl solution (3 mL). The two phases were separated, and the aqueous layer was extracted with EtOAc (2 × 20 mL). The combined organic phases were washed with 2N NaOH solution, dried over anh. Na2SO4, filtered, and concentrated under vacuum. Column chromatography (SiO2, Hexane/EtOAc mixtures) gave 6a as a white solid (134 mg, 60% yield), mp 85–86 °C. IR (NaCl disk): 3315, 3060, 3017, 2916, 2860, 1631, 1544, 1493, 1450, 1361, 1304, 1273, 1241, 1197, 1165, 1138, 1096, 1048 cm−1. 1H-NMR (400 MHz, CDCl3) δ: 0.91 (s, 3 H, C9′′-CH3), 1.10 [m, 2 H, 3′(5′)-Hax], 1.53 [d, J = 13.6 Hz, 2 H, 10′′(13′′)-Hax], 1.65 [dm, J = 12.8 Hz, 10′′(13′′)-Heq], 1.71 (d, J = 12.8 Hz, 1 H, 5′-Heq or 3′-Heq), 1.76 (d, J = 12.4 Hz, 1 H, 3′-Heq or 5′-Heq), 1.83 (s, 2 H, 8′′-H), 1.96–2.04 [complex signal, 5 H, 2-H2, 4′-H, 6′′(12′′)-Hax], 2.06 (s, 3 H, COCH3), 2.15 [m, 2 H, 6′′(12′′)-Heq], 2.53 (t, J = 12.4 Hz, 1 H, 2′-Hax or 6′-Hax), 3.03 (m, 1 H, 6′-Hax or 2′-Hax), 3.08 [broad t, J = 6.0 Hz, 2 H, 5′′(11′′)-H], 3.76 (d, J = 13.6 Hz, 1 H, 6′-Heq or 2′-Heq), 4.58 (d, J = 13.2 Hz, 1 H, 2′-Heq or 6′-Heq), 5.25 (s, 1H, NH), 7.03 [m, 2 H, 1′′(4′′)-H], 7.06 [m, 2 H, 2′′(3′′)-H]. 13C-NMR (100.6 MHz, CDCl3) δ: 21.5 (CH3, COCH3), 31.6 (CH2, C5′ or C3′), 32.2 (CH3, C9-CH3), 32.4 (CH2, C3′ or C5′), 33.56 (C or CH, C9′′ or C4′), 33.57 (CH or C, C4′ or C9′′), 39.0 (CH2, C6′′ or C12′′), 39.1 (CH2, C12′′ or C6′′), 40.9 [CH, C5′′(11′′)], 41.1 [CH2, C10′′(13′′)], 41.7 (CH2, C2′ or C6′), 44.4 (CH2, C2), 46.5 (CH2, C6′ or C2′), 47.2 (CH2, C8′′), 54.7 (C, C7′′), 126.3 [CH, C2′′(3′′)], 128.0 [CH, C1′′(4′′)], 146.1 [C, C4a’’(11a’’)], 168.7 (C, COCH3), 170.4 (C, NHCO). Anal. Calcd for C25H34N2O2 · 0.5 H2O: C 74.41, H 8.74, N 6.94. Found: C 74.36, H 8.79, N 6.74. HRMS: Calcd for [C25H34N2O2 + H]+: 395.2693; Found: 395.2691.
3.1.9. Synthesis of 2-[1-(Isopropylsulfonyl)piperidin-4-yl]-N-(9-methyl-5,6,8,9,10,11-hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-yl)acetamide, 6b
To a solution of 5a (185 mg, 0.52 mmol) in DCM (5 mL) was added Et3N (63 mg, 0.63 mmol). The mixture was cooled down to 0 °C and propane-2-sulfonyl chloride (74 mg, 0.52 mmol) was added dropwise. Then, the reaction mixture was stirred at room temperature overnight and quenched by an addition of 2N HCl solution (3 mL). The two phases were separated, and the aqueous phase was extracted with EtOAc (2 × 20 mL). The combined organic phases were washed with 5N NaOH solution, dried over anh. Na2SO4, filtered, and concentrated under vacuum to give a yellow solid. Column chromatography (SiO2, Hexane/EtOAc mixtures) gave 6b as a white solid (145 mg, 60% yield). The analytical sample was obtained by crystallization from hot EtOAc, mp 172 -173 °C. IR (NaCl disk): 3365, 3319, 3059, 3018, 2916, 2852, 1648, 1536, 1493, 1452, 1361, 1323, 1309, 1265, 1190, 1167, 1138, 1091, 1044, 1011, 993, 945, 905, 881, 801, 759, 732, 702, 665 cm−1. 1H-NMR (400 MHz, CDCl3) δ: 0.91 (s, 3 H, C9′′-CH3), 1.25 [dq, J = 12.0 Hz, J’ = 4.0 Hz, 2 H, 3′(5′)-Hax], 1.31 [d, J = 6.8 Hz, 6 H, CH(CH3)2], 1.53 [d, J = 13.6 Hz, 2 H, 10′′(13′′)-Hax], 1.65 [dm, J = 13.6 Hz, 2 H, 10′′(13′′)-Heq], 1.74 [dm, J = 12.0 Hz, 2 H, 3′(5′)-Heq], 1.82 (s, 2 H, 8′′-H2), 1.93 (m, 1 H, 4′-H), 1.97–2.04 [complex signal, 4 H, 2-H2, 6′′(12′′)-Hax], 2.15 [dd, J = 11.6 Hz, J’ = 6.0 Hz, 6′′(12′′)-Heq], 2.85 [dt, J = 12.4 Hz, J’ = 2.4 Hz, 2 H, 2′(6′)-Hax], 3.06 [t, J = 6.0 Hz, 2 H, 5′′(11′′)-H], 3.15 [hept, J = 6.8 Hz, 1 H, CH(CH3)2], 3.80 [dt, J = 12.8 Hz, J’ = 2.0 Hz, 2 H, 2′(6′)-Heq], 5.22 (s, 1 H, NH), 7.03 [m, 2 H, 1′′(4′′)-H], 7.07 [m, 2 H, 2′′(3′′)-H]. 13C-NMR (100.6 MHz, CDCl3) δ: 16.8 [CH3, CH(CH3)2], 32.2 (CH3, C9-CH3), 32.3 [CH2, C3′(5′)], 33.1 (CH, C4′), 33.6 (C, C9′′), 39.1 [CH2, C6′′(12′′)], 41.0 [CH, C5′′(11′′)], 41.1 [CH2, C10′′(13′′)], 44.4 (CH2, C2), 46.5 [CH2, C2′(6′)], 47.2 (CH2, C8′′), 53.2 [CH, CH(CH3)2], 54.7 (C, C7′′), 126.3 [CH, C2′′(3′′)], 128.0 [CH, C1′′(4′′)], 146.1 [C, C4a’’(11a’’)], 170.3 (C, CO). Anal. Calcd for C26H38N2O3S: C 68.09, H 8.35, N 6.11. Found: C 67.75 H 8.62, N 5.74. HRMS: Calcd for [C26H38N2O3S + H]+: 459.2676; Found: 459.2675.
3.1.10. Synthesis of N-(9-Fluoro-5,6,8,9,10,11-hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-yl)-2-(1-(isopropylsulfonyl)piperidin-4-yl)acetamide, 6c
To a solution of 5d (186 mg, 0.52 mmol) and triethylamine (63 mg, 0.63 mmol) in anh. DCM (2 mL) was added 2-propanesulfonyl chloride (89 mg, 0.63 mmol). Then, the mixture was stirred at room temperature for 24 h. NaHCO3 sat. (15 mL) was added followed by EtOAc (15 mL) and the mixture was partitioned. The aqueous layer was extracted again with EtOAc (2 x 15 mL). Both organic phases were joined, dried over Na2SO4 anh., filtered, and solvents were concentrated under vacuum. The resulting crude was purified by column chromatography in silica gel (SiO2, Hexane/EtOAc mixtures) and gave 6c as a white solid (123 mg, 51% yield), mp: 192–193 °C. IR (ATR) v: 3342, 2914, 2855, 1663, 1536, 1449, 1316, 1138, 1045, 1017, 998, 953, 940, 865, 761, 753, 732, 652 cm−1. 1H-NMR (400 MHz, CDCl3) δ: 1.24 [m, 2 H, 3′′(5′′)-Hax], 1.31 [d, J = 6.9 Hz, 6 H, 2′′′(3′′′)-H], 1.73 [m, 2 H, 3′′(5′′)-Heq], 1.88–2.04 [complex signal, 7 H, 2-H, 6′(12′)-Hax, 10′(13′)-Hax, 4′′-H], 2.10–2.23 [complex signal, 4 H, 6′(12′)-Heq, 10′(13′)-Heq], 2.26 (d, J = 6.3 Hz, 2 H, 8′-H), 2.85 [m, 2 H, 2′′(6′′)-Hax], 3.14 (hept, J = 6.9 Hz, 1 H, 1′′′-H), 3.23 [m, 2 H, 5′(11′)-H], 3.80 [m, 2 H, 2′′(6′′)-Heq], 5.42 (broad s, 1 H, NH), 7.07 [m, 2 H, 1′(4′)-H], 7.13 [m, 2 H, 2′(3′)-H]. 13C-NMR (100.6 MHz, CDCl3) δ: 16.9 [CH3, C2′′′(3′′′)], 32.4 [CH2, C3′′(5′′)], 33.2 (CH, C4′′), 38.7 [CH2, C6′(12′)], 39.6 [d, 3JCF = 13.3 Hz, CH, C5′(11′)], 40.2 [d, 2JCF = 20.2 Hz, CH2, C10′(13′)], 44.3 (CH2, C2), 46.1 (d, 2JCF = 18.5 Hz, CH2, C8′), 46.7 [CH2, C2′′(6′′)], 53.4 (CH, C1′′′), 58.0 (d, 3JCF = 11.4 Hz, C, C7′), 94.2 (d, 1JCF = 177.6 Hz, C, C9′), 127.1 [CH, C2′(3′)], 128.3 [CH, C1′(4′)], 144.8 [C, C4a(11a)], 170.6 (C, CO). 19F-NMR (376 MHz, CDCl3) δ: −124.92 (m, 1 F). Anal. Calcd for C25H35FN2O3S: C 64.91, H 7.63, N 6.06. Found: C 65.08, H 7.97, N 5.74. HRMS: Calcd for [C25H35FN2O3S + H]+: 463.2425, found: 463.2425.
3.1.11. Synthesis of N-(9-Chloro-5,6,8,9,10,11-hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-yl)-2-(1-(isopropylsulfonyl)piperidin-4-yl)acetamide, 6d
To a solution of 5d (75 mg, 0.18 mmol) and triethylamine (73 mg, 0.72 mmol) in anh. DCM (2 mL) was added 2-propanesulfonyl chloride (39 mg, 0.27 mmol). Then, the mixture was stirred at room temperature for 24 h. NaHCO3 sat. (15 mL) was added followed by EtOAc (10 mL) and the mixture was partitioned. The aqueous layer was extracted again with EtOAc (2 × 10 mL). Both organic phases were joined, dried over Na2SO4 anh., filtered, and solvents were concentrated under vacuum. The resulting crude was purified by column chromatography in silica gel (SiO2, Hexane/EtOAc mixtures) and gave 6d as a reddish solid (37 mg, 40% yield), mp: 163–164 °C. IR (ATR) v: 3305, 2922, 2906, 2858, 1643, 1548, 1356, 1321, 1276, 1196, 1138, 1058, 1047, 952, 935, 799, 764, 738, 658 cm−1. 1H-NMR (400 MHz, CD3OD) δ: 1.22 [m, 2 H, 3′′(5′′)-Hax], 1.29 [d, J = 6.8 Hz, 6 H, 2′′′(3′′′)-H], 1.72 [m, 2 H, 3′′(5′′)-Heq], 1.88 (m, 1 H, 4-H’’), 2.02–2.12 [complex signal, 6 H, 2-H, 6′(12′)-Hax, 10′(13′)-Hax], 2.22 [m, 2 H, 6′(12′)-Heq], 2.40 [m, 2 H, 10′(13′)-Heq], 2.48 (s, 2 H, 8′-H), 2.90 [m, 2 H, 2′′(6′′)-Hax], 3.19 [m, 2 H, 5′(11′)-H], 3.28 (hept, J = 6.9 Hz, 1 H, 1′′′-H), 3.76 [m, 2 H, 2′′(6′′)-Heq], 7.05–7.14 [complex signal, 4 H, 1′(4′), 2′(3′)-H]. 13C NMR (100.6 MHz, CD3OD) δ: 17.0 [CH3, C2′′′(3′′′)], 33.3 [CH2, C3′′(5′′)], 34.6 (CH, C4′′), 38.9 [CH2, C6′(12′)], 42.6 [CH, C5′(11′)], 44.4 (CH2, C2), 45.9 [CH2, C10′(13′)], 47.4 [CH2, C2′′(6′′)], 51.0 (CH2, C8′), 54.0 (CH, C1′′′), 57.6 (C, C7′), 70.3 (C, C9′), 128.0 [CH, C2′(3′)], 129.1 [CH, C1′(4′)], 146.2 [C, C4a(11a)], 173.8 (C, CO). Anal. Calcd for C25H35ClN2O3S: C 62.68, H 7.36, N 5.85. Found: C 62.63, H 7.31, N 5.68. HRMS: Calcd for [C25H35ClN2O3S + H]+: 479.2130, found: 479.2143.
3.1.12. Synthesis of N-(5,6,8,9,10,11-Hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-yl)-2-(1-(isopropylsulfonyl)piperidin-4-yl)acetamide, 6e
To a solution of 5b (90 mg, 0.27 mmol) and triethylamine (109 mg, 1.08 mmol) in anh. acetonitrile (2 mL) was added 2-propanesulfonyl chloride (76 mg, 0.53 mmol). Then, the mixture was stirred at room temperature for 24 h. NaHCO3 sat. (10 mL) was added followed by EtOAc (10 mL) and the mixture was partitioned. The aqueous phase was extracted again with EtOAc (10 mL). Both organic phases were joined, dried over anh. Na2SO4, filtered, and concentrated under vacuum. The resulting crude was purified by column chromatography in silica gel (SiO2, Hexane/EtOAc mixtures) and gave 6e as a white solid (68 mg, 55% yield), mp: 136–137 °C. IR (ATR) v: 3301, 2922, 2858, 1642, 1548, 1355, 1320, 1276, 1195, 1137, 1047, 951, 935, 799, 764, 738, 684, 657, 618 cm−1. 1H-NMR (400 MHz, CD3OD) δ: 1.24 [m, 2 H, 3′′(5′′)-Hax], 1.29 [d, J = 6.9 Hz, 6 H, 2′′′(3′′′)-H], 1.68–1.77 [complex signal, 4 H, 10′(13′)-Hax, 3′′(5′′)-Heq], 1.86 (m, 1 H, 4′′-H), 2.00 [m, 2 H, 10′(13′)-Heq], 2.02–2.09 (complex signal, 4 H, 2-H, 8′-H), 2.16 [m, 2 H, 6′(12′)-Hax], 2.23 [m, 2 H, 6′(12′)-Heq], 2.32 (m, 1 H, 9′-H), 2.90 [m, 2 H, 2′′(6′′)-Hax], 3.04 [m, 2 H, 5′(11′)-H], 3.26 (hept, J = 6.9 Hz, 1 H, 1′′′-H), 3.76 [m, 2 H, 2′′(6′′)-Heq], 7.03 [s, 4 H, 1′(4′)-H, 2′(3′)-H]. 13C-NMR (100.6 MHz, CD3OD) δ: 17.0 [CH3, C2′′′(3′′′)], 32.6 (CH, C9′), 33.3 [CH2, C3′′(5′′)], 34.6 (CH, C4′′), 35.7 [CH2, C10′(13′)], 40.4 [CH2, C6′(12′)], 41.4 (CH2, C2), 42.6 [CH, C5′(11′)], 44.5 (CH2, C8′), 47.4 [CH2, 2′′(6′′)], 53.9 (CH, 1′′′C), 54.0 (C, C7′), 127.3 [CH, C2′(3′)], 129.0 [CH, C1′(4′)], 148.0 [C, C4a’(11a’)], 173.6 (C, CO). Anal. Calcd for C25H36N2O3S: C 67.53, H 8.16, N 6.30. Calcd for C25H36N2O3S · 0.2 H2O: C 66.99, H 8.19, N 6.25. Found: C 67.04, H 8.12, N 6.09. HRMS: Calcd for [C25H36N2O3S + H]+: 445.2528, found: 445.2519.
3.1.13. Synthesis of N-(5,6,8,9,10,11-Hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-yl-9-d)-2-(1-(isopropylsulfonyl)piperidin-4-yl)acetamide, 6f
To a solution of 5c (111 mg, 0.33 mmol) and triethylamine (134 mg, 1.32 mmol) in anh. Acetonitrile (2 mL) was added 2-propanesulfonyl chloride (93 mg, 0.65 mmol). Then, the mixture was stirred at room temperature for 24 h. NaHCO3 sat. (20 mL) was added followed by EtOAc (15 mL) and the mixture was partitioned. The aqueous layer was extracted again with EtOAc (15 mL). Both organic phases were joined, dried over Na2SO4 anh., filtered, and solvents were concentrated under vacuum. The resulting crude was purified by column chromatography in silica gel (SiO2, Hexane/EtOAc mixtures) and gave 6f as a white solid (97 mg, 63% yield), mp: 138–139 °C. IR (ATR) v: 3299, 2922, 2857, 1643, 1549, 1321, 1276, 1138, 1047, 952, 934, 764, 738, 656 cm−1. 1H-NMR (400 MHz, CD3OD) δ: 1.22 [m, 2 H, 3′′(5′′)-Hax], 1.29 [d, J = 6.9 Hz, 6 H, 2′′′(3′′′)-H], 1.68–1.76 [complex signal, 4 H, 10′(13′)-Hax, 3′′(5′′)-Heq], 1.86 (m, 1 H, 4′′-H), 1.99 [m, 2 H, 10′(13′)-Heq], 2.03–2.08 (complex signal, 4 H, 2-H, 8′-H), 2.16 [m, 2 H, 6′(12′)-Hax], 2.23 [m, 2 H, 6′(12′)-Heq], 2.89 [m, 2 H, 2′′(6′′)-Hax], 3.03 [m, 2 H, 5′(11′)-H], 3.25 (hept, J = 6.9 Hz, 1 H, 1′′′-H), 3.76 [m, 2 H, 2′′(6′′)-Heq], 7.03 [s, 4 H, 1′(4′)-H, 2′(3′)-H]. 13C-NMR (100.6 MHz, CD3OD) δ: 17.0 [CH3, C2′′′(3′′′)], 32.1 (CD, C9′), 33.3 [CH2, C3′′(5′′)], 34.6 (CH, C4′′), 35.6 [CH2, C10′(13′)], 40.4 [CH2, C6′(12′)], 41.3 (CH2, C2), 42.6 [CH, C5′(11′)], 44.5 (CH2, C8′), 47.4 [CH2, 2′′(6′′)], 53.9 (CH, 1′′′C), 54.0 (C, C7′), 127.3 [CH, C2′(3′)], 129.0 [CH, C1′(4′)], 148.0 [C, C4a’(11a’)], 173.6 (C, CO). Anal. Calcd for C25H35DN2O3S: C 67.38, H 8.37, N 6.29. Calcd for C25H35DN2O3S · 0.3 H2O: C 66.58, H 7.96, N 6.21. Found: C 66.68, H 8.05, N 6.10. HRMS: Calcd for [C25H35DN2O3S + H]+: 446.2582, found: 446.2589.
3.1.14. Synthesis of 2-(1-Benzylpiperidin-4-yl)-N-(9-methyl-5,6,8,9,10,11-hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-yl)acetamide, 6g
To a suspension of 9-methyl-5,6,8,9,10,11-hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-amine hydrochloride (250 mg, 0.95 mmol), 2a, in EtOAc (5 mL), 2-(1-benzylpiperidin-4-yl)acetic acid hydrochloride (255 mg, 0.95 mmol), 7, HOBt (192 mg, 1.42 mmol), EDC·HCl (220 mg, 1.42 mmol) and Et3N (480 mg, 4.74 mmol) were added. The mixture was stirred at room temperature for 24 h. Water (10 mL) and DCM (10 mL) were added to the resulting suspension and the 2 phases were separated. The organic phase was washed with sat. NaHCO3 aqueous solution (10 mL), brine (10 mL), dried over anh. Na2SO4, filtered, and concentrated under vacuum to give a yellow gum (479 mg). Column chromatography (SiO2, DCM/Methanol mixtures) gave 6g as a white solid (280 mg, 67% yield). The analytical sample was obtained by crystallization from hot EtOAc and Et2O, mp 145–146 °C. IR (NaCl disk): 3302, 3060, 3025, 2917, 2842, 2799, 2756, 1641, 1545, 1493, 1452, 1361, 1343, 1309, 1279, 1211, 1185, 1144, 1078, 1009, 974, 944, 917, 794, 757, 737, 698 cm−1. 1H-NMR (400 MHz, CDCl3) δ: 0.91 (s, 3 H, C9′′-CH3), 1.27 [dq, J = 12.4 Hz, J’ = 3.6 Hz, 2 H, 3′(5′)-Hax], 1.53 [d, J = 13.2 Hz, 2 H, 10′′(13′′)-Hax], 1.62–1.70 [complex signal, 4 H, 3′(5′)-Heq, 10′’(13′’)-Heq], 1.77 (m, 1 H, 4′-H), 1.84 (s, 2 H, 8′′-H), 1.94–2.02 (complex signal, 6 H, 6′′(12′′)-Hax, 2-H, 6′(2′)-Hax], 2.16 [dd, J = 12 Hz, J’ = 6 Hz, 6′′(12′′)-Heq], 2.86 [dt, J = 11.6 Hz, J’ = 2.8 Hz, 2 H, 2′(6′)-Heq], 3.06 [t, J = 6 Hz, 2 H, 5′′(11′′)-H], 3.48 (s, 2 H, CH2-C6H5), 5.20 (s, 1 H, NH), 7.03 [m, 2 H, 1′′(4′′)-H], 7.06 [m, 2 H, 2′′(3′′)-H], 7.23 (m, 1 H, 4′′′-H), 7.27–7.32 [complex signal, 4 H, 2′′′(6′′′)-H, 3′′′(5′′′)-H]. 13C-NMR (100.6 MHz, CDCl3) δ: 32.0 [CH2, C3′(5′)], 32.2 (CH3, C9′′-CH3), 33.4 (C, C9′′), 33.5 (CH, C4′), 39.1 [CH2, C6′′(12′′)], 40.9 [CH, C5′′(11′′)], 41.1 [CH2, C10′′(13′′)], 44.9 (CH2, C2), 47.1 (CH2, C8′′), 53.5 (CH2, C2′(6′)], 54.4 (C, C7′′), 63.3 (CH2, CH2-C6H5), 126.2 [CH, C2′′(3′′)], 126.9 (CH, Ar-CHpara), 127.9 [CH, C1′′(4′′)], 128.1 [CH, C3′′′(5′′′)], 129.2 [CH, C2′′′(6′′′)], 138.3 (C, Ar-Cipso), 146.1 [C, C4a’’(11a’’)], 171.0 (C, NHCO). HRMS: Calcd for [C30H38N2O + H]+: 443.3057; Found: 443.3061.
3.1.15. Synthesis of 2-(1-Benzylpiperidin-4-yl)-N-(9-fluoro-5,6,8,9,10,11-hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-yl)acetamide, 6h
To a solution of 9-fluoro-5,6,8,9,10,11-hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-amine (71 mg, 0.31 mmol), 2d, in DMF (2 mL), 2-(1-benzylpiperidin-4-yl)acetic acid hydrochloride (100 mg, 0.37 mmol), 7, HATU (176 mg, 0.46 mmol), and DIPEA (119 mg, 0.92 mmol) were added. The mixture was stirred at room temperature for 24 h. Solvent was concentrated in vacuo and EtOAc (10 mL) was added. The mixture was washed with NaHCO3 sat. (2 × 20 mL). The organic phase was dried over anh. Na2SO4, filtered, and concentrated under vacuum. The resulting crude was purified by column chromatography in silica gel (SiO2, DCM/MeOH mixtures) and gave 6h as a brown solid (72 mg, 52% yield), mp: 164–165 °C IR (ATR) v: 3317, 2938, 2857, 2808, 2766, 1637, 1548, 1447, 1423, 1360, 1317, 1281, 1135, 1068, 1000, 974, 864, 840, 764, 733, 697, 663, 643, 571, 592 cm−1. 1H-NMR (400 MHz, CD3OD) δ: 1.30 [m, 2 H, 3′′(5′′)-Hax], 1.68 [dm, J = 12.7 Hz, 2 H, 3′′(5′′)-Heq], 1.74 (m, 1 H, 4′′-H), 1.81 [broad d, J = 12.8 Hz, 2 H, 10′(13′)-Hax], 2.04 (d, J = 7.1 Hz, 2 H, 2-H), 2.06–2.17 [complex signal, 8 H, 6′(12′)-H2, 10′(13′)-Heq, 2′′(6′′)-Hax], 2.19 (d, 2JHF = 6.5 Hz, 2 H, 8′-H2), 2.94 [broad d, J = 12.1 Hz, 2 H, 2′′(6′′)-Heq], 3.23 [broad s, 2 H, 5′(11′)-H], 3.57 (s, 2 H, CH2-C6H5), 7.06–7.10 [complex signal, 4 H, 1′(4′)-H, 2′(3′)-H], 7.28 (m, 1 H, 4′′′-H), 7.31–7.36 [complex signal, 4 H, 2′′′(6′′′)-H, 3′′′(5′′′)-H]. 13C-NMR (100.6 MHz, CD3OD) δ: 32.2 [CH2, C3′′(5′′)], 34.5 (CH, C4′′), 39.3 [CH2, C6′(12′)], 41.0 [d, 3JCF = 13.1 Hz, CH, C5′(11′)], 41.4 [d, 2JCF = 20.1 Hz, CH2, C10′(13′)], 44.5 (CH2, C2), 46.9 (d, 2JCF = 18.3 Hz, CH2, C8′), 54.4 [CH2, C2′′(6′′)], 58.8 (d, 3JCF = 11.2 Hz, C, C7′), 64.1 (CH2, CH2-C6H5), 94.7 (d, 1JCF = 177.1 Hz, C, C9′), 127.9 [CH, C2′(3′)], 128.7 (CH, C4′′′), 129.2 [CH, C1′(4′)], 129.4 [CH, C3′′′(5′′′)], 131.0 [CH, C2′′′(6′′′)], 137.5 (C, C1′′′), 146.3 [C, C4a(11a)], 174.1 (C, CONH). Anal. Calcd for C29H35FN2O: C 77.99, H 7.90, N 6.27; Calcd for C29H35FN2O · 0.6 H2O: C 76.15, H 7.98, N 6.12. Found: C 75.99, H 7.81, N 6.06. HRMS: Calcd for [C29H35FN2O + H]+: 447.2806, found: 447.2804.
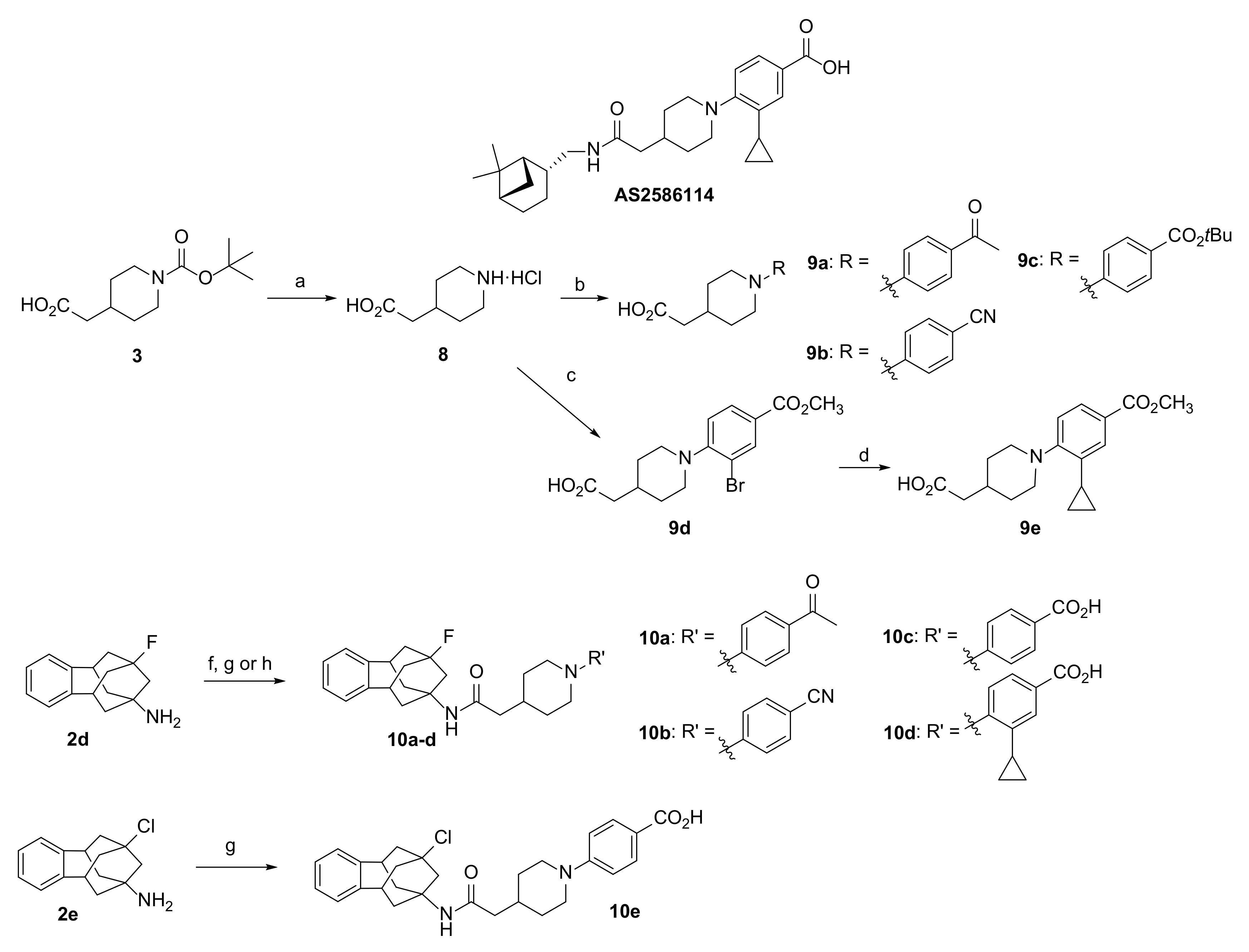
3.1.16. Synthesis of 2-(1-(4-Acetylphenyl)piperidin-4-yl)acetic Acid, 9a
To a solution of 2-(piperidin-4-yl)acetic acid hydrochloride (200 mg, 1.11 mmol),
8, and 4′-fluoroacetophenone (163 µL, 185 mg, 1.34 mmol) in DMSO (3 mL) was added K
2CO
3 (612 mg, 4.44 mmol) and the mixture was stirred at 100 °C overnight. Water was added (20 mL) followed by HCl 1 M until pH = 4. The mixture was extracted with EtOAc (20 mL). The organic layer was washed again with water at pH = 4 (20 mL). Then, the organic layer was dried over anh. Na
2SO
4, filtered, and solvents were concentrated
in vacuo to afford
9a as an orangish solid that was used as such without further purification (132 mg, 45% yield) [
33].
1H-NMR (400 MHz, CD
3OD) δ: 1.34 (qd,
J = 12.3 Hz,
J’ = 4.0 Hz, 2 H), 1.86 (m, 2 H), 2.01 (m, 1 H), 2.26 (d,
J = 7.1 Hz, 2 H), 2.49 (s, 3 H), 2.91 (td,
J = 12.8 Hz,
J’ = 2.6 Hz, 2 H), 3.98 (m, 2 H), 6.94 (d,
J = 9.1 Hz, 2 H), 7.86 (d,
J = 9.0 Hz, 2 H).
3.1.17. Synthesis of 2-(1-(4-Cyanophenyl)piperidin-4-yl)acetic Acid, 9b
To a solution of 2-(piperidin-4-yl)acetic acid hydrochloride (800 mg, 4.45 mmol),
8, and 4-fluorobenzonitrile (647 mg, 5.34 mmol) in DMSO (15 mL) was added K
2CO
3 (2.45 g, 17.76 mmol) and the mixture was stirred at 100 °C overnight. Water was added (50 mL) followed by HCl 1 M until pH = 4. The mixture was extracted with EtOAc (30 mL). The organic layer was washed again with water at pH = 4 (50 mL). Then, the organic layer was dried over anh. Na
2SO
4, filtered, and solvents were concentrated
in vacuo to afford
9b as a beige solid that was used as such without further purification (708 mg, 65% yield) [
34].
1H-NMR (400 MHz, CD
3OD) δ: 1.33 (qd,
J = 12.7 Hz,
J’ = 4.0 Hz, 2 H), 1.86 (m, 2 H), 2.00 (m, 1 H), 2.26 (d,
J = 7.1 Hz, 2 H), 2.89 (td,
J = 12.7 Hz,
J’ = 2.6 Hz, 2 H), 3.94 (m, 2 H), 6.98 (d,
J = 9.1 Hz, 2 H), 7.48 (d,
J = 9.0 Hz, 2 H).
3.1.18. Synthesis of 2-(1-(4-(t-Butoxycarbonyl)phenyl)piperidin-4-yl)acetic Acid, 9c
To a solution of 2-(piperidin-4-yl)acetic acid hydrochloride (369 mg, 2.06 mmol), 8, and t-butyl 4-fluorobenzoate (443 mg, 2.26 mmol) in DMSO (8 mL) was added K2CO3 (1.14 g, 8.22 mmol) and the mixture was stirred at 130 °C for 48 h. Water was added (20 mL) followed by HCl 1 M until pH = 5. The mixture was extracted with EtOAc (20 mL). The organic layer was washed again with water at pH = 5 (20 mL). Then, the organic layer was dried over anh. Na2SO4, filtered, and solvents were concentrated under vacuum. The resulting crude was purified by column chromatography in silica gel (SiO2, Hexane/EtOAc mixtures) and gave 9c as a white solid (255 mg, 39% yield). 1H-NMR (400 MHz, CD3OD) δ: 1.40 (qd, J = 12.6 Hz, J’ = 4.0 Hz, 2 H), 1.57 (s, 9 H), 1.87 (d, J = 13.1 Hz, 2 H), 2.02 (m, 1 H), 2.33 (d, J = 7.0 Hz, 2 H), 2.86 (td, J = 12.4 Hz, J’ = 2.6 Hz, 2 H), 3.84 (d, J = 12.5 Hz, 2 H), 6.85 (d, J = 8.9 Hz, 2 H), 7.86 (d, J = 9.1 Hz, 2 H).
3.1.19. Synthesis of 2-(1-(2-Bromo-4-(methoxycarbonyl)phenyl)piperidin-4-yl)acetic Acid, 9d
To a solution of 2-(piperidin-4-yl)acetic acid hydrochloride (200 mg, 1.11 mmol), 8, and methyl 3-bromo-4-fluorobenzoate (311 mg, 1.34 mmol) in DMF (3 mL) was added K2CO3 (612 mg, 4.44 mmol) and the mixture was stirred at 100 °C for 24 h. Water was added (20 mL) followed by HCl 1 M until pH = 4. The mixture was extracted with EtOAc (20 mL). The organic layer was washed again with water at pH = 4 (20 mL). Then, the organic layer was dried over anh. Na2SO4, filtered, and solvents were concentrated under vacuum. The resulting crude was purified by column chromatography in silica gel (SiO2, DCM/MeOH mixtures) and gave 9d as a white solid (200 mg, 50% yield). 1H-NMR (400 MHz, CDCl3) δ: 1.51 (m, 2 H), 1.85 (d, J = 13.0 Hz, 3 H), 2.32 (d, J = 6.9 Hz, 2 H), 2.71 (t, J = 11.7 Hz, 2 H), 3.46 (d, J = 11.5 Hz, 2 H), 3.88 (s, 3 H), 7.01 (d, J = 8.5 Hz, 1 H), 7.91 (dd, J = 8.4 Hz, J’ = 2.0 Hz, 1 H), 8.21 (d, J = 2.0 Hz, 1 H).
3.1.20. Synthesis of 2-(1-(2-Cyclopropyl-4-(methoxycarbonyl)phenyl)piperidin-4-yl)acetic Acid, 9e
A suspension of 2-(1-(2-bromo-4-(methoxycarbonyl)phenyl)piperidin-4-yl)acetic acid (200 mg, 0.56 mmol), 9d, cyclopropylboronic acid (96 mg, 1.12 mmol) and K3PO4 (360 mg, 1.70 mmol) in dioxane (5 mL) was degassed bubbling with N2 for 10 min. Then, tetrakis(triphenylphosphine)palladium(0) (65 mg, 0.06 mmol) was added and the mixture was heated at 100 °C and stirred for 24 h. Water was added (20 mL) followed by HCl 1 M until pH = 4. The mixture was extracted with EtOAc (20 mL). The aqueous layer was extracted again with EtOAc (20 mL). Both organic layers were dried over anh. Na2SO4, filtered, and solvents were concentrated under vacuum. The resulting crude was purified by column chromatography in silica gel (SiO2, DCM/MeOH mixtures) and gave 9e as a green solid. 1H-NMR (400 MHz, CDCl3) δ: 0.79 [m, 2 H, 2′′′(3′′′)-Hax], 1.02 [m, 2 H, 2′′′(3′′′)-Heq], 1.52 [qd, J = 12.0 Hz, J’ = 3.8 Hz, 2 H, 3′(5′)-Hax], 1.89 [m, 2 H, 3′(5′)-Hax], 1.99 (m, 1 H, 4′-H), 2.13 (m, 1 H, 1′′′-H), 2.39 [d, J = 7.0 Hz, 2 H, 3′(5′)-Heq], 2.73 [td, J = 12.0 Hz, J’ = 2.2 Hz, 2 H, 2′(6′)-Hax], 3.45 [d, J = 11.9 Hz, 2 H, 2′(6′)-Heq], 3.86 (s, 3 H, CH3), 6.97 (d, J = 8.4 Hz, 1 H, 6′′-H), 7.45 (d, J = 2.1 Hz, 1 H, 3′′-H), 7.78 (dd, J = 8.4 Hz, J’ = 2.1 Hz, 1 H, 5′′-H).
3.1.21. Synthesis of 2-(1-(4-Acetylphenyl)piperidin-4-yl)-N-(9-fluoro-5,6,8,9,10,11-hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-yl)acetamide, 10a
To a solution of 9-fluoro-5,6,8,9,10,11-hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-amine (50 mg, 0.22 mmol), 2d, in DMF (1 mL), 2-(1-(4-acetylphenyl)piperidin-4-yl)acetic acid (68 mg, 0.26 mmol), 9a, HATU (123 mg, 0.32 mmol), and DIPEA (85 mg, 0.66 mmol) were added. The mixture was stirred at room temperature for 24 h. Solvent was concentrated in vacuo and EtOAc (10 mL) was added. The mixture was washed with NaHCO3 sat. (2 x 20 mL). The organic phase was dried over anh. Na2SO4, filtered, and concentrated under vacuum. The resulting crude was purified by column chromatography in silica gel (SiO2, Hexane/EtOAc mixtures) and gave 10a as an off-white solid (33 mg, 32% yield), mp: 229–230 °C. IR (ATR) v: 3318, 2931, 2854, 1659, 1644, 1605, 1551, 1514, 1443, 1380, 1359, 1282, 1233, 1180, 1169, 1080, 1011, 998, 960, 865, 835, 818, 767, 632, 603, 593, 569 cm−1. 1H-NMR (400 MHz, CDCl3) δ: 1.30 [m, 2H, 3′′(5′′)-Hax], 1.79 [dm, J = 12.7 Hz, 2 H, 3′′(5′′)-Heq], 1.93 [m, 2 H, 10′(13′)-Hax], 1.97–2.06 [complex signal, 5 H, 2-H2, 6′(12′)-Hax, 4′′-H], 2.14–2.26 [complex signal, 4 H, 6′(12′)-Heq, 10′(13′)-Heq], 2.29 (d, 2JHF = 6.4 Hz, 2 H, 8′-H2), 2.50 (s, 3 H, CH3), 2.88 [td, J = 12.7 Hz, J’ = 2.6 Hz, 2 H, 2′′(6′′)-Hax], 3.24 [m, 2 H, 5′(11′)-H], 3.87 [m, 2 H, 2′′(6′′)-Heq], 5.30 (broad s, 1 H, NH), 6.84 [d, J = 9.0 Hz, 2 H, 2′′′(6′′′)-H], 7.08 [m, 2 H, 1′(4′)-H], 7.13 [m, 2 H, 2′(3′)-H], 7.84 [d, J = 9.0 Hz, 2 H, 3′′′(5′′′)-H]. 13C-NMR (100.6 MHz, CDCl3) δ: 26.2 (CH3, COCH3), 31.4 [CH2, C3′′(5′′)], 33.6 (CH, C4′′), 38.7 [CH2, C6′(12′)], 39.6 [d, 3JCF = 13.3 Hz, CH, C5′(11′)], 40.2 [d, 2JCF = 20.1 Hz, CH2, C10′(13′)], 44.6 (CH2, C2), 46.1 (d, 2JCF = 18.4 Hz, CH2, C8′), 48.0 [CH2, C2′′(6′′)], 58.0 (d, 3JCF = 11.4 Hz, C, C7′), 94.2 (d, 1JCF = 177.6 Hz, C, C9′), 113.6 [CH, C2′′′(6′′′)], 127.1 [CH, C2′(3′)], 128.3 [CH, C1′(4′)], 130.6 [CH, C3′′′(5′′′)], 144.8 [C, C4a(11a)], 154.1 (C, C1′′′), 170.8 (C, CONH), 196.6 (C, COCH3). The signal from C4′′′ was not observed. Anal. Calcd for C30H35FN2O2: C 75.92, H 7.43, N 5.90; Calcd for C30H35FN2O2 · 0.25 H2O: C 75.21, H 7.47, N 5.85. Found: 75.34, H 7.31, N 5.69. HRMS: Calcd for [C30H35FN2O2 + H]+: 475.2755, found: 475.2763.
3.1.22. Synthesis of 2-(1-(4-Cyanophenyl)piperidin-4-yl)-N-(9-fluoro-5,6,8,9,10,11-hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-yl)acetamide, 10b
To a solution of 9-fluoro-5,6,8,9,10,11-hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-amine (200 mg, 0.86 mmol), 2d, in DMF (4 mL), 2-(1-(4-cyanophenyl)piperidin-4-yl)acetic acid (252 mg, 1.03 mmol), 9b, HATU (490 mg, 1.29 mmol), and DIPEA (333 mg, 2.58 mmol) were added. The mixture was stirred at room temperature for 24 h. Solvent was concentrated in vacuo and EtOAc (20 mL) was added. The mixture was washed with brine (2 × 40 mL). The organic phase was dried over anh. Na2SO4, filtered, and concentrated under vacuum. The resulting crude was purified by column chromatography in silica gel (SiO2, Hexane/EtOAc mixtures) and gave 10b as an off-white solid (23 mg, 6% yield), mp: 180–181 °C. IR (ATR) v: 3317, 2926, 2857, 2212, 1645, 1603, 1515, 1445, 1359, 1306, 1239, 1177, 1112, 1089, 1010, 863, 819, 762, 732, 680, 645, 569, 560 cm−1. 1H-NMR (400 MHz, CDCl3) δ: 1.29 [m, 2 H, 3′′(5′′)-Hax], 1.80 [broad d, J = 10.3 Hz, 2 H, 3′′(5′′)-Heq], 1.93 [m, 2 H, 10′(13′)-Hax], 1.97–2.08 [complex signal, 5 H, 2-H2, 6′(12′)-Hax, 4′′-H], 2.11–2.25 [complex signal, 4 H, 6′(12′)-Heq, 10′(13′)-Heq], 2.28 (d, 2JHF = 6.3 Hz, 2 H, 8′-H2), 2.87 [td, J = 12.7 Hz, J’ = 2.6 Hz, 2 H, 2′′(6′′)-Hax], 3.23 [m, 2 H, 5′(11′)-H], 3.82 [broad dt, J = 12.9 Hz, J’ =2.5 Hz, 2 H, 2′′(6′′)-Heq], 5.33 (broad s, 1 H, NH), 6.83 [d, J = 9.0 Hz, 2 H, 2′′′(6′′′)-H], 7.08 [m, 2 H, 1′(4′)-H], 7.12 [m, 2 H, 2′(3′)-H], 7.45 [d, J = 9.0 Hz, 2 H, 3′′′(5′′′)-H]. 13C-NMR (100.6 MHz, CDCl3) δ: 31.3 [CH2, C3′′(5′′)], 33.5 (CH, C4′′), 38.7 [CH2, C6′(12′)], 39.6 [d, 3JCF = 13.2 Hz, CH, C5′(11′)], 40.1 [d, 2JCF = 20.2 Hz, CH2, C10′(13′)], 44.4 (CH2, C2), 46.0 (d, 2JCF = 18.4 Hz, CH2, C8′), 47.9 [CH2, C2′′(6′′)], 58.0 (d, 3JCF = 11.4 Hz, C, C7′), 94.2 (d, 1JCF = 177.6 Hz, C, C9′), 99.5 (C, CN), 114.4 [CH, C2′′′(6′′′)], 120.3 (C, C4′′′), 127.1 [CH, C2′(3′)], 128.3 [CH, C1′(4′)], 133.6 [CH, C3′′′(5′′′)], 144.7 [C, C4a(11a)], 153.3 (C, C1′′′), 170.7 (C, CONH). Anal. Calcd for C29H32FN3O: C 76.12, H 7.05, N 9.18; Calcd for C29H32FN3O · 0.5 CH2Cl2: C 70.86, H 6.65, N 8.40. Found: C 70.78, H 6.67, N 8.17. HRMS: Calcd for [C29H32FN3O + H]+: 458.2602, found: 458.2593.
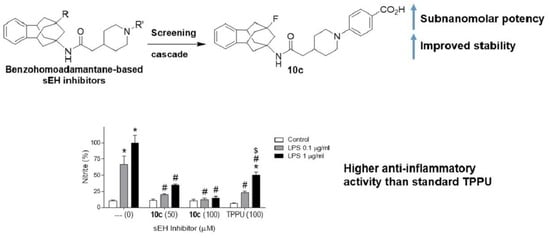
3.1.23. Synthesis of 4-(4-(2-((9-Fluoro-5,6,8,9,10,11-hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-yl)amino)-2-oxoethyl)piperidin-1-yl)benzoic Acid, 10c
To a solution of 9-fluoro-5,6,8,9,10,11-hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-amine (82 mg, 0.36 mmol), 2d, in DMF (2 mL) were added 2-(1-(4-(t-butoxycarbonyl)phenyl)piperidin-4-yl)acetic acid (125 mg, 0.39 mmol), 9c, HATU (203 mg, 0.53 mmol), and DIPEA (92 mg, 0.71 mmol). The mixture was stirred at room temperature for 24 h. Solvent was concentrated in vacuo and EtOAc (15 mL) was added. The mixture was washed with brine (2 × 30 mL). The organic phase was dried over anh. Na2SO4, filtered, and concentrated under vacuum. The resulting crude was purified by column chromatography in silica gel (SiO2, Hexane/EtOAc mixtures). Fractions containing the desired product were collected and concentrated in vacuo. Then, HCl 4 M in dioxane (2 mL) with some drops of water were added to the solid and the mixture was stirred at RT overnight. EtOAc (10 mL) was added, and the mixture was washed with water acidified at pH = 4 with HCl 2 M (2 x 20 mL). The organic layer was dried over anh. Na2SO4, filtered, and solvents were concentrated under vacuum. The resulting crude was purified by column chromatography in silica gel (SiO2, DCM/MeOH mixtures) and gave 10c as an off-white solid (40 mg, 24% yield), mp: 267–268 °C. IR (ATR) v: 2923, 2853, 1665, 1634, 1601, 1519, 1431, 1418, 1318, 1283, 1211, 1189, 1089, 993, 827, 767, 697, 632, 553 cm−1. 1H-NMR (400 MHz, CD3OD) δ: 1.34 [qd, J = 12.6 Hz, J’ = 4.1 Hz, 2 H, 3′(5′)-Hax], 1.76 [broad d, J = 12.1 Hz, 2 H, 3′(5′)-Heq], 1.82 [broad d, J = 10.4 Hz, 2 H, 10′′′(13′′′)-Hax], 1.95 (m, 1 H, 4′-H), 2.05–2.13 [complex signal, 4 H, 1′′-H, 6′′′(12′′′)-Hax], 2.13–2.19 [complex signal, 4 H, 6′′′(12′′′)-Heq, 10′′′(13′′′)-Hax], 2.21 (d, 2JHF = 6.4 Hz, 2 H, 8′′′-H), 2.84 [td, J = 12.7 Hz, J’ = 2.6 Hz, 2 H, 2′(6′)-Hax], 3.24 (broad s, 2 H, 5′′′(11′′′)-H], 3.92 [broad d, J = 13.3 Hz, 2 H, 2′(6′)-Heq], 6.93 [d, J = 9.1 Hz, 2 H, 3(5)-H], 7.07–7.13 [complex signal, 4 H, 1′′′(4′′′)-H, 2′′′(3′′′)-H], 7.85 [d, J = 9.1 Hz, 2 H, 2(6)-H]. 13C-NMR (100.6 MHz, CD3OD) δ: 32.4 [CH2, C3′(5′)], 35.1 (CH, C4′), 39.3 [CH2, C6′′′(12′′′)], 41.0 [d, 3JCF = 13.1 Hz, CH, C5′′′(11′′′)], 41.4 [d, 2JCF = 20.2 Hz, CH2, C10′′′(13′′′)], 44.7 (CH2, C1′′), 46.9 (d, 2JCF = 18.4 Hz, CH2, C8′′′), 49.1 [CH2, C2′′(6′′)], 58.8 (d, 3JCF = 11.4 Hz, C, C7′′′), 94.8 (d, 1JCF = 177.1 Hz, C, C9′′′), 114.9 [CH, C3(5)], 120.1 (C, C1), 128.0 [CH, C2′′′(3′′′)], 129.2 [CH, C1′′′(4′′′)], 132.5 [CH, C2(6)], 146.3 [C, C4a’’(11a’’)], 155.9 (C, C4), 170.4 (C, CONH), 174.1 (C, CO2H). Anal. Calcd for C29H33FN2O3: C 73.09, H 6.98, N 5.88. Found: C 72.64, H 7.16, N 5.39. HRMS: Calcd for [C29H33FN2O3−H]−: 475.2402, found: 475.2400.
3.1.24. Synthesis of 3-Cyclopropyl-4-(4-(2-((9-fluoro-5,6,8,9,10,11-hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-yl)amino)-2-oxoethyl)piperidin-1-yl)benzoic Acid, 10d
To a solution of 9-fluoro-5,6,8,9,10,11-hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-amine (100 mg, 0.45 mmol), 2d, in DMF (2 mL) were added 2-(1-(2-cyclopropyl-4-(methoxycarbonyl)phenyl)piperidin-4-yl)acetic acid (125 mg, 0.39 mmol), 9e, HATU (203 mg, 0.53 mmol), and DIPEA (92 mg, 0.71 mmol). The mixture was stirred at room temperature for 24 h. Solvent was concentrated in vacuo and EtOAc (10 mL) was added. The mixture was washed with NaHCO3 sat. (2 × 20 mL). The organic phase was dried over anh. Na2SO4, filtered, and concentrated under vacuum. The resulting crude was purified by column chromatography in silica gel (SiO2, Hexane/EtOAc mixtures). Fractions containing the desired product were collected and concentrated in vacuo. MeOH (1 mL) and KOH (116 mg, 2.07 mmol) were added, and the mixture was stirred at 50 °C for 4 h. Amberlite® 120 H+ was added until pH = 4 and the mixture was filtered, using MeOH as an eluting agent. Solvents were concentrated in vacuo to afford a white solid that was purified by column chromatography in silica gel (SiO2, DCM/MeOH mixtures) and gave 10d as a reddish solid (16 mg, 7% yield), mp: 190–191 °C. IR (ATR) v: 2920, 2854, 1645, 1602, 1539, 1495, 1442, 1382, 1359, 1303, 1228, 1180, 1116, 1089, 1011, 937, 864, 758, 715, 641, 569 cm−1. 1H-NMR (400 MHz, CD3OD) δ: 0.73 [m, 2 H, 2′(3′)-Hax], 1.02 [m, 2 H, 2′(3′)-Heq], 1.47 [qd, J = 12.0 Hz, J’ = 3.8 Hz, 2 H, 3′′(5′′)-Hax], 1.76–1.86 [complex signal, 4 H, 3′′(5′′)-Heq, 10′′′′(13′′′′)-Hax], 1.90 (m, 1 H, 4′′-H), 2.06–2.21 [complex signal, 9 H, 1′-H, 1′′′-H, 6′′′′(12′′′′)-H2, 10′′′′(13′′′′)-Heq], 2.22 (d, 2JHF = 6.4 Hz, 2 H, 8′′′′-H), 2.71 [td, J = 11.9 Hz, J’ = 2.2 Hz, 2 H, 2′′(6′′)-Hax], 3.25 [broad t, J = 5.7 Hz, 2 H, 5′′′′(11′′′′)-H], 3.43 [broad d, J = 12.1 Hz, 2 H, 2′′(6′′)-Heq], 7.03 (d, J = 8.4 Hz, 1 H, 5-H), 7.10 [broad signal, 4 H, 1′′′′(4′′′′)-H, 2′′′′(3′′′′)-H], 7.44 (d, J = 2.0 Hz, 1 H, 2-H), 7.75 (dd, J = 8.4 Hz, J’ = 2.1 Hz, 1 H, 6-H). 13C-NMR (100.6 MHz, CD3OD) δ: 9.9 [CH2, C2′(3′)], 12.2 (CH, C1′), 33.6 [CH2, C3′′(5′′)], 35.0 (CH, C4′′), 39.4 [CH2, C6′′′′(12′′′′)], 41.0 [d, 3JCF = 13.2 Hz, CH, C5′′′′(11′′′′)], 41.4 [d, 2JCF = 20.1 Hz, CH2, C10′′′′(13′′′′)], 44.9 (CH2, C1′′′), 46.9 (d, 2JCF = 18.3 Hz, CH2, C8′′′′), 53.3 [CH2, C2′′(6′′)], 58.8 (d, 3JCF = 11.3 Hz, C, C7′′′′), 94.8 (d, 1JCF = 177.2 Hz, C, C9′′′′), 119.1 (CH, C5), 125.6 (C, C1), 126.4 (CH, C2), 128.0 [CH, C2′′′′(3′′′′)], 129.0 (CH, C6), 129.2 [CH, C1′′′′(4′′′′)], 138.1 (C, C3), 146.3 [C, C4a’’’(11a’’’)], 158.3 (C, C4), 170.5 (C, CONH), 174.3 (C, CO2H). HRMS: Calcd for [C32H37FN2O3 + H]+: 517.2861, found: 517.2847.
3.1.25. Synthesis of 4-(4-(2-(9-Chloro-5,6,8,9,10,11-hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-yl)ureido)piperidin-1-yl)benzoic Acid, 10e
To a solution of 9-chloro-5,6,8,9,10,11-hexahydro-7H-5,9:7,11-dimethanobenzo[9]annulen-7-amine (88 mg, 0.36 mmol), 2e, in DMF (2 mL), 2-(1-(4-(t-butoxycarbonyl)phenyl)piperidin-4-yl)acetic acid (125 mg, 0.39 mmol), 9c, HATU (203 mg, 0.53 mmol), and DIPEA (124 µL, 92 mg, 0.71 mmol) were added. The mixture was stirred at room temperature overnight. EtOAc (15 mL) was added, and the mixture was washed with brine (2 x 30 mL). The organic layer was dried over anh. Na2SO4, filtered, and solvents were concentrated. The resulting crude was purified by column chromatography in silica gel (using as eluent mixtures of EtOAc in hexane from 0% to 25%). Fractions containing the desired product were collected and concentrated in vacuo. HCl 4 M in dioxane (2 mL) with some drops of water were added and the mixture was stirred at room temperature overnight. EtOAc (10 mL) was added, and the mixture was washed with water acidified at pH = 4 with HCl 2 M (2 × 20 mL). The organic layer was dried over anh. Na2SO4, filtered, and solvents were concentrated in vacuo. The crude was purified by column chromatography in silica gel (using as eluent mixtures of MeOH in DCM from 0% to 4%). Fractions containing the desired product were collected and concentrated in vacuo to afford 10e as a pink solid (23 mg, 13% yield), mp: 242–243 °C. IR (ATR) v: 2920, 2855, 1664, 1638, 1600, 1518, 1415, 1391, 1357, 1283, 1230, 1184, 1110, 1082, 978, 947, 931, 900, 830, 800, 773, 762, 697, 645 cm−1. 1H-NMR (400 MHz, CD3OD) δ: 1.35 [m, 2 H, 3′(5′)-Hax], 1.77 [broad d, J = 12.9 Hz, 2 H, 3′(5′)-Heq], 1.95 (m, 1 H, 4′-H), 2.02–2.11 [complex signal, 6 H, 1′′-H, 6′′′(12′′′)-Hax, 10′′′(13′′′)-Hax], 2.23 [m, 2 H, 6′′′(12′′′)-Heq], 2.40 [m, 2 H, 10′′′(13′′′)-Heq], 2.49 (s, 2 H, 8′′′-H), 2.85 [td, J = 12.6 Hz, J’ = 2.6 Hz, 2 H, 2′(6′)-Hax], 3.19 [broad t, J = 6.5 Hz, 2 H, 5′′′(11′′′)-H], 3.92 [broad d, J = 13.4 Hz, 2 H, 2′(6′)-Heq], 6.93 [d, J = 9.1 Hz, 2 H, 3(5)-H], 7.06–7.12 [complex signal, 4 H, 1′′′(4′′′)-H, 2′′′(3′′′)-H], 7.85 [d, J = 9.0 Hz, 2 H, 2(6)-H]. 13C-NMR (101 MHz, CD3OD) δ: 32.4 [CH2, C3′(5′)], 35.1 (CH, C4′), 39.0 [CH2, C6′′′(12′′′)], 42.6 [CH, C5′′′(11′′′)], 44.7 (CH2, C1′′), 45.9 [CH2, C10′′′(13′′′)], 49.1 [signal overlapped, CH2, C2′(6′)], 51.0 (CH2, C8′′′), 57.6 (C, C7′′′), 70.3 (C, C9′′′), 114.9 [CH, C3(5)], 120.1 (C, C1), 128.0 [CH, C2′′′(3′′′)], 129.1 [CH, C1′′′(4′′′)], 132.5 [CH, C2(6)], 146.2 [C, C4a’’’(11a’’’)], 155.9 (C, C4), 170.4 (C, CONH), 174.1 (C, CO2H). Anal. Calcd for C29H33ClN2O3: C 70.65, H 6.75, N 5.68; Calcd for C29H33ClN2O3 · ¾ H2O: 68.76, H 6.86, N 5.53. Found: C 68.77, H 6.66, N 5.18. HRMS: Calcd for [C29H33ClN2O3−H]−: 491.2107, found: 491.2106.




 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
