
Species Differences in Microsomal Metabolism of Xanthine-Derived A1 Adenosine Receptor Ligands


Abstract
:1. Introduction
2. Results
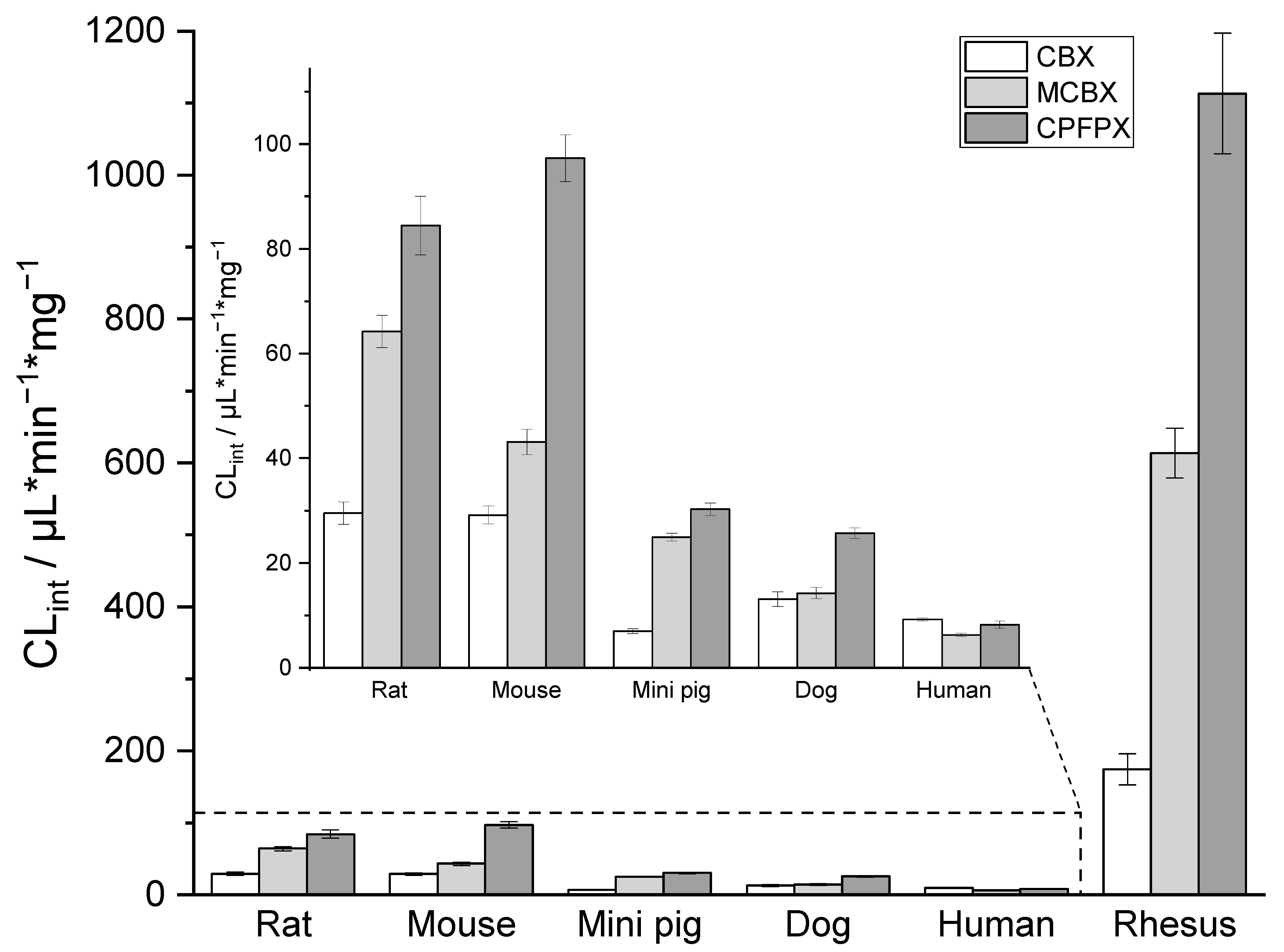
2.1. In Vitro Intrinsic Clearance
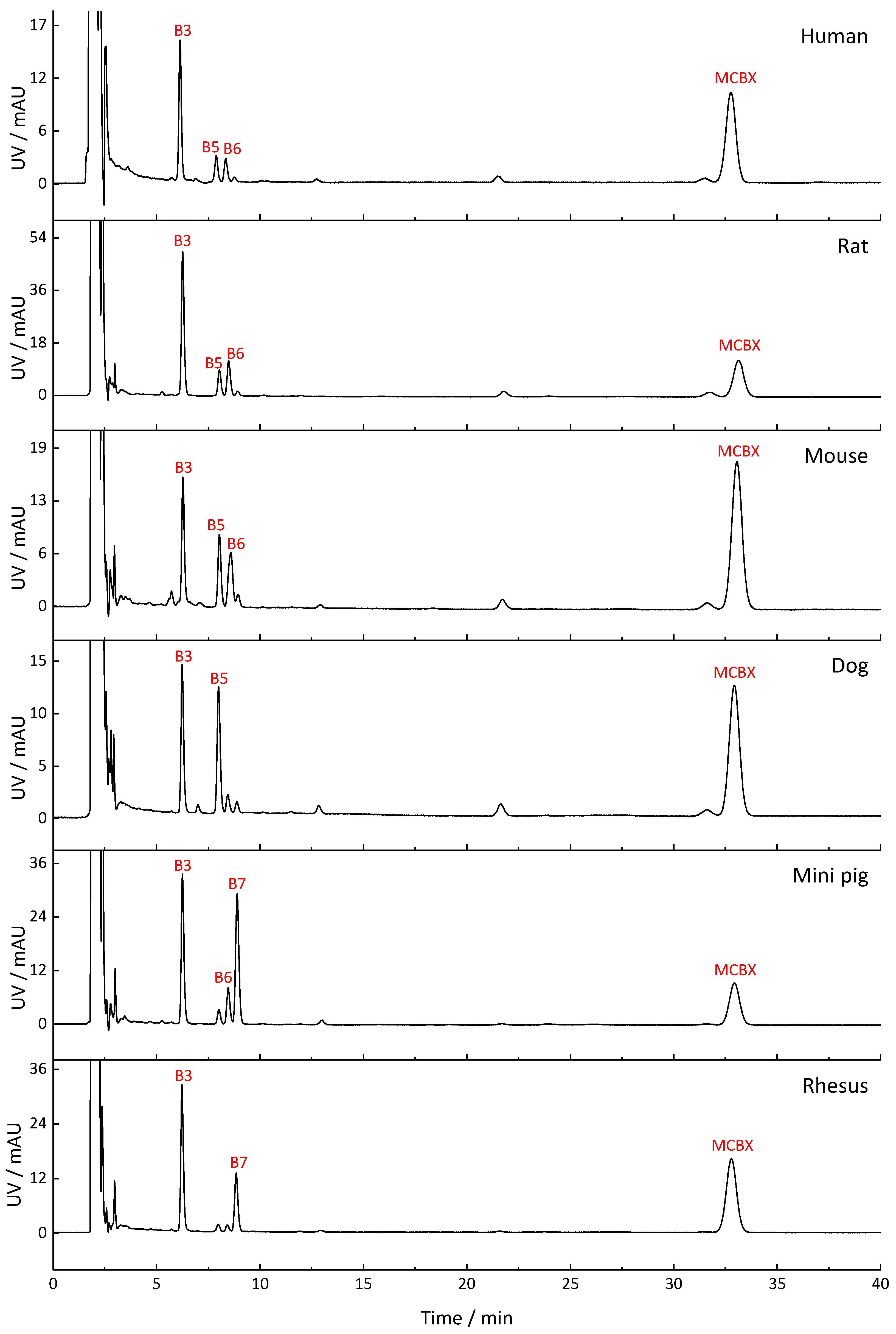
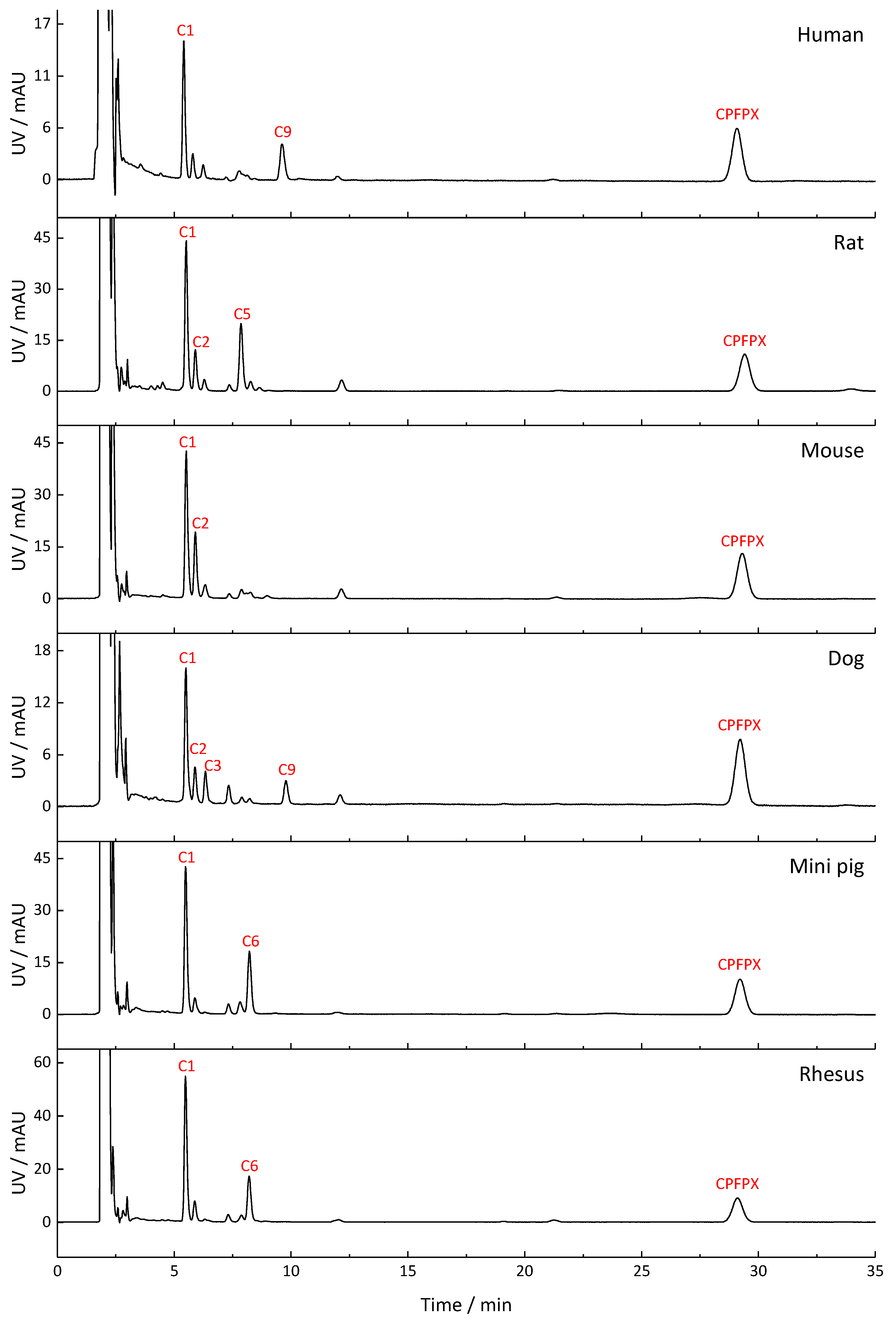
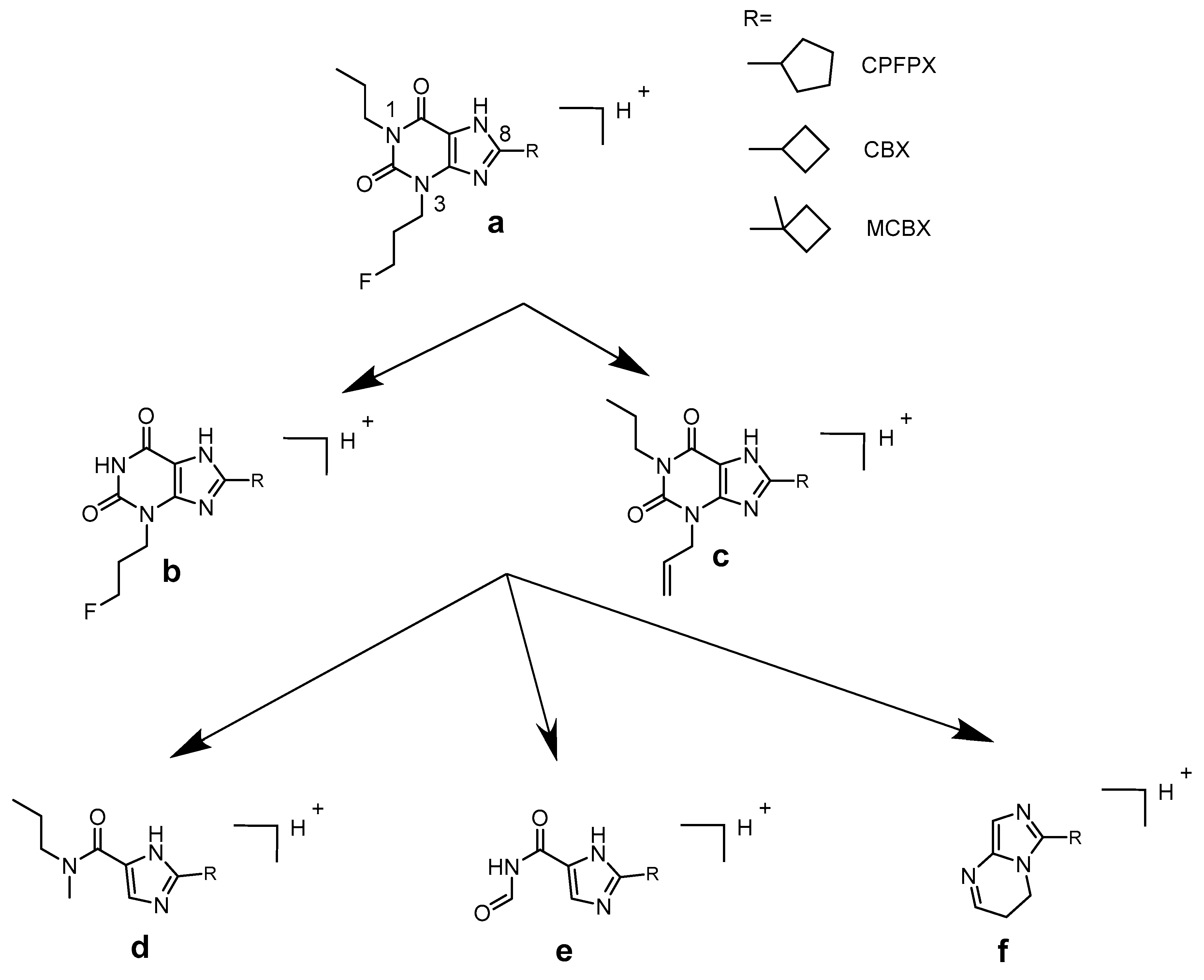
2.2. Metabolite Profiles
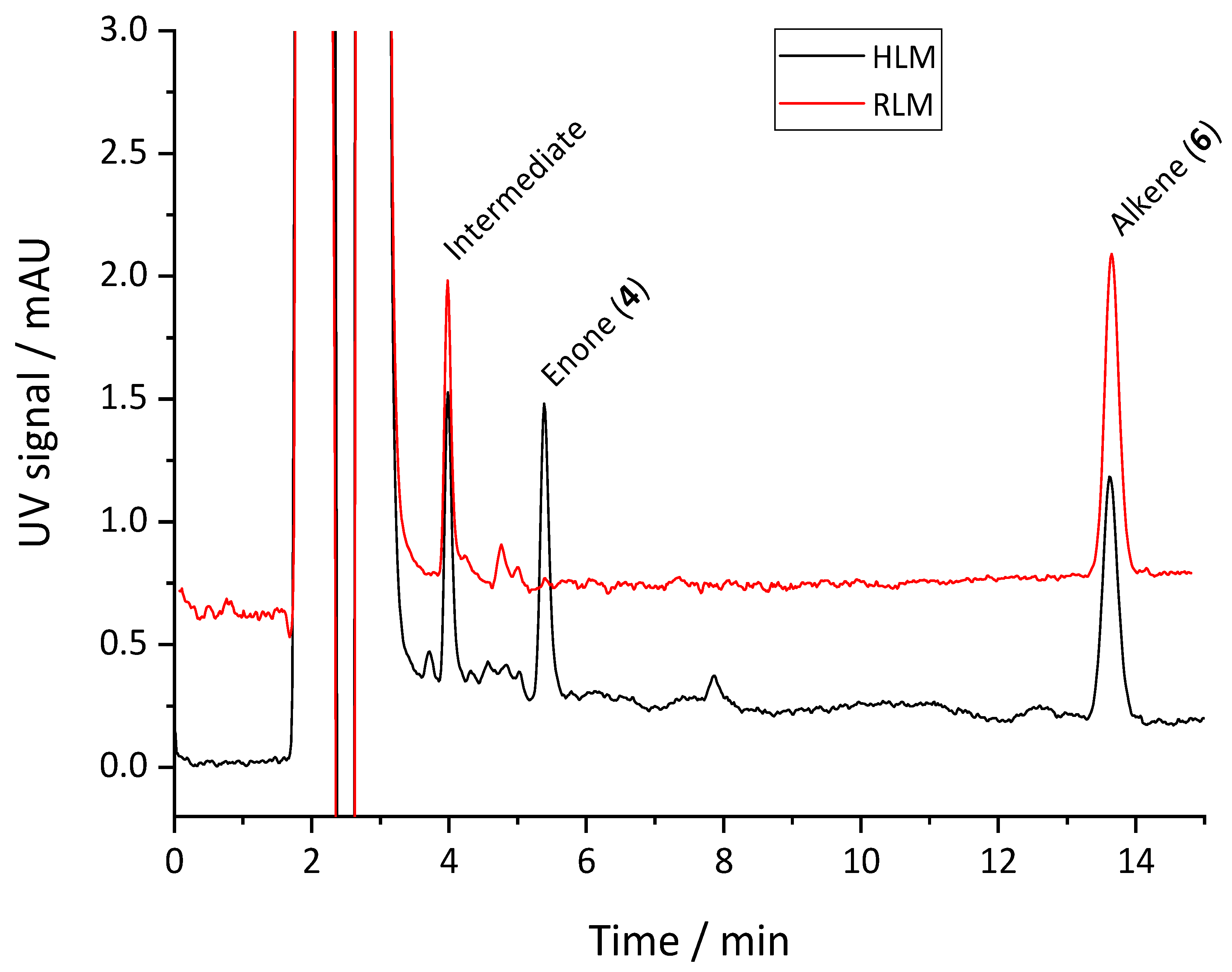
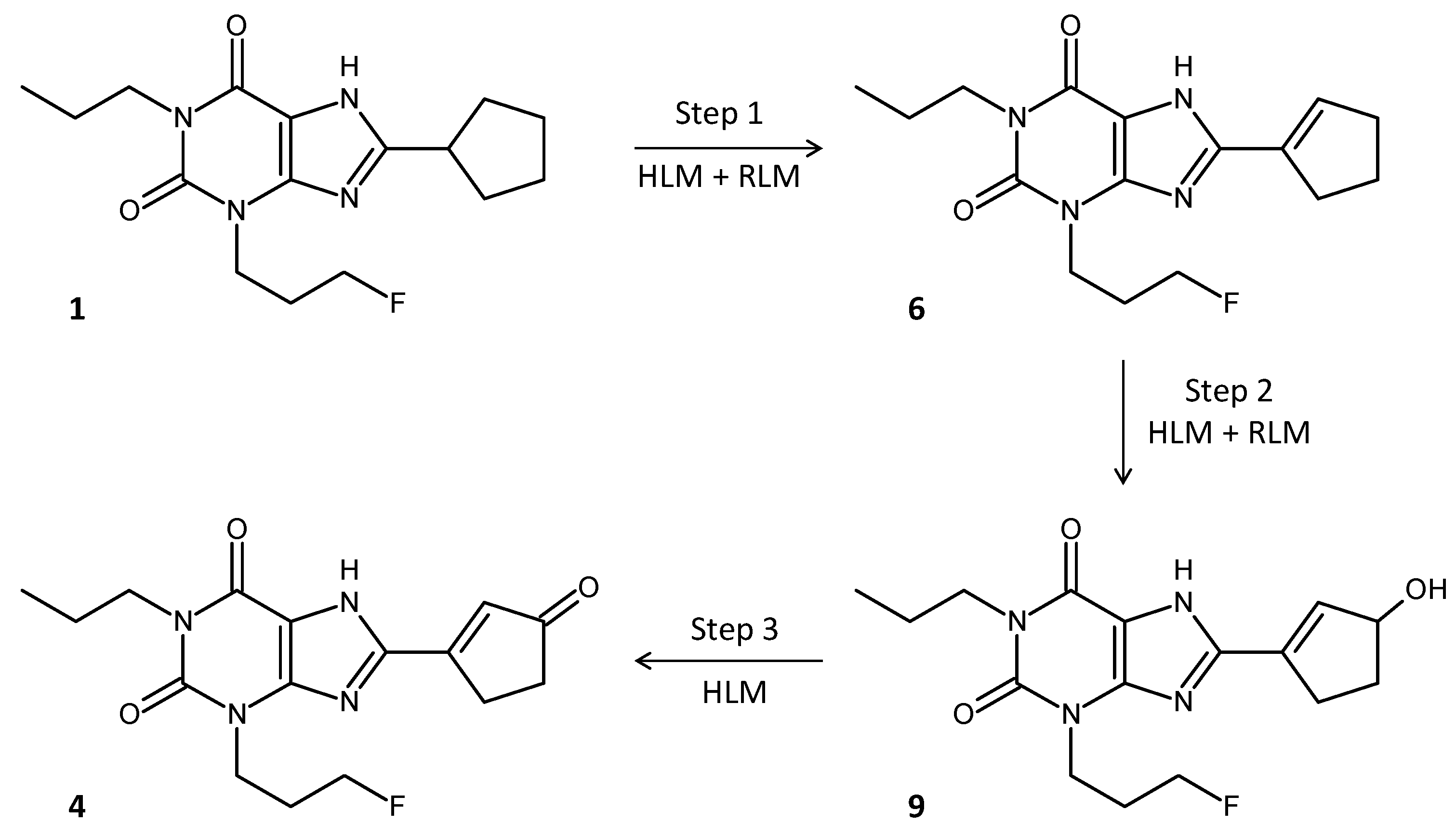
2.3. Enone Metabolite Formation in Liver Microsomes
3. Discussion
4. Materials and Methods
4.1. Compounds
4.2. Reagents and Solvents
4.3. In Vitro Studies
4.3.1. Determination of In Vitro Intrinsic Clearance
4.3.2. Metabolite Analysis
4.3.3. Enone Metabolite Formation
4.4. Data Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pike, V.W. PET radiotracers: Crossing the blood–brain barrier and surviving metabolism. Trends Pharmacol. Sci. 2009, 30, 431–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laruelle, M. Relationships between radiotracer properties and image quality in molecular imaging of the brain with positron emission tomography. Mol. Imaging Biol. 2003, 5, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Eberlein, U.; Bröer, J.H.; Vandevoorde, C.; Dos Santos, P.M.P.; Bardiès, M.; Bacher, K.; Nosske, D.; Lassmann, M. Biokinetics and dosimetry of commonly used radiopharmaceuticals in diagnostic nuclear medicine—A review. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 2269–2281. [Google Scholar] [CrossRef] [Green Version]
- Holschbach, M.H.; Olsson, R.A.; Bier, D.; Wutz, W.; Sihver, W.; Schüller, M.; Palm, B.; Coenen, H.H. Synthesis and Evaluation of No-Carrier-Added 8-Cyclopentyl-3-(3-[18F]fluoropropyl)-1-propylxanthine ([18F]CPFPX): A Potent and Selective A1-Adenosine Receptor Antagonist for in Vivo Imaging. J. Med. Chem. 2002, 45, 5150–5156. [Google Scholar] [CrossRef]
- Bauer, A.; Holschbach, M.H.; Cremer, M.; Weber, S.; Boy, C.; Shah, N.J.; Olsson, R.A.; Halling, H.; Coenen, H.H.; Zilles, K. Evaluation of 18F-CPFPX, a novel adenosine A1 receptor ligand: In vitro autoradiography and high-resolution small animal PET. J. Nucl. Med. 2003, 44, 1682–1689. [Google Scholar] [PubMed]
- Bier, D.; Holschbach, M.H.; Wutz, W.; Olsson, R.A.; Coenen, H.H. Metabolism of the A1 Adenosine Receptor Positron Emission Tomography Ligand [18F]8-Cyclopentyl-3-(3-Fluoropropyl)-1-Propylxanthine ([18F]CPFPX) in Rodents and Humans. Drug Metab. Dispos. 2006, 34, 570–576. [Google Scholar] [CrossRef]
- Schneider, D.; Oskamp, A.; Holschbach, M.; Neumaier, B.; Bier, D.; Bauer, A. Influence of binding affinity and blood plasma level on cerebral pharmacokinetics and PET imaging characteristics of two novel xanthine PET radioligands for the A1 adenosine receptor. Nucl. Med. Biol. 2020, 82–83, 1–8. [Google Scholar] [CrossRef]
- Bier, D.; Hartmann, R.; Holschbach, M. Collision-induced dissociation studies of caffeine in positive electrospray ionisation mass spectrometry using six deuterated isotopomers and one N1-ethylated homologue. Rapid Commun. Mass Spectrom. 2013, 27, 885–895. [Google Scholar] [CrossRef]
- Matusch, A.; Meyer, P.T.; Bier, D.; Holschbach, M.H.; Woitalla, D.; Elmenhorst, D.; Winz, O.H.; Zilles, K.; Bauer, A. Metabolism of the A1 adenosine receptor PET ligand [18F]CPFPX by CYP1A2: Implications for bolus/infusion PET studies. Nucl. Med. Biol. 2006, 33, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Schneider, D.; Oskamp, A.; Holschbach, M.; Neumaier, B.; Bauer, A.; Bier, D. Relevance of In Vitro Metabolism Models to PET Radiotracer Development: Prediction of In Vivo Clearance in Rats from Microsomal Stability Data. Pharmaceuticals 2019, 12, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, J.A.; Houston, J.B.; Hallifax, D. Comparison of intrinsic clearances in human liver microsomes and suspended hepatocytes from the same donor livers: Clearance-dependent relationship and implications for prediction ofin vivoclearance. Xenobiotica 2010, 41, 124–136. [Google Scholar] [CrossRef]
- Di, L.; Keefer, C.; Scott, D.O.; Strelevitz, T.J.; Chang, G.; Bi, Y.-A.; Lai, Y.; Duckworth, J.; Fenner, K.; Troutman, M.D.; et al. Mechanistic insights from comparing intrinsic clearance values between human liver microsomes and hepatocytes to guide drug design. Eur. J. Med. Chem. 2012, 57, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Mimura, M.; Inoue, K.; Nakamura, S.-I.; Oda, H.; Ohmori, S.; Yamazaki, H. Cytochrome P450-dependent drug oxidation activities in liver microsomes of various animal species including rats, guinea pigs, dogs, monkeys, and humans. Arch. Toxicol. 1997, 71, 401–408. [Google Scholar] [CrossRef]
- Tanaka, E.; Ishikawa, A.; Horie, T. In vivo and in vitro trimethadione oxidation activity of the liver from various animal species including mouse, hamster, rat, rabbit, dog, monkey and human. Hum. Exp. Toxicol. 1999, 18, 12–16. [Google Scholar] [CrossRef]
- Amri, H.S.-E.; Batt, A.M.; Siest, G. Comparison of cytochrome P-450 content and activities in liver microsomes of seven animal species, including man. Xenobiotica 1986, 16, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Skaanild, M.T.; Friis, C. Cytochrome P450 Sex Differences in Minipigs and Conventional Pigs. Pharmacol. Toxicol. 1999, 85, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Stevens, J.C.; Shipley, L.A.; Cashman, J.R.; Vandenbranden, M.; Wrighton, S.A. Comparison of human and rhesus monkey in vitro phase I and phase II hepatic drug metabolism activities. Drug Metab. Dispos. 1993, 21, 753–760. [Google Scholar] [PubMed]
- Shimada, T.; Yamazaki, H.; Mimura, M.; Inui, Y.; Guengerich, F.P. Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: Studies with liver microsomes of 30 Japanese and 30 Caucasians. J. Pharmacol. Exp. Ther. 1994, 270, 414–423. [Google Scholar]
- Nedelcheva, V.; Gut, I. P450 in the rat and man: Methods of investigation, substrate specificities and relevance to cancer. Xenobiotica 1994, 24, 1151–1175. [Google Scholar] [CrossRef]
- Eguchi, K.; Nishibe, Y.; Baba, T.; Ohno, K. Quantitation of cytochrome P450 enzymes (CYP1A1/2, 2B11, 2C21 and 3A12) in dog liver microsomes by enzyme-linked immunosorbent assay. Xenobiotica 1996, 26, 755–763. [Google Scholar] [CrossRef]
- Edwards, R.J.; Murray, B.P.; Murray, S.; Schulz, T.; Neubert, D.; Gant, T.W.; Thorgeirsson, S.S.; Boobis, A.R.; Davies, D.S. Contribution of CYP1A1 and CYP1A2 to the activation of heterocyclic amines in monkeys and human. Carcinogenesis 1994, 15, 829–836. [Google Scholar] [CrossRef]
- Sakuma, T.; Hieda, M.; Igarashi, T.; Ohgiya, S.; Nagata, R.; Nemoto, N.; Kamataki, T. Molecular cloning and functional analysis of cynomolgus monkey CYP1A2. Biochem. Pharmacol. 1998, 56, 131–139. [Google Scholar] [CrossRef]
- Uehara, S.; Murayama, N.; Nakanishi, Y.; Zeldin, D.C.; Yamazaki, H.; Uno, Y. Immunochemical Detection of Cytochrome P450 Enzymes in Liver Microsomes of 27 Cynomolgus Monkeys. J. Pharmacol. Exp. Ther. 2011, 339, 654–661. [Google Scholar] [CrossRef] [Green Version]
- Bullock, P.; Pearce, R.; Draper, A.; Podval, J.; Bracken, W.; Veltman, J.; Thomas, P.; Parkinson, A. Induction of liver micro-somal cytochrome P450 in cynomolgus monkeys. Drug Metab. Dispos. 1995, 23, 736–748. [Google Scholar]
- Sadrieh, N.; Snyderwine, E.G. Cytochromes P450 in cynomolgus monkeys mutagenically activate 2-amino-3-methylimidazo(4, 5-f)quinoline (IQ) but not 2-amino-3, 8-dimethylimidazo[4,5-f]quinoxaline(MeIQx). Carcinogenesis 1995, 16, 1549–1555. [Google Scholar] [CrossRef] [PubMed]
- Berthou, F.; Guillois, B.; Riche, C.; Dreano, Y.; Jacqz-Aigrain, E.; Beaune, P.H. Interspecies variations in caffeine metabolism related to cytochrome P4501A enzymes. Xenobiotica 1992, 22, 671–680. [Google Scholar] [CrossRef]
- Bogaards, J.J.P.; Bertrand, M.; Jackson, P.; Oudshoorn, M.J.; Weaver, R.J.; Van Bladeren, P.J.; Walther, B. Determining the best animal model for human cytochrome P450 activities: A comparison of mouse, rat, rabbit, dog, micropig, monkey and man. Xenobiotica 2000, 30, 1131–1152. [Google Scholar] [CrossRef] [PubMed]
- Chauret, N.; Gauthier, A.; Martin, J.; Nicoll-Griffith, D.A. In vitro comparison of cytochrome P450-mediated metabolic ac-tivities in human, dog, cat, and horse. Drug Metab. Dispos. 1997, 25, 1130–1136. [Google Scholar] [PubMed]
- Nishimuta, H.; Nakagawa, T.; Nomura, N.; Yabuki, M. Species differences in hepatic and intestinal metabolic activities for 43 human cytochrome P450 substrates between humans and rats or dogs. Xenobiotica 2013, 43, 948–955. [Google Scholar] [CrossRef] [PubMed]
- Nishimuta, H.; Sato, K.; Mizuki, Y.; Yabuki, M.; Komuro, S. Species differences in intestinal metabolic activities of cytochrome P450 isoforms between cynomolgus monkeys and humans. Drug Metab. Pharmacokinet. 2011, 26, 300–306. [Google Scholar] [CrossRef] [Green Version]
- Sharer, J.E.; Shipley, L.A.; Vandenbranden, M.R.; Binkley, S.N.; Wrighton, S.A. Comparisons of phase I and phase II in vitro hepatic enzyme activities of human, dog, rhesus monkey, and cynomolgus monkey. Drug Metab. Dispos. 1995, 23, 1231–1241. [Google Scholar] [PubMed]
- Weaver, R.J.; Thompson, S.; Smith, G.; Dickins, M.; Elcombe, C.R.; Mayer, R.T.; Burke, M. A comparative study of constitutive and induced alkoxyresorufin O-dealkylation and individual cytochrome P450 forms in cynomolgus monkey (Macaca fascicularis), human, mouse, rat and hamster liver microsomes. Biochem. Pharmacol. 1994, 47, 763–773. [Google Scholar] [CrossRef]
- Berthou, F.; Flinois, J.P.; Ratanasavanh, D.; Beaune, P.; Riche, C.; Guillouzo, A. Evidence for the involvement of several cy-tochromes P-450 in the first steps of caffeine metabolism by human liver microsomes. Drug Metab. Dispos. 1991, 19, 561–567. [Google Scholar] [PubMed]
- Yamazaki, M.; Wakasugi, C. Postmortem changes in drug-metabolizing enzymes of rat liver microsome. Forensic Sci. Int. 1994, 67, 155–168. [Google Scholar] [CrossRef]
- MacLeod, S.M.; Renton, K.W.; Eade, N.R. Post mortem characteristics of the hepatic microsomal drug oxidising enzyme system. Chem. Interact. 1973, 7, 29–37. [Google Scholar] [CrossRef]
- Leadbeater, L.; Davies, D. The stability of the drug metabolising enzymes of liver microsomal preparations. Biochem. Pharmacol. 1964, 13, 1607–1617. [Google Scholar] [CrossRef]
- Jakobsson, S.W.; Okita, R.T.; Mock, N.I.; Masters, B.S.S.; Buja, L.M.; Prough, R.A. Monooxygenase Activities of Human Liver, Lung, and Kidney Microsomes—A Study of 42 post mortem Cases. Acta Pharmacol. Toxicol. 1982, 50, 332–341. [Google Scholar] [CrossRef]
- Jondorf, W.R.; Donahue, J.D. Post-mortem changes in liver microsomal protein-synthesizing activity. Biochem. J. 1970, 119, 50P–51P. [Google Scholar] [CrossRef] [Green Version]
- Holschbach, M.H.; Bier, D.; Wutz, W.; Willbold, S.; Olsson, R.A. Synthesis of the Main Metabolite in Human Blood of the A1Adenosine Receptor Ligand [18F]CPFPX. Org. Lett. 2009, 11, 4266–4269. [Google Scholar] [CrossRef]
- Holschbach, M.H.; Bier, D.; Sihver, W.; Schulze, A.; Neumaier, B. Synthesis and Pharmacological Evaluation of Identified and Putative Metabolites of the A1 Adenosine Receptor Antagonist 8-Cyclopentyl-3-(3-fluoropropyl)-1-propylxanthine (CPFPX). ChemMedChem 2017, 12, 770–784. [Google Scholar] [CrossRef]
- Schneider, D.; Bier, D.; Bauer, A.; Neumaier, B.; Holschbach, M. Influence of incubation conditions on microsomal metabolism of xanthine-derived A1 adenosine receptor ligands. J. Pharmacol. Toxicol. Methods 2019, 95, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Obach, R.S.; Baxter, J.G.; Liston, T.E.; Silber, B.M.; Jones, B.C.; MacIntyre, F.; Rance, D.J.; Wastall, P. The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J. Pharmacol. Exp. Ther. 1997, 283, 46–58. [Google Scholar] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Numbering | Structural Formula | Name, Molecular Weight (MW) |
|---|---|---|
| 1 |  | 8-Cyclopentyl-3-(3-fluoropropyl)-1-propylxanthine (CPFPX) MW: 322.38 g/mol |
| 2 |  | 8-Cyclobutyl-3-(3-fluoropropyl)-1-propylxanthine (CBX) MW: 308.35 g/mol |
| 3 |  | 3-(3-Fluoropropyl)-8-(1-methylcyclobutyl)-1-propylxanthine (MCBX) MW: 322.38 g/mol |
| 4 |  | 3-(3-Fluoropropyl)-8-(3-oxocyclopent-1-en-1-yl)-1-propylxanthine (CPFPX metabolite, enone metabolite) MW: 334.35 g/mol |
| 5 |  | 8-(Cyclopent-3-en-1-yl)-3-(3-fluoropropyl)-1-propylxanthine (CPFPX metabolite) MW: 320.36 g/mol |
| 6 |  | 8-(Cyclopent-1-en-1-yl)-3-(3-fluoropropyl)-1-propylxanthine (CPFPX metabolite) MW: 320.36 g/mol |
| 7 |  | 3-(3-Fluoropropyl)-8-(3-hydroxycyclopentyl)-1-propylxanthine (CPFPX metabolite) MW: 338.38 g/mol |
| 8 |  | 3-(3-Fluoropropyl)-8-(2-hydroxycyclopentyl)-1-propylxanthine (CPFPX metabolite) MW: 338.38 g/mol |
| Peak | Retention Time (min) | Retention Factor | Functionalization | Site of Functionalization | Interpretation of Fragments 1 | Species |
|---|---|---|---|---|---|---|
| A1 | 4.8 | 0.9 | “–OH” | R | b, c, e, f: + OH − H | h, r, m, d, mp, rh |
| A2 | 5.3 | 1.1 | n.s. | n.s. | n.s. | (h), r, (m), mp, rh |
| A3 2 | 5.6 | 1.2 | “=”: “–OH” 3 human 1: 0.5 mini pig 0.05: 1 mouse 0.7: 1 rhesus 0: 1 rat 0.4: 1 dog 0.4: 1 | “=” @ R “–OH” @ F(Pr) | “=” d, e: − 2H e,f | h, r, m, d, mp, rh |
| A4 | 6.0 | 1.4 | n.s. | n.s. | n.s. | (r), (m), mp, (rh) |
| A5 | 9.6 | 2.9 | “–OH” | R | c, e, f: + OH − H | h, r, m, d, mp, rh |
| A6 | 10.4 | 3.2 | n.s. | n.s. | n.s. | (h), (r), d, (mp) |
| A7 | 12.1 | 3.9 | n.s | n.s. | n.s. | (h), r, m, d, (mp), (rh) |
| CBX | 16.6 | 5.7 | n.a. | n.a | n.a. | (h), r, m, d, (mp), rh |
| Peak | Retention Time (min) | Retention Factor | Functionalization | Site of Functionalization | Interpretation of Fragments 1 | Species |
|---|---|---|---|---|---|---|
| B1 | 5.3 | 1.1 | n.s. | n.s. | n.s. | r, mp |
| B2 | 5.7 | 1.3 | n.s. | n.s. | n.s. | (r), m, (d), (mp), (rh) |
| B3 | 6.3 | 1.5 | “–OH” | R | d: + OH − 2 H | h, r, m, d, mp, rh |
| B4 | 7.1 | 1.8 | n.s. | n.s. | n.s. | (h), m, d, (rh) |
| B5 | 8.0 | 2.2 | “–OH” | R | b, d: + OH − 2 H | h, r, m, d, mp, rh |
| B6 | 8.6 | 2.4 | “–OH” | F(Pr) | d, e | h, r, m, d, mp, rh |
| B7 | 8.9 | 2.6 | “–OH” | R | c, d: + OH − 2H | h, r, m, d, mp, rh |
| B8 | 12.9 | 4.2 | n.s. | n.s. | n.s. | (h), m, d, mp, (rh) |
| B9 | 21.7 | 7.7 | n.s | n.s. | n.s. | h, r, m, d, (mp), (rh) |
| B10 | 31.6 | 11.7 | n.s | n.s. | n.s. | h, r, m, d, (mp), (rh) |
| MCBX | 33.1 | 12.3 | n.a. | n.a. | n.a. | h, r, m, d, mp, rh |
| Peak | Retention Time (min) | Retention Factor | Functionalization | Site of Functionalization | Interpretation of Fragments 1 | Species |
|---|---|---|---|---|---|---|
| C1 | 5.5 | 1.2 | “–OH” | R | b, e, f: + OH − H | h, r, m, d, mp, rh |
| C2 | 5.9 | 1.4 | “–OH” | R | f: + OH − H | h, r, m, d, mp, rh |
| C3 | 6.3 | 1.5 | “=O” | R | b, e, f: + O − H | h, r, m, d, (mp), (rh) |
| C4 | 7.4 | 2.0 | n.s. | n.s. | n.s. | (h), r, m, d, mp, rh |
| C5 | 7.9 | 2.2 | “=” | (F)Pr | d, e, f | h, r, m, d, mp, rh |
| C6 | 8.3 | 2.3 | “=” | (F)Pr | e, f | h, r, m, d, mp, rh |
| C7 | 8.7 | 2.5 | n.s. | n.s. | n.s. | (h), r, (m), (d), (rh) |
| C8 | 9.0 | 2.6 | n.s. | n.s. | n.s. | (r), m, (d), (rh) |
| C9 | 9.8 | 2.9 | “=O”/“=” | R | b, e, f: + OH − 4H | h, d |
| C10 | 12.2 | 3.9 | n.s | n.s. | n.s. | h, r, m, d, mp, rh |
| C11 | 21.4 | 7.6 | n.s. | n.s. | n.s. | (h), r, m, (mp), rh |
| C12 | 34.0 | 12.6 | n.s | n.s. | n.s. | r, (m), (d) |
| CPFPX | 29.4 | 10.8 | n.a. | n.a. | n.a. | h, r, m, d, mp, rh |
| Species | Total Microsomal P450 Content (nmol/mg Microsomal Protein) | ||||
|---|---|---|---|---|---|
| [13] | [14] | [15] | [16] | [17] | |
| Human | 0.307 ± 0.160 | 0.231 ± 0.013 | 0.31 ± 0.09 | n.d. | 0.29 ± 0.06 |
| Rat | 0.673 ± 0.050 | 0.444 ± 0.016 | 0.58 ± 0.02 | n.d. | n.d. |
| Mouse | n.d. | 0.719 ± 0.041 | 0.48 ± 0.04 | n.d. | n.d. |
| Mini pig | n.d. | n.d. | n.d. | 0.798 ± 0.145 | n.d. |
| Dog | 0.385 ± 0.036 | 0.685 ± 0.031 | n.d. | n.d. | n.d. |
| Monkey | 1.030 ± 0.106 1 | 1.195 ± 0.089 2 | 0.74 ± 0.02 1 | n.d. | 0.95 ± 0.08 2 |
| Microsomes | Substrate | Microsomal Protein Concentration (mg/mL) | Incubation Time (min) |
|---|---|---|---|
| HLM | CBX, MCBX, CPFPX | 0.8 | 180 |
| RLM | CBX, MCBX, CPFPX | 0.4 | 30 |
| MLM | CBX, MCBX, CPFPX | 0.4 | 30 |
| DLM | CBX, MCBX, CPFPX | 0.8 | 45 |
| MPLM | CBX | 0.8 | 45 |
| MCBX, CPFPX | 0.8 | 30 | |
| RMLM | CBX | 0.04 | 45 |
| MCBX, CPFPX | 0.04 | 30 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schneider, D.; Bier, D.; Holschbach, M.; Bauer, A.; Neumaier, B. Species Differences in Microsomal Metabolism of Xanthine-Derived A1 Adenosine Receptor Ligands. Pharmaceuticals 2021, 14, 277. https://doi.org/10.3390/ph14030277
Schneider D, Bier D, Holschbach M, Bauer A, Neumaier B. Species Differences in Microsomal Metabolism of Xanthine-Derived A1 Adenosine Receptor Ligands. Pharmaceuticals. 2021; 14(3):277. https://doi.org/10.3390/ph14030277
Chicago/Turabian StyleSchneider, Daniela, Dirk Bier, Marcus Holschbach, Andreas Bauer, and Bernd Neumaier. 2021. "Species Differences in Microsomal Metabolism of Xanthine-Derived A1 Adenosine Receptor Ligands" Pharmaceuticals 14, no. 3: 277. https://doi.org/10.3390/ph14030277
APA StyleSchneider, D., Bier, D., Holschbach, M., Bauer, A., & Neumaier, B. (2021). Species Differences in Microsomal Metabolism of Xanthine-Derived A1 Adenosine Receptor Ligands. Pharmaceuticals, 14(3), 277. https://doi.org/10.3390/ph14030277