
Development and Validation of an Up-to-Date Highly Sensitive UHPLC-MS/MS Method for the Simultaneous Quantification of Current Anti-HIV Nucleoside Analogues in Human Plasma

 ,
,  , ,
, ,  , , , ,
, , , ,
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Method Development and Preliminary Experiments
2.2. Calibration Curve and Dilution Integrity
2.3. Specificity and Selectivity
2.4. Accuracy and Precision
2.5. Lower Limit of Quantification (LLOQ) and Limit of Detection (LOD)
2.6. Recovery
2.7. Matrix Effect
2.8. Carry-Over
2.9. Stability
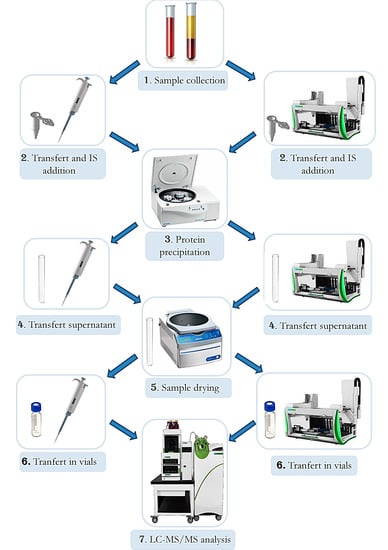
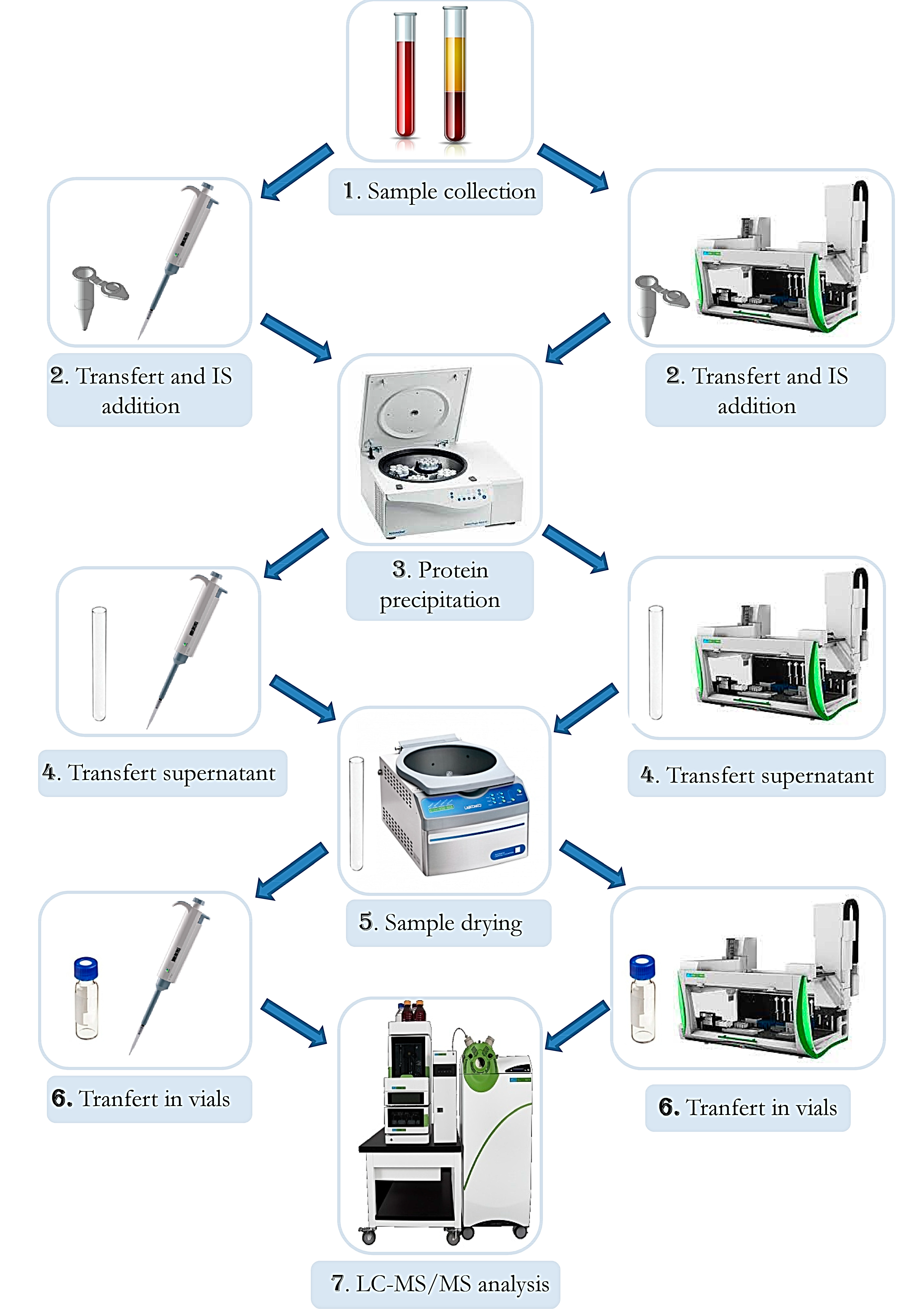
2.10. Automation
2.11. Testing of Patients’ Samples
3. Materials and Methods
3.1. Chemicals
3.2. Stock Solutions, Standards and Quality Controls
3.3. Method Development and Preliminary Experiments
3.4. Chromatographic Conditions
3.5. Mass Spectrometry Conditions
3.6. STDs, QCs and Patients’ Samples Extraction
3.7. Specificity and Selectivity
3.8. Accuracy, Precision, Calibration and Limit of Quantification
3.9. Recovery
3.10. Stability
3.11. Matrix Effect
3.12. Carry-Over
3.13. Automation
3.14. Clinical Application and Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Younai, F.S. Thirty years of the human immunodeficiency virus epidemic and beyond. Int. J. Oral Sci. 2013, 5, 191–199. [Google Scholar] [CrossRef] [Green Version]
- WHO. Updated recommendations on first-line and second-line antiretroviral regimens and post-exposure prophylaxis and recommendations on early infant diagnosis of HIV. In Interim Guidelines; Supplement to the 2016 Consolidated Guidelines on the Use of Antiretroviral Drugs for Treating and Preventing HIV Infection; World Health Organization: Geneva, Switzerland, 2018. [Google Scholar]
- Chargin, A.; Yin, F.; Song, M.; Subramaniam, S.; Knutson, G.; Patterson, B.K. Identification and characterization of HIV-1 latent viral reservoirs in peripheral blood. J. Clin. Microbiol. 2015, 53, 60–66. [Google Scholar] [CrossRef] [Green Version]
- Olabode, D.; Rong, L.; Wang, X. Optimal control in HIV chemotherapy with termination viral load and latent reservoir. Math. Biosci. Eng. 2019, 16, 619–635. [Google Scholar] [CrossRef] [PubMed]
- Rothenberger, M.K.; Keele, B.F.; Wietgrefe, S.W.; Fletcher, C.V.; Beilman, G.J.; Chipman, J.G.; Khoruts, A.; Estes, J.D.; Anderson, J.; Callisto, S.P.; et al. Large number of rebounding/founder HIV variants emerge from multifocal infection in lymphatic tissues after treatment interruption. Proc. Natl. Acad. Sci. USA 2015, 112, E1126–E1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Novoa, S.; Alvarez, E.; Labarga, P.; Soriano, V. Renal toxicity associated with tenofovir use. Expert Opin. Drug Saf. 2010, 9, 545–559. [Google Scholar] [CrossRef] [PubMed]
- Margolis, A.M.; Heverling, H.; Pham, P.A.; Stolbach, A. A review of the toxicity of HIV medications. J. Med. Toxicol. 2013, 10, 26–39. [Google Scholar] [CrossRef] [Green Version]
- Karris, M.Y.; Jain, S.; Bowman, V.Q.; Rieg, G.; Goicoechea, M.; Dube, M.P.; Kerkar, S.; Kemper, C.; Diamond, C.; Sun, X.; et al. Nucleoside-Sparing Regimens With Raltegravir and a Boosted Protease Inhibitor: An Unsettled Issue. J. Acquir. Immune Defic. Syndr. 2016, 72, e48–e50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calcagno, A.; Gonzalez de Requena, D.; Simiele, M.; D’Avolio, A.; Tettoni, M.C.; Salassa, B.; Orofino, G.; Bramato, C.; Libanore, V.; Motta, I.; et al. Tenofovir plasma concentrations according to companion drugs: A cross-sectional study of HIV-positive patients with normal renal function. Antimicrob. Agents Chemother. 2013, 57, 1840–1843. [Google Scholar] [CrossRef] [Green Version]
- Calcagno, A.; Di Perri, G.; Bonora, S. What do we know about tailoring treatment with tenofovir? Pharmacogenomics 2016, 17, 531–534. [Google Scholar] [CrossRef] [Green Version]
- Jotwani, V.; Scherzer, R.; Glidden, D.V.; Mehrotra, M.; Defechereux, P.; Liu, A.; Gandhi, M.; Bennett, M.; Coca, S.G.; Parikh, C.R.; et al. Pre-exposure Prophylaxis With Tenofovir Disoproxil Fumarate/Emtricitabine and Kidney Tubular Dysfunction in HIV-Uninfected Individuals. J. Acquir. Immune Defic. Syndr. 2018, 78, 169–174. [Google Scholar] [CrossRef]
- Podany, A.T.; Bares, S.H.; Havens, J.; Dyavar, S.R.; O’Neill, J.; Lee, S.; Fletcher, C.V.; Swindells, S.; Scarsi, K.K. Plasma and intracellular pharmacokinetics of tenofovir in patients switched from tenofovir disoproxil fumarate to tenofovir alafenamide. AIDS 2018, 32, 761–765. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.; Hughes, S.L.; Gotham, D.; Pozniak, A.L. Tenofovir alafenamide versus tenofovir disoproxil fumarate: Is there a true difference in efficacy and safety? J. Virus Erad. 2018, 4, 72–79. [Google Scholar] [CrossRef]
- EASL. EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J. Hepatol. 2017, 67, 370–398. [Google Scholar] [CrossRef] [Green Version]
- Sutton, S.S.; Ahuja, D.; Magagnoli, J. What is the effect of pill burden on adherence to HIV antiretroviral therapy? JAAPA 2016, 29, 16–17. [Google Scholar] [CrossRef] [PubMed]
- Moosa, A.; Gengiah, T.N.; Lewis, L.; Naidoo, K. Long-term adherence to antiretroviral therapy in a South African adult patient cohort: A retrospective study. BMC Infect. Dis. 2019, 19, 775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boussari, O.; Subtil, F.; Genolini, C.; Bastard, M.; Iwaz, J.; Fonton, N.; Etard, J.F.; Ecochard, R. Impact of variability in adherence to HIV antiretroviral therapy on the immunovirological response and mortality. BMC Med. Res. Methodol. 2015, 15, 10. [Google Scholar] [CrossRef] [Green Version]
- Gervasoni, C.; Meraviglia, P.; Landonio, S.; Riva, A.; Galli, M.; Rizzardini, G.; Cattaneo, D. Tenofovir plasma concentrations in post-menopausal versus pre-menopausal HIV-infected women. J. Antimicrob. Chemother. 2013, 68, 1206–1207. [Google Scholar] [CrossRef] [Green Version]
- Gervasoni, C.; Meraviglia, P.; Landonio, S.; Baldelli, S.; Fucile, S.; Castagnoli, L.; Clementi, E.; Riva, A.; Galli, M.; Rizzardini, G.; et al. Low body weight in females is a risk factor for increased tenofovir exposure and drug-related adverse events. PLoS ONE 2013, 8, e80242. [Google Scholar] [CrossRef]
- Avataneo, V.; De Nicolò, A.; Rabbia, F.; Perlo, E.; Burrello, J.; Berra, E.; Pappaccogli, M.; Cusato, J.; D’Avolio, A.; Di Perri, G.; et al. Therapeutic drug monitoring-guided definition of adherence profiles in resistant hypertension and identification of predictors of poor adherence. Br. J. Clin. Pharmacol. 2018, 84, 2535–2543. [Google Scholar] [CrossRef]
- De Nicolò, A.; Avataneo, V.; Rabbia, F.; Sciandra, M.; Tosello, F.; Cusato, J.; Perlo, E.; Mulatero, P.; Veglio, F.; Di Perri, G.; et al. UHPLC-MS/MS method with sample dilution to test therapeutic adherence through quantification of ten antihypertensive drugs in urine samples. J. Pharm. Biomed. Anal. 2017, 142, 279–285. [Google Scholar] [CrossRef]
- Illamola, S.M.; Valade, E.; Hirt, D.; Dulioust, E.; Zheng, Y.; Wolf, J.P.; Treluyer, J.M. Development and validation of a LC-MS/MS method for the quantification of tenofovir and emtricitabine in seminal plasma. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2016, 1033–1034, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Hummert, P.; Parsons, T.L.; Ensign, L.M.; Hoang, T.; Marzinke, M.A. Validation and implementation of liquid chromatographic-mass spectrometric (LC-MS) methods for the quantification of tenofovir prodrugs. J. Pharm. Biomed. Anal. 2018, 152, 248–256. [Google Scholar] [CrossRef]
- Ma, Z.; Li, S.; He, D.; Wang, Y.; Jiang, H.; Zhou, H.; Jin, J.; Lin, N. Rapid quantification of tenofovir in umbilical cord plasma and amniotic fluid in hepatitis B mono-infected pregnant women during labor by ultra-performance liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2020, 34, e8728. [Google Scholar] [CrossRef]
- Yamada, E.; Takagi, R.; Sudo, K.; Kato, S. Determination of abacavir, tenofovir, darunavir, and raltegravir in human plasma and saliva using liquid chromatography coupled with tandem mass spectrometry. J. Pharm. Biomed. Anal. 2015, 114, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Nirogi, R.; Bhyrapuneni, G.; Kandikere, V.; Mudigonda, K.; Komarneni, P.; Aleti, R.; Mukkanti, K. Simultaneous quantification of a non-nucleoside reverse transcriptase inhibitor efavirenz, a nucleoside reverse transcriptase inhibitor emtricitabine and a nucleotide reverse transcriptase inhibitor tenofovir in plasma by liquid chromatography positive ion electrospray tandem mass spectrometry. Biomed. Chromatogr. 2009, 23, 371–381. [Google Scholar] [CrossRef]
- Le Saux, T.; Chhun, S.; Rey, E.; Launay, O.; Weiss, L.; Viard, J.P.; Pons, G.; Jullien, V. Quantification of seven nucleoside/nucleotide reverse transcriptase inhibitors in human plasma by high-performance liquid chromatography with tandem mass-spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2008, 865, 81–90. [Google Scholar] [CrossRef]
- Kromdijk, W.; Pereira, S.A.; Rosing, H.; Mulder, J.W.; Beijnen, J.H.; Huitema, A.D. Development and validation of an assay for the simultaneous determination of zidovudine, abacavir, emtricitabine, lamivudine, tenofovir and ribavirin in human plasma using liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2013, 919–920, 43–51. [Google Scholar] [CrossRef]
- De Nicolò, A.; Simiele, M.; Pensi, D.; Boglione, L.; Allegra, S.; Di Perri, G.; D’Avolio, A. UPLC-MS/MS method for the simultaneous quantification of anti-HBV nucleos(t)ides analogs: Entecavir, lamivudine, telbivudine and tenofovir in plasma of HBV infected patients. J. Pharm. Biomed. Anal. 2015, 114, 127–132. [Google Scholar] [CrossRef]
- De Clercq, E. Tenofovir alafenamide (TAF) as the successor of tenofovir disoproxil fumarate (TDF). Biochem. Pharmacol. 2016, 119, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Birkus, G.; Kutty, N.; He, G.X.; Mulato, A.; Lee, W.; McDermott, M.; Cihlar, T. Activation of 9-[(R)-2-[[(S)-[[(S)-1-(Isopropoxycarbonyl)ethyl]amino] phenoxyphosphinyl]-methoxy]propyl]adenine (GS-7340) and other tenofovir phosphonoamidate prodrugs by human proteases. Mol. Pharmacol. 2008, 74, 92–100. [Google Scholar] [CrossRef]
- Eisenberg, E.J.; He, G.X.; Lee, W.A. Metabolism of GS-7340, a novel phenyl monophosphoramidate intracellular prodrug of PMPA, in blood. Nucleosides Nucleotides Nucleic Acids 2001, 20, 1091–1098. [Google Scholar] [CrossRef] [PubMed]
- Custodio, J.M.; Fordyce, M.; Garner, W.; Vimal, M.; Ling, K.H.; Kearney, B.P.; Ramanathan, S. Pharmacokinetics and Safety of Tenofovir Alafenamide in HIV-Uninfected Subjects with Severe Renal Impairment. Antimicrob. Agents Chemother. 2016, 60, 5135–5140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Nicolò, A.; Ianniello, A.; Ferrara, M.; Avataneo, V.; Cusato, J.; Antonucci, M.; De Vivo, E.; Waitt, C.; Calcagno, A.; Trentalange, A.; et al. Validation of a UHPLC-MS/MS Method to Quantify Twelve Antiretroviral Drugs within Peripheral Blood Mononuclear Cells from People Living with HIV. Pharmaceuticals 2020, 14, 12. [Google Scholar] [CrossRef] [PubMed]
- EMA. Guideline on Bioanalytical Method Validation. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/08/WC500109686.pdf (accessed on 4 March 2021).
- FDA. Guidance for Industry: Bioanalytical Method Validation. Available online: https://www.fda.gov/files/drugs/published/Bioanalytical-Method-Validation-Guidance-for-Industry.pdf (accessed on 4 March 2021).
- De Nicolò, A.; Avataneo, V.; Rabbia, F.; Bonifacio, G.; Cusato, J.; Tomasello, C.; Perlo, E.; Mulatero, P.; Veglio, F.; Di Perri, G.; et al. UHPLC-MS/MS method with protein precipitation extraction for the simultaneous quantification of ten antihypertensive drugs in human plasma from resistant hypertensive patients. J. Pharm. Biomed. Anal. 2016, 129, 535–541. [Google Scholar] [CrossRef]
- De Nicolò, A.; Cantu, M.; D’Avolio, A. Matrix effect management in liquid chromatography mass spectrometry: The internal standard normalized matrix effect. Bioanalysis 2017, 9, 1093–1105. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.A.; He, G.X.; Eisenberg, E.; Cihlar, T.; Swaminathan, S.; Mulato, A.; Cundy, K.C. Selective intracellular activation of a novel prodrug of the human immunodeficiency virus reverse transcriptase inhibitor tenofovir leads to preferential distribution and accumulation in lymphatic tissue. Antimicrob. Agents Chemother. 2005, 49, 1898–1906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- EMA. Genvoya: EPAR—Product Information. Available online: https://www.ema.europa.eu/en/documents/product-information/genvoya-epar-product-information_en.pdf (accessed on 9 March 2021).
- Lepist, E.I.; Phan, T.K.; Roy, A.; Tong, L.; Maclennan, K.; Murray, B.; Ray, A.S. Cobicistat boosts the intestinal absorption of transport substrates, including HIV protease inhibitors and GS-7340, in vitro. Antimicrob. Agents Chemother. 2012, 56, 5409–5413. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, C.V.; Podany, A.T.; Thorkelson, A.; Winchester, L.C.; Mykris, T.; Anderson, J.; Jorstad, S.; Baker, J.V.; Schacker, T.W. The Lymphoid Tissue Pharmacokinetics of Tenofovir Disoproxil Fumarate and Tenofovir Alafenamide in HIV-Infected Persons. Clin. Pharmacol. Ther. 2020, 108, 971–975. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| DRUGs | RT (min) | STD 9 (ULOQ) (ng/mL) | Calibration Range (ng/mL) | LLOQ (ng/mL) | LOD (ng/mL) | [M + H]+ (m/z) | Dwell Time (ms) | Entrance Voltage (V) | FIRST Trace (m/z) | Collision Energy First Ion Trace (eV) | SECOND Trace (m/z) | Collision Energy Second Ion Trace (eV) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 13C5-TFV | 2.05 | - | - | - | - | 293.1 | 25 | 30 | 181.1 | −30 | 164.1 | −45 |
| TFV | 2.05 | 400 | 1.6–400 | 1.6 | <0.8 | 288.1 | 25 | 30 | 176.1 | −30 | 159.1 | −45 |
| 13C1-2H2-3TC | 2.62 | - | - | - | - | 233.1 | 25 | 10 | 113.1 | −42 | - | - |
| 3TC | 2.66 | 2500 | 9.8–2500 | 9.8 | <5.0 | 230.1 | 25 | 10 | 112.1 | −42 | - | - |
| THY * | 3.66 | - | - | - | - | 241.1 * | 25 | −26 | 42.0 | 74 | 151.0 | 15 |
| 2H3-15N-FTC | 3.87 | - | - | - | - | 252.0 | 25 | 15 | 132.1 | −37 | 114.0 | −65 |
| FTC | 3.88 | 2500 | 1.6–400 | 9.8 | <5.0 | 248.0 | 25 | 15 | 130.1 | −37 | 113.0 | −65 |
| 2H5-ABV | 4.00 | - | - | - | - | 292.2 | 25 | 24 | 196.1 | −37 | - | - |
| ABV | 4.01 | 2500 | 9.8–2500 | 9.8 | <5.0 | 287.2 | 25 | 24 | 191.1 | −37 | - | - |
| AZT * | 4.32 | 2500 | 9.8–2500 | 9.8 | <5.0 | 266.1 * | 25 | −17 | 223.0 | 15 | 193.0 | 19 |
| 2H6-TAF | 4.55 | - | - | - | - | 483.2 | 25 | 40 | 271.0 | −42 | 346.1 | −33 |
| TAF | 4.55 | 400 | 1.6–400 | 1.6 | <0.8 | 477.2 | 25 | 40 | 270.0 | −42 | 345.1 | −33 |
| DRUG | QC High | QC Medium | QC Low | OVERALL | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Conc. ng/mL | Acc. % | Precision RSD% | Conc. ng/mL | Acc. % | Precision RSD% | Conc. ng/mL | Acc. % | Precision RSD% | Acc. % | RSD Intra-Day % | RSD Inter-Day % | ||||
| Intra-Day % | Inter-Day % | Intra-Day % | Inter-Day % | Intra-Day % | Inter-Day % | ||||||||||
| TFV | 320 | 102.4 | 3.4 | 3.5 | 20 | 99.2 | 2.4 | 4.4 | 4 | 100.9 | 6.7 | 6.3 | 100.9 | 4.5 | 4.7 |
| 3TC | 2000 | 104.0 | 3.4 | 1.9 | 100 | 106.1 | 1.0 | 1.5 | 20 | 106.8 | 1.6 | 5.2 | 105.6 | 2.4 | 4.7 |
| FTC | 2000 | 104.6 | 3.2 | 5.8 | 100 | 109.6 | 1.5 | 6.9 | 20 | 94.3 | 4.4 | 3.0 | 102.9 | 7.1 | 6.5 |
| ABV | 2000 | 104.3 | 2.6 | 3.2 | 100 | 106.9 | 5.7 | 2.6 | 20 | 95.8 | 4.1 | 3.7 | 102.3 | 6.3 | 3.1 |
| AZT | 2000 | 106.5 | 2.8 | 3.5 | 100 | 101.6 | 8.0 | 3.9 | 20 | 100.2 | 7.3 | 4.6 | 102.4 | 6.5 | 4.0 |
| TAF | 320 | 107.6 | 2.9 | 5.2 | 20 | 101.0 | 6.2 | 5.4 | 4 | 98.4 | 9.7 | 6.3 | 102.3 | 7.2 | 5.6 |
| Lithium/Heparin Samples (n = 6) | ||||
| Drug | Mean REC % (RSD %) | Mean IS-nREC % (RSD %) | Mean ME % (RSD %) | Mean IS-nME % (RSD %) |
| TFV | 82.4 (5.6) | 106.9 (0.3) | +19.6 (4.4) | −1.6 (5.1) |
| 3TC | 103.2 (2.7) | 103.7 (1.4) | +23.5 (1.1) | −2.2 (1.3) |
| FTC | 103.2 (4.0) | 104.2 (1.9) | +25.8 (2.4) | −4.1 (2.3) |
| ABV | 91.2 (1.7) | 110.3 (0.4) | +49.8 (2.8) | −1.2 (2.5) |
| AZT | 68.7 (7.0) | 72.2 (6.2) | −9.4 (1.3) | −2.2 (1.5) |
| TAF | 74.0 (2.0) | 96.2 (5.1) | +17.5 (3.4) | +4.5 (3.4) |
| Sodium Citrate Samples (n = 6) | ||||
| Drug | Mean Rec. % (RSD %) | Mean IS-nREC % (RSD %) | Mean ME% (RSD %) | Mean IS-nME % (RSD %) |
| TFV | 94.1 (1.7) | 101.1 (9.7) | +33.1 (6.2) | +8.3 (4.4) |
| 3TC | 110.5 (2.7) | 105.9 (0.8) | +21.3 (1.5) | −1.6 (0.2) |
| FTC | 107.2 (3.3) | 105.0 (1.2) | +21.0 (2.1) | −3.3 (2.4) |
| ABV | 90.1 (1.6) | 105.8 (2.5) | +42.0 (3.2) | +1.8 (2.8) |
| AZT | 71.3 (3.8) | 79.7 (4.0) | −8.9 (2.2) | −2.5 (2.3) |
| TAF | 92.3 (5.1) | 101.2 (3.8) | +23.0 (2.1) | −2.9 (3.2) |
| OVERALL | ||||
| Drug | Mean Rec. % (RSD %) | Mean IS-nREC % (RSD %) | Mean ME% (RSD %) | Mean IS-nME % (RSD %) |
| TFV | 88.2 (8.3) | 104.0 (7.8) | +26.3 (6.2) | +3.4 (5.5) |
| 3TC | 106.8 (4.3) | 104.8 (1.5) | +22.4 (1.0) | −1.9 (0.3) |
| FTC | 105.2 (3.7) | 104.6 (2.5) | +23.4 (2.3) | −3.7 (4.5) |
| ABV | 90.6 (1.5) | 108.1 (2.2) | +45.9 (3.1) | +0.3 (1.7) |
| AZT | 70.0 (5.0) | 78.4 (5.9) | −9.1 (0.3) | −2.3 (1.8) |
| TAF | 83.1 (13.2) | 98.7 (5.9) | +20.2 (2.6) | −0.8 (5.4) |
| Room Temperature Stability in Quality Control Samples | ||||||
|---|---|---|---|---|---|---|
| 2 Days | 4 Days | 7 Days | 10 Days | 12 Days | 14 Days | |
| TFV | 120.9% | 138.4% | 156.8% | 142.4% | 148.2% | 149.0% |
| 3TC | 111.1% | 102.1% | 106.0% | 98.8% | 102.0% | 102.4% |
| ABV | 110.6% | 103.4% | 106.0% | 101.7% | 101.8% | 102.4% |
| FTC | 111.9% | 101.1% | 103.2% | 101.0% | 97.8% | 105.1% |
| TAF | 53.8% | 23.1% | 8.2% | 2.7% | 7.0% | 0.0% |
| AZT | 93.0% | 100.0% | 102.0% | 103.7% | 89.3% | 93.7% |
| Room Temperature Stability in Patients Samples at the End of Dosing Interval (Ctrough) | ||||||
| TFV | 99% | 99% | 109% | 81% | 89% | n.a. |
| FTC | 90% | 92% | 99% | 121% | 97% | n.a. |
| TAF | n.d. | n.d. | n.d. | n.d. | n.d. | n.a. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Nicolò, A.; Manca, A.; Ianniello, A.; Palermiti, A.; Calcagno, A.; Ferrara, M.; Antonucci, M.; Cusato, J.; Avataneo, V.; De Vivo, E.; et al. Development and Validation of an Up-to-Date Highly Sensitive UHPLC-MS/MS Method for the Simultaneous Quantification of Current Anti-HIV Nucleoside Analogues in Human Plasma. Pharmaceuticals 2021, 14, 460. https://doi.org/10.3390/ph14050460
De Nicolò A, Manca A, Ianniello A, Palermiti A, Calcagno A, Ferrara M, Antonucci M, Cusato J, Avataneo V, De Vivo E, et al. Development and Validation of an Up-to-Date Highly Sensitive UHPLC-MS/MS Method for the Simultaneous Quantification of Current Anti-HIV Nucleoside Analogues in Human Plasma. Pharmaceuticals. 2021; 14(5):460. https://doi.org/10.3390/ph14050460
Chicago/Turabian StyleDe Nicolò, Amedeo, Alessandra Manca, Alice Ianniello, Alice Palermiti, Andrea Calcagno, Micol Ferrara, Miriam Antonucci, Jessica Cusato, Valeria Avataneo, Elisa De Vivo, and et al. 2021. "Development and Validation of an Up-to-Date Highly Sensitive UHPLC-MS/MS Method for the Simultaneous Quantification of Current Anti-HIV Nucleoside Analogues in Human Plasma" Pharmaceuticals 14, no. 5: 460. https://doi.org/10.3390/ph14050460
APA StyleDe Nicolò, A., Manca, A., Ianniello, A., Palermiti, A., Calcagno, A., Ferrara, M., Antonucci, M., Cusato, J., Avataneo, V., De Vivo, E., Bonora, S., De Rosa, F. G., Di Perri, G., & D’Avolio, A. (2021). Development and Validation of an Up-to-Date Highly Sensitive UHPLC-MS/MS Method for the Simultaneous Quantification of Current Anti-HIV Nucleoside Analogues in Human Plasma. Pharmaceuticals, 14(5), 460. https://doi.org/10.3390/ph14050460