
Lung Cancer Management with Silibinin: A Historical and Translational Perspective

 , , ,
, , ,  ,
, 
 and
and 
Abstract
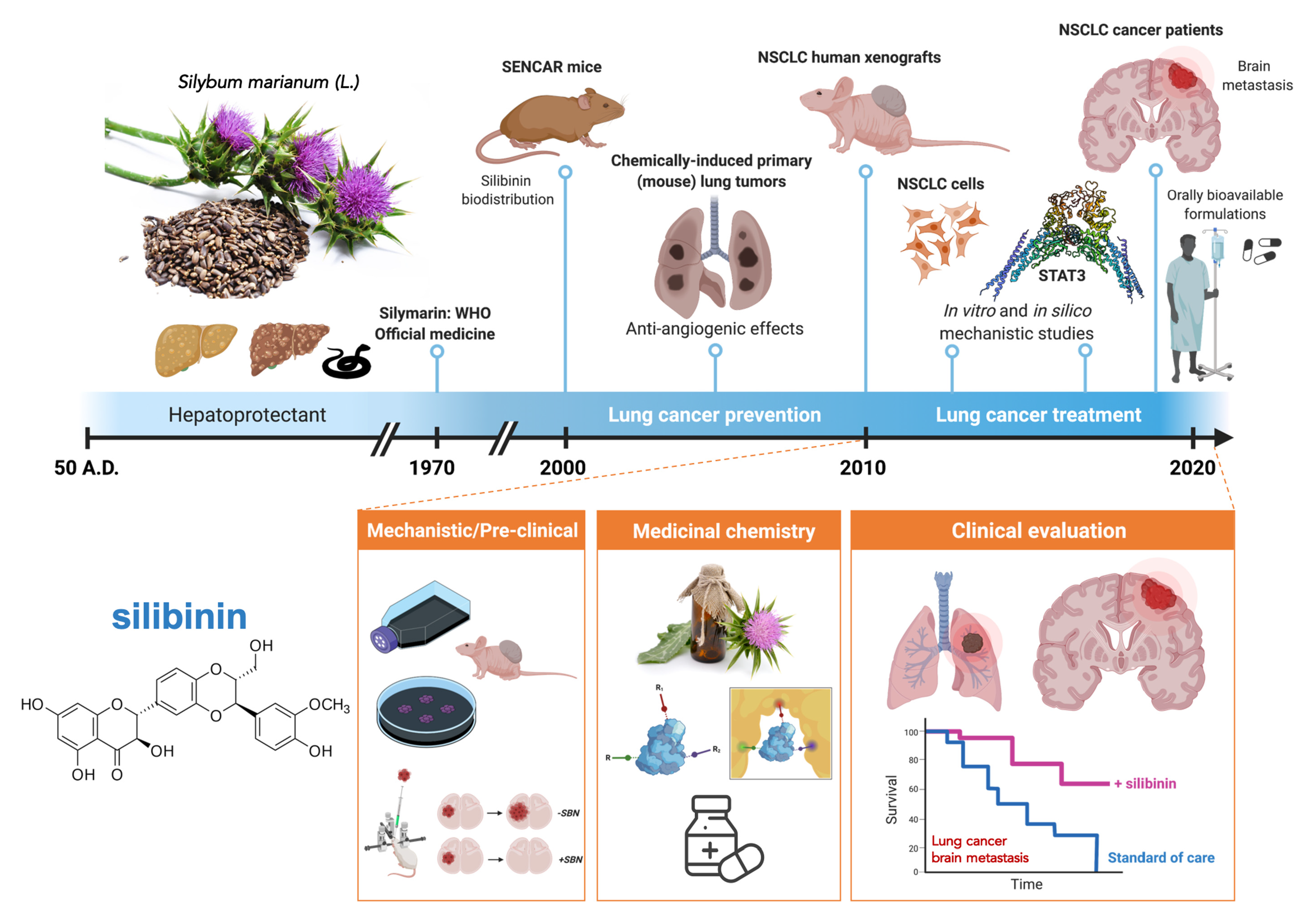
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
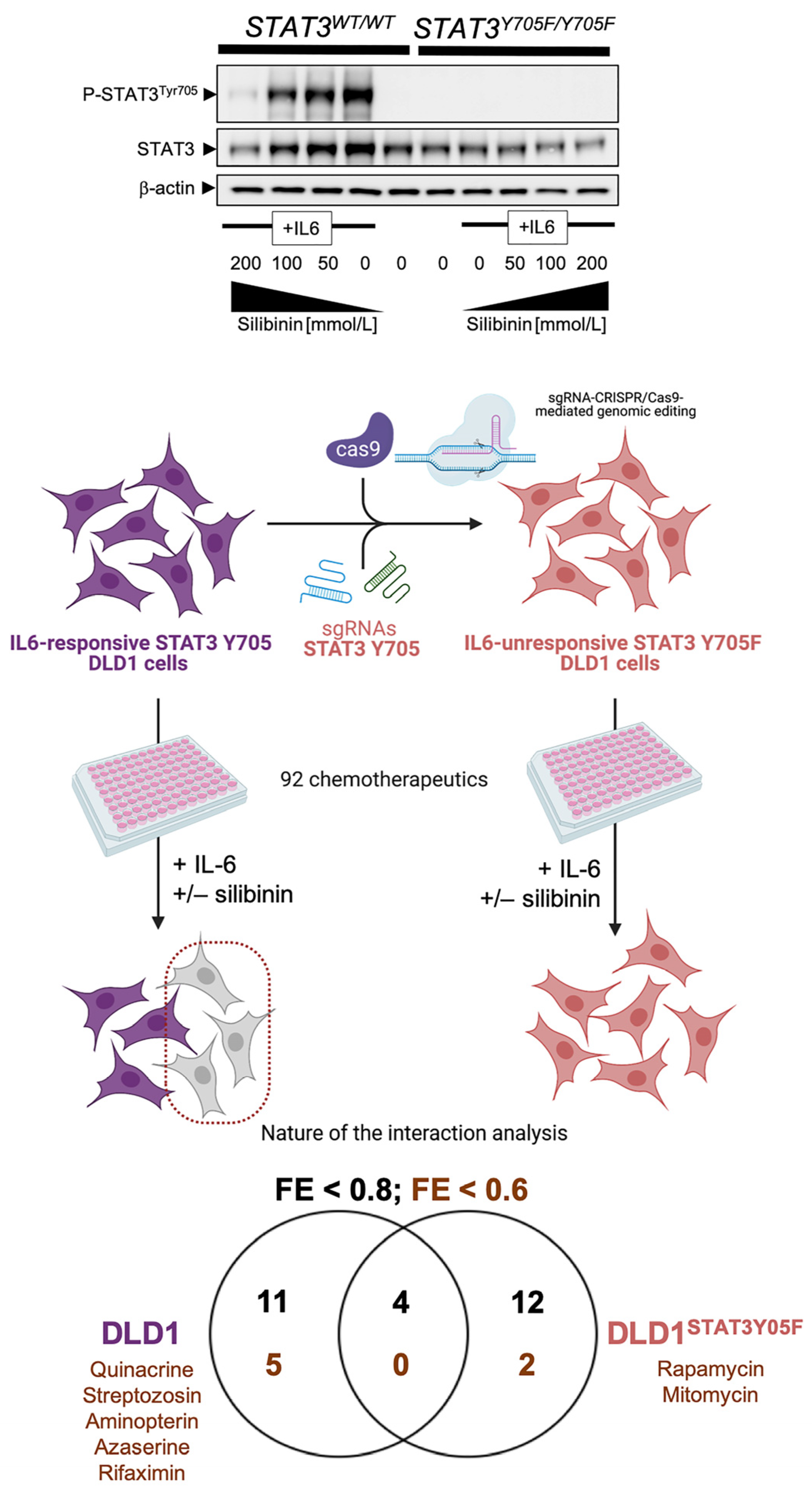{kind=link}
1. Introduction
2. Silibinin-Containing Milk Thistle Fruits and Human Health: A Brief Historical Overview
3. Silibinin to Therapeutically Manage Lung Cancer: Pioneering Studies
4. Silibinin and Lung Cancer Prevention: Evidence from Chemically Induced Primary Lung Tumors
5. Silibinin and Lung Cancer Treatment: Evidence from Laboratory In Vitro and Animal Models
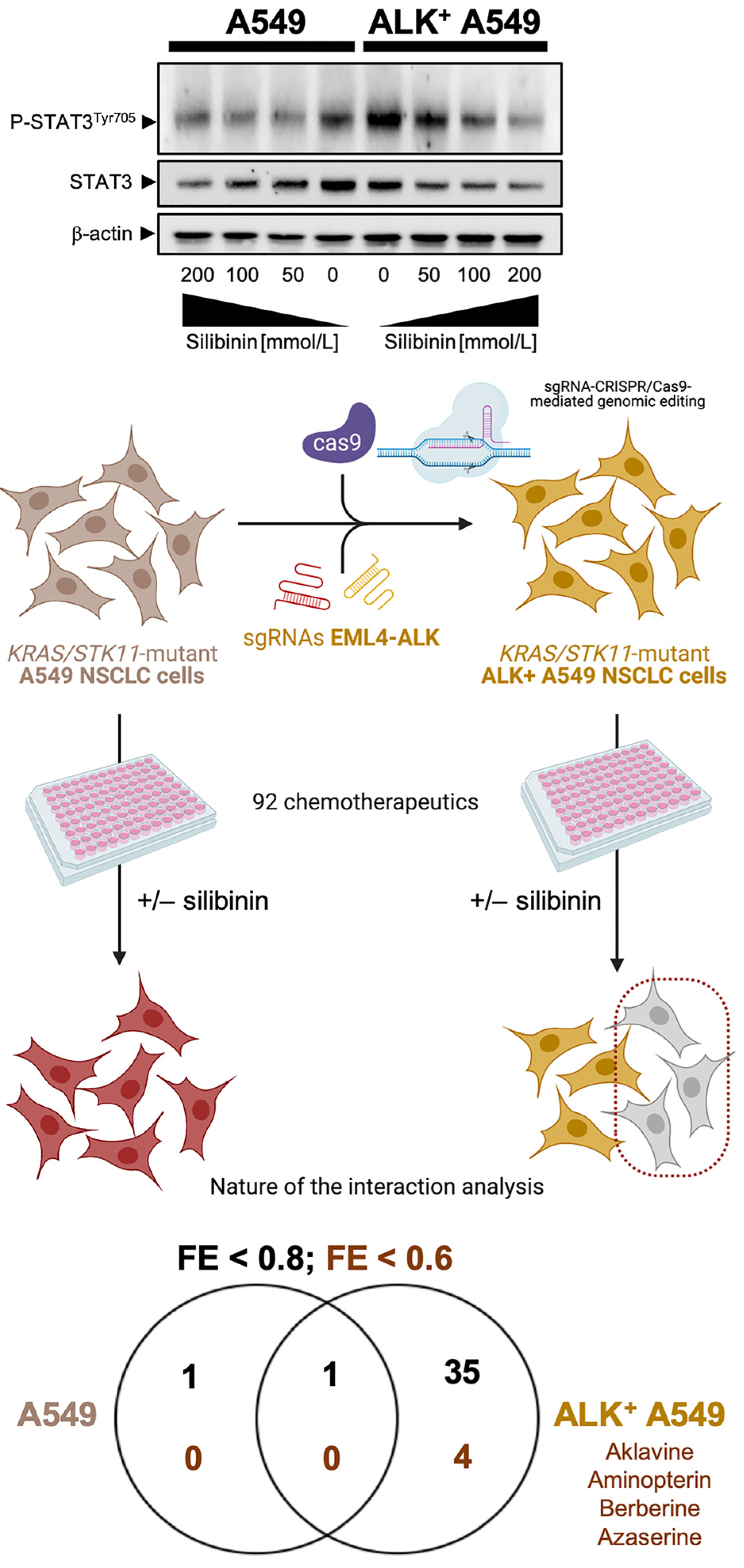
5.1. Silibinin and Lung Cancer Drug Resistance
5.2. Silibinin and Lung Cancer Metastatic Traits
5.2.1. Inhibition of Cell Invasion
5.2.2. Inhibition of Epithelial-to-Mesenchymal Transition
5.2.3. Inhibition of Brain Metastasis
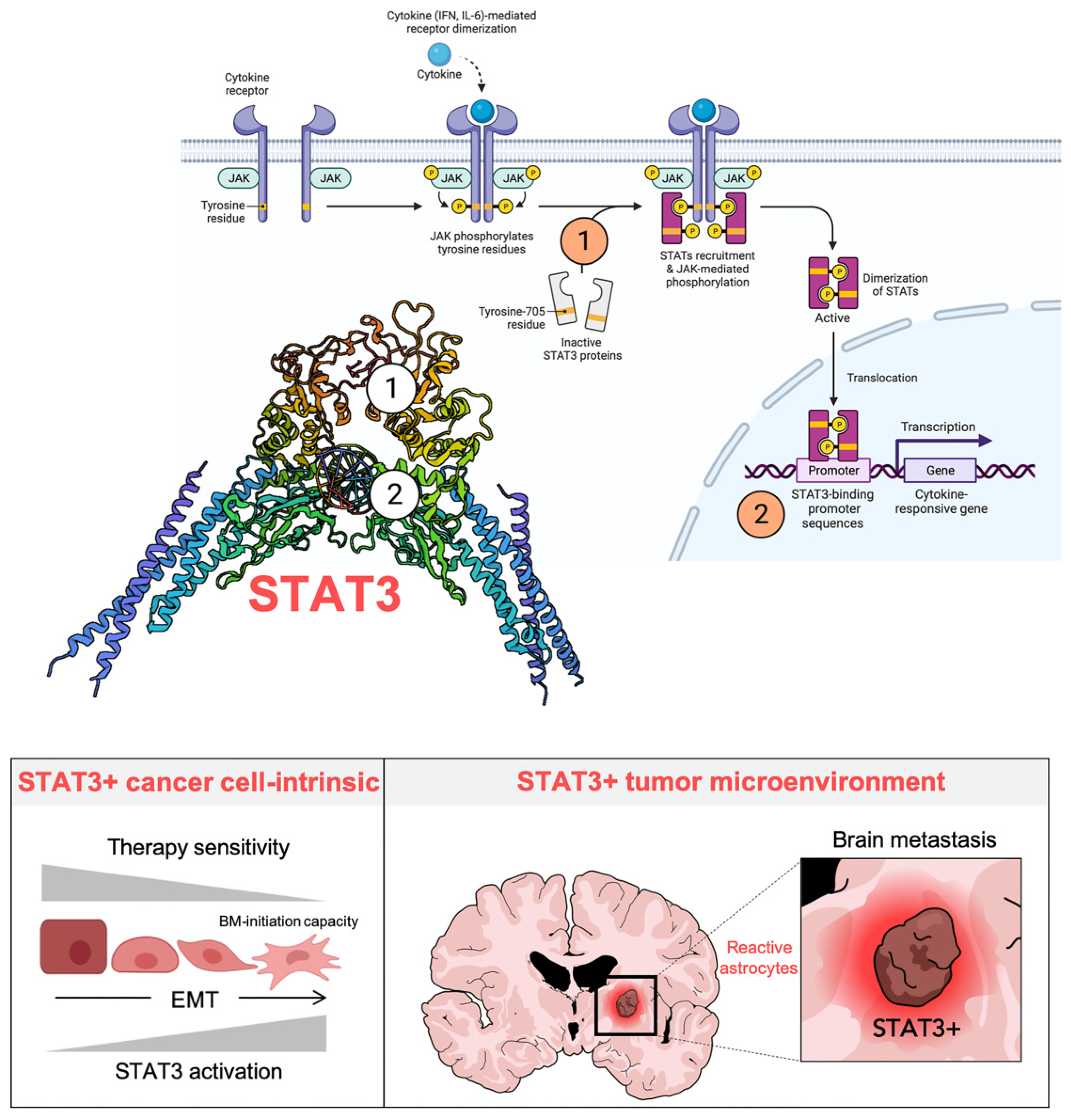
6. STAT3: A primary Tumor-Cell Intrinsic and Microenvironmental Target of Silibinin in Lung Cancer
6.1. Identification of Silibinin as a Direct STAT3 Inhibitor
6.2. STAT3-Targeted Cancer Cell-Intrinsic and Microenvironmental Effects of Silibinin
6.3. Silibinin versus Other Natural Products Exhibiting STAT3 Inhibitory Activity
7. Silibinin and Lung Cancer: The Past, Present, and Future (a Corollary)
8. Conclusions
- -
- The deconstruction and validation of a central mechanism of action of silibinin (i.e., STAT3) has enabled this natural compound to reach clinical development in lung cancer;
- -
- Silibinin is capable of reaching target cancer tissues and groundbreakingly provides survival advantages to lung cancer patients with brain metastasis when used as part of formulations with an optimized oral bioavailability;
- -
- Critical drivers for silibinin responsiveness versus resistance in specific lung cancer molecular subtypes can be identified using CRISPR-based functional genomics;
- -
- Lessons from natural chemistry of silibinin can offer novel approaches for synthetic chemistry in lung cancer drug discovery.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Middleton, E., Jr.; Kandaswami, C.; Theoharides, T.C. The effects of plant flavonoids on mammalian cells: Implications for inflammation, heart disease, and cancer. Pharmacol. Rev. 2000, 52, 673–751. [Google Scholar] [PubMed]
- Romano, B.; Pagano, E.; Montanaro, V.; Fortunato, A.L.; Milic, N.; Borrelli, F. Novel insights into the pharmacology of flavonoids. Phytother. Res. 2013, 27, 1588–1596. [Google Scholar] [CrossRef] [PubMed]
- Barrajón-Catalán, E.; Herranz-López, M.; Joven, J.; Segura-Carretero, A.; Alonso-Villaverde, C.; Menéndez, J.A.; Micol, V. Molecular promiscuity of plant polyphenols in the management of age-related diseases: Far beyond their antioxidant properties. Adv. Exp. Med. Biol. 2014, 824, 141–159. [Google Scholar]
- Howitz, K.T.; Sinclair, D.A. Xenohormesis: Sensing the chemical cues of other species. Cell 2008, 133, 387–391. [Google Scholar] [CrossRef] [Green Version]
- Menendez, J.A.; Joven, J.; Aragonès, G.; Barrajón-Catalán, E.; Beltrán-Debón, R.; Borrás-Linares, I.; Camps, J.; Corominas-Faja, B.; Cufí, S.; Fernández-Arroyo, S.; et al. Xenohormetic and anti-aging activity of secoiridoid polyphenols present in extra virgin olive oil: A new family of gerosuppressant agents. Cell Cycle 2013, 12, 555–578. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.C.; Graf, T.N.; Sparacino, C.M.; Wani, M.C.; Wall, M.E. Complete isolation and characterization of silybins and isosilybins from milk thistle (Silybum marianum). Org. Biomol. Chem. 2003, 1, 1684–1689. [Google Scholar] [CrossRef]
- Gazák, R.; Walterová, D.; Kren, V. Silybin and silymarin—New and emerging applications in medicine. Curr. Med. Chem. 2007, 14, 315–338. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.I.; Narayan, M.; Barrett, J.S. Analysis and comparison of active constituents in commercial standardized silymarin extracts by liquid chromatography-electrospray ionization mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2007, 845, 95–103. [Google Scholar] [CrossRef]
- Abenavoli, L.; Capasso, R.; Milic, N.; Capasso, F. Milk thistle in liver diseases: Past, present, future. Phytother. Res. 2010, 24, 1423–1432. [Google Scholar] [CrossRef]
- Hackett, E.S.; Twedt, D.C.; Gustafson, D.L. Milk thistle and its derivative compounds: A review of opportunities for treatment of liver disease. J. Vet. Intern. Med. 2013, 27, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Bijak, M. Silybin, a Major Bioactive Component of Milk Thistle (Silybum marianum L. Gaernt.)—Chemistry, Bioavailability, and Metabolism. Molecules 2017, 22, 1942. [Google Scholar]
- Abenavoli, L.; Izzo, A.A.; Milić, N.; Cicala, C.; Santini, A.; Capasso, R. Milk thistle (Silybum marianum): A concise overview on its chemistry, pharmacological, and nutraceutical uses in liver diseases. Phytother. Res. 2018, 32, 2202–2213. [Google Scholar] [CrossRef] [PubMed]
- Biedermann, D.; Vavříková, E.; Cvak, L.; Křen, V. Chemistry of silybin. Nat. Prod. Rep. 2014, 31, 1138–1157. [Google Scholar] [CrossRef]
- Vargas-Mendoza, N.; Madrigal-Santillán, E.; Morales-González, A.; Esquivel-Soto, J.; Esquivel-Chirino, C.; García-Luna, Y.; González-Rubio, M.; Gayosso-de-Lucio, J.A.; Morales-González, J.A. Hepatoprotective effect of silymarin. World J. Hepatol. 2014, 6, 144–149. [Google Scholar] [CrossRef]
- Saller, R.; Meier, R.; Brignoli, R. The use of silymarin in the treatment of liver diseases. Drugs 2001, 61, 2035–2063. [Google Scholar] [CrossRef] [PubMed]
- Saller, R.; Melzer, J.; Reichling, J.; Brignoli, R.; Meier, R. An updated systematic review of the pharmacology of silymarin. Forsch. Komplementmed. 2007, 14, 70–80. [Google Scholar] [CrossRef]
- Loguercio, C.; Festi, D. Silybin and the liver: From basic research to clinical practice. World J. Gastroenterol. 2011, 17, 2288–2301. [Google Scholar] [CrossRef]
- Federico, A.; Dallio, M.; Loguercio, C. Silymarin/Silybin and Chronic Liver Disease: A Marriage of Many Years. Molecules 2017, 22, 191. [Google Scholar] [CrossRef] [Green Version]
- Tajmohammadi, A.; Razavi, B.M.; Hosseinzadeh, H. Silybum marianum (milk thistle) and its main constituent, silymarin, as a potential therapeutic plant in metabolic syndrome: A review. Phytother. Res. 2018, 32, 1933–1949. [Google Scholar] [CrossRef]
- Gillessen, A.; Schmidt, H.H. Silymarin as Supportive Treatment in Liver Diseases: A Narrative Review. Adv. Ther. 2020, 37, 1279–1301. [Google Scholar] [CrossRef] [Green Version]
- Wellington, K.; Jarvis, B. Silymarin: A review of its clinical properties in the management of hepatic disorders. BioDrugs 2001, 15, 465–489. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.P.; Agarwal, R. Mechanisms and preclinical efficacy of silibinin in preventing skin cancer. Eur. J. Cancer 2005, 41, 1969–1979. [Google Scholar] [CrossRef]
- Prasad, R.R.; Paudel, S.; Raina, K.; Agarwal, R. Silibinin and non-melanoma skin cancers. J. Tradit. Complement. Med. 2020, 10, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.P.; Agarwal, R. Prostate cancer prevention by silibinin. Curr. Cancer Drug Targets 2004, 4, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.P.; Agarwal, R. Prostate cancer chemoprevention by silibinin: Bench to bedside. Mol. Carcinog. 2006, 45, 436–442. [Google Scholar] [CrossRef]
- Zhao, J.; Agarwal, R. Tissue distribution of silibinin, the major active constituent of silymarin, in mice and its association with enhancement of phase II enzymes: Implications in cancer chemoprevention. Carcinogenesis 1999, 20, 2101–2108. [Google Scholar] [CrossRef] [Green Version]
- Mateen, S.; Raina, K.; Agarwal, R. Chemopreventive and anti-cancer efficacy of silibinin against growth and progression of lung cancer. Nutr. Cancer 2013, 65 (Suppl. 1), 3–11. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.P.; Agarwal, R. Cosmeceuticals and silibinin. Clin. Dermatol. 2009, 27, 479–484. [Google Scholar] [CrossRef] [Green Version]
- Slaga, T.J. SENCAR mouse skin tumorigenesis model versus other strains and stocks of mice. Environ. Health Perspect. 1986, 68, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Ewing, M.W.; Conti, C.J.; Kruszewski, F.H.; Slaga, T.J.; DiGiovanni, J. Tumor progression in Sencar mouse skin as a function of initiator dose and promoter dose, duration, and type. Cancer Res. 1988, 48, 7048–7054. [Google Scholar]
- Sharma, G.; Singh, R.P.; Chan, D.C.; Agarwal, R. Silibinin induces growth inhibition and apoptotic cell death in human lungcarcinoma cells. Anticancer Res. 2003, 23, 2649–2655. [Google Scholar]
- Singh, R.P.; Mallikarjuna, G.U.; Sharma, G.; Dhanalakshmi, S.; Tyagi, A.K.; Chan, D.C.; Agarwal, C.; Agarwal, R. Oral silibinin inhibits lung tumor growth in athymic nude mice and forms a novel chemocombination with doxorubicin targeting nuclear factor kappaB-mediated inducible chemoresistance. Clin. Cancer Res. 2004, 10, 8641–8647. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Wang, Y.; Tan, Q.; Lubet, R.A.; You, M. Efficacy of deguelin and silibinin on benzo(a)pyrene-induced lung tumorigenesis in A/J mice. Neoplasia 2005, 7, 1053–1057. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.P.; Deep, G.; Chittezhath, M.; Kaur, M.; Dwyer-Nield, L.D.; Malkinson, A.M.; Agarwal, R. Effect of silibinin on the growth and progression of primary lung tumors in mice. J. Natl. Cancer Inst. 2006, 98, 846–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chittezhath, M.; Deep, G.; Singh, R.P.; Agarwal, C.; Agarwal, R. Silibinin inhibits cytokine-induced signaling cascades and down-regulates inducible nitric oxide synthase in human lung carcinoma A549 cells. Mol. Cancer Ther. 2008, 7, 1817–1826. [Google Scholar] [CrossRef] [Green Version]
- Tyagi, A.; Singh, R.P.; Ramasamy, K.; Raina, K.; Redente, E.F.; Dwyer-Nield, L.D.; Radcliffe, R.A.; Malkinson, A.M.; Agarwal, R. Growth inhibition and regression of lung tumors by silibinin: Modulation of angiogenesis by macrophage-associated cytokines and nuclear factor-kappaB and signal transducers and activators of transcription 3. Cancer Prev. Res. (Phila) 2009, 2, 74–83. [Google Scholar] [CrossRef] [Green Version]
- Ramasamy, K.; Dwyer-Nield, L.D.; Serkova, N.J.; Hasebroock, K.M.; Tyagi, A.; Raina, K.; Singh, R.P.; Malkinson, A.M.; Agarwal, R. Silibinin prevents lung tumorigenesis in wild-type but not in iNOS−/− mice: Potential of real-time micro-CT in lung cancer chemoprevention studies. Clin. Cancer Res. 2011, 17, 753–761. [Google Scholar] [CrossRef] [Green Version]
- Tyagi, A.; Agarwal, C.; Dwyer-Nield, L.D.; Singh, R.P.; Malkinson, A.M.; Agarwal, R. Silibinin modulates TNF-alpha and IFN-gamma mediated signaling to regulate COX2 and iNOS expression in tumorigenic mouse lung epithelial LM2 cells. Mol. Carcinog. 2012, 51, 832–842. [Google Scholar] [CrossRef] [PubMed]
- Bosch-Barrera, J.; Menendez, J.A. Silibinin and STAT3: A natural way of targeting transcription factors for cancer therapy. Cancer Treat. Rev. 2015, 41, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Bosch-Barrera, J.; Queralt, B.; Menendez, J.A. Targeting STAT3 with silibinin to improve cancer therapeutics. Cancer Treat. Rev. 2017, 58, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Mateen, S.; Tyagi, A.; Agarwal, C.; Singh, R.P.; Agarwal, R. Silibinin inhibits human nonsmall cell lung cancer cell growth through cell-cycle arrest by modulating expression and function of key cell-cycle regulators. Mol. Carcinog. 2010, 49, 247–258. [Google Scholar] [CrossRef] [Green Version]
- Mateen, S.; Raina, K.; Jain, A.K.; Agarwal, C.; Chan, D.; Agarwal, R. Epigenetic modifications and p21-cyclin B1 nexus in anticancer effect of histone deacetylase inhibitors in combination with silibinin on non-small cell lung cancer cells. Epigenetics 2012, 7, 1161–1172. [Google Scholar] [CrossRef] [Green Version]
- Corominas-Faja, B.; Oliveras-Ferraros, C.; Cuyàs, E.; Segura-Carretero, A.; Joven, J.; Martin-Castillo, B.; Barrajón-Catalán, E.; Micol, V.; Bosch-Barrera, J.; Menendez, J.A. Stem cell-like ALDH(bright) cellular states in EGFR-mutant non-small cell lung cancer: A novel mechanism of acquired resistance to erlotinib targetable with the natural polyphenol silibinin. Cell Cycle 2013, 12, 3390–3404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuyàs, E.; Pérez-Sánchez, A.; Micol, V.; Menendez, J.A.; Bosch-Barrera, J. STAT3-targeted treatment with silibinin overcomes the acquired resistance to crizotinib in ALK-rearranged lung cancer. Cell Cycle 2016, 15, 3413–3418. [Google Scholar] [CrossRef]
- Liang, Z.; Yang, Y.; Wang, H.; Yi, W.; Yan, X.; Yan, J.; Li, Y.; Feng, Y.; Yu, S.; Yang, J.; et al. Inhibition of SIRT1 signaling sensitizes the antitumor activity of silybin against human lung adenocarcinoma cells in vitro and in vivo. Mol. Cancer Ther. 2014, 13, 1860–1872. [Google Scholar] [CrossRef] [Green Version]
- Rho, J.K.; Choi, Y.J.; Jeon, B.S.; Choi, S.J.; Cheon, G.J.; Woo, S.K.; Kim, H.R.; Kim, C.H.; Choi, C.M.; Lee, J.C. Combined treatment with silibinin and epidermal growth factor receptor tyrosine kinase inhibitors overcomes drug resistance caused by T790M mutation. Mol. Cancer Ther. 2010, 9, 3233–3243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vazquez-Martin, A.; Cufí, S.; Oliveras-Ferraros, C.; Torres-Garcia, V.Z.; Corominas-Faja, B.; Cuyàs, E.; Bonavia, R.; Visa, J.; Martin-Castillo, B.; Barrajón-Catalán, E.; et al. IGF-1R/epithelial-to-mesenchymal transition (EMT) crosstalk suppresses the erlotinib-sensitizing effect of EGFR exon 19 deletion mutations. Sci. Rep. 2013, 3, 2560. [Google Scholar] [CrossRef] [PubMed]
- Cufí, S.; Bonavia, R.; Vazquez-Martin, A.; Corominas-Faja, B.; Oliveras-Ferraros, C.; Cuyàs, E.; Martin-Castillo, B.; Barrajón-Catalán, E.; Visa, J.; Segura-Carretero, A.; et al. Silibinin meglumine, a water-soluble form of milk thistle silymarin, is an orally active anti-cancer agent that impedes the epithelial-to-mesenchymal transition (EMT) in EGFR-mutant non-small-cell lung carcinoma cells. Food Chem. Toxicol. 2013, 60, 360–368. [Google Scholar]
- Cufí, S.; Bonavia, R.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Corominas-Faja, B.; Cuyàs, E.; Martin-Castillo, B.; Barrajón-Catalán, E.; Visa, J.; Segura-Carretero, A.; et al. Silibinin suppresses EMT-driven erlotinib resistance by reversing the high miR-21/low miR-200c signature in vivo. Sci. Rep. 2013, 3, 2459. [Google Scholar] [CrossRef]
- Shien, K.; Toyooka, S.; Yamamoto, H.; Soh, J.; Jida, M.; Thu, K.L.; Hashida, S.; Maki, Y.; Ichihara, E.; Asano, H.; et al. Acquired resistance to EGFR inhibitors is associated with a manifestation of stem cell-like properties in cancer cells. Cancer Res. 2013, 73, 3051–3061. [Google Scholar] [CrossRef] [Green Version]
- Maitrejean, M.; Comte, G.; Barron, D.; El Kirat, K.; Conseil, G.; Di Pietro, A. The flavanolignan silybin and its hemisynthetic derivatives, a novel series of potential modulators of P-glycoprotein. Bioorg. Med. Chem. Lett. 2000, 10, 157–160. [Google Scholar] [CrossRef]
- Dzubák, P.; Hajdúch, M.; Gazák, R.; Svobodová, A.; Psotová, J.; Walterová, D.; Sedmera, P.; Kren, V. New derivatives of silybin and 2,3-dehydrosilybin and their cytotoxic and P-glycoprotein modulatory activity. Bioorg. Med. Chem. 2006, 14, 3793–3810. [Google Scholar] [CrossRef] [PubMed]
- Sadava, D.; Kane, S.E. Silibinin reverses drug resistance in human small-cell lung carcinoma cells. Cancer Lett. 2013, 339, 102–106. [Google Scholar] [CrossRef] [Green Version]
- Dinic, J.; Podolski-Renic, A.; Stankovic, T.; Bankovic, J.; Pesic, M. New Approaches With Natural Product Drugs for Overcoming Multidrug Resistance in Cancer. Curr. Pharm. Des. 2015, 21, 5589–5604. [Google Scholar] [CrossRef] [PubMed]
- Dobiasová, S.; Řehořová, K.; Kučerová, D.; Biedermann, D.; Káňová, K.; Petrásková, L.; Koucká, K.; Václavíková, R.; Valentová, K.; Ruml, T.; et al. Multidrug Resistance Modulation Activity of Silybin Derivatives and Their Anti-inflammatory Potential. Antioxidants 2020, 9, 455. [Google Scholar] [CrossRef]
- Lee, C.K.; Choi, J.S. Effects of silibinin, inhibitor of CYP3A4 and P-glycoprotein in vitro, on the pharmacokinetics of paclitaxel after oral and intravenous administration in rats. Pharmacology 2010, 85, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.C.; Chiou, H.L.; Chen, P.N.; Yang, S.F.; Hsieh, Y.S. Silibinin inhibits the invasion of human lung cancer cells via decreased productions of urokinase-plasminogen activator and matrix metalloproteinase-2. Mol. Carcinog. 2004, 40, 143–149. [Google Scholar] [CrossRef]
- Chen, P.N.; Hsieh, Y.S.; Chiou, H.L.; Chu, S.C. Silibinin inhibits cell invasion through inactivation of both PI3K-Akt and MAPK signaling pathways. Chem. Biol. Interact. 2005, 156, 141–150. [Google Scholar] [CrossRef]
- Chen, P.-N.; Hsieh, Y.-S.; Chiang, C.-L.; Chiou, H.-L.; Yang, S.-F.; Chu, S.-C. Silibinin inhibits invasion of oral cancer cells by suppressing the MAPK pathway. J. Dent. Res. 2006, 85, 220–225. [Google Scholar] [CrossRef]
- Byun, H.J.; Darvin, P.; Kang, D.Y.; Sp, N.; Joung, Y.H.; Park, J.H.; Kim, S.J.; Yang, Y.M. Silibinin downregulates MMP2 expression via Jak2/STAT3 pathway and inhibits the migration and invasive potential in MDA-MB-231 cells. Oncol. Rep. 2017, 37, 3270–3278. [Google Scholar] [CrossRef] [Green Version]
- Davis, F.M.; Stewart, T.A.; Thompson, E.W.; Monteith, G.R. Targeting EMT in cancer: Opportunities for pharmacological intervention. Trends Pharmacol. Sci. 2014, 35, 479–488. [Google Scholar] [CrossRef]
- Marcucci, F.; Stassi, G.; De Maria, R. Epithelial-mesenchymal transition: A new target in anticancer drug discovery. Nat. Rev. Drug Discov. 2016, 15, 311–325. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [Green Version]
- Frederick, B.A.; Helfrich, B.A.; Coldren, C.D.; Zheng, D.; Chan, D.; Bunn, P.A., Jr.; Raben, D. Epithelial to mesenchymal transition predicts gefitinib resistance in cell lines of head and neck squamous cell carcinoma and non-small cell lung carcinoma. Mol. Cancer Ther. 2007, 6, 1683–1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byers, L.A.; Diao, L.; Wang, J.; Saintigny, P.; Girard, L.; Peyton, M.; Shen, L.; Fan, Y.; Giri, U.; Tumula, P.K.; et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin. Cancer Res. 2013, 19, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Guo, F.; Liu, X.; Qing, Q.; Sang, Y.; Feng, C.; Li, X.; Jiang, L.; Su, P.; Wang, Y. EML4-ALK induces epithelial-mesenchymal transition consistent with cancer stem cell properties in H1299 non-small cell lung cancer cells. Biochem. Biophys. Res. Commun. 2015, 459, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.R.; Kim, W.S.; Choi, Y.J.; Choi, C.M.; Rho, J.K.; Lee, J.C. Epithelial-mesenchymal transition leads to crizotinib resistance in H2228 lung cancer cells with EML4-ALK translocation. Mol. Oncol. 2013, 7, 1093–1102. [Google Scholar] [CrossRef] [PubMed]
- Gower, A.; Hsu, W.H.; Hsu, S.T.; Wang, Y.; Giaccone, G. EMT is associated with, but does not drive resistance to ALK inhibitors among EML4-ALK non-small cell lung cancer. Mol. Oncol. 2016, 10, 601–609. [Google Scholar] [CrossRef] [Green Version]
- Kogita, A.; Togashi, Y.; Hayashi, H.; Sogabe, S.; Terashima, M.; De Velasco, M.A.; Sakai, K.; Fujita, Y.; Tomida, S.; Takeyama, Y.; et al. Hypoxia induces resistance to ALK inhibitors in the H3122 non-small cell lung cancer cell line with an ALK rearrangement via epithelial-mesenchymal transition. Int. J. Oncol. 2014, 45, 1430–1436. [Google Scholar] [CrossRef] [Green Version]
- Nakamichi, S.; Seike, M.; Miyanaga, A.; Chiba, M.; Zou, F.; Takahashi, A.; Ishikawa, A.; Kunugi, S.; Noro, R.; Kubota, K.; et al. Overcoming drug-tolerant cancer cell subpopulations showing AXL activation and epithelial-mesenchymal transition is critical in conquering ALK-positive lung cancer. Oncotarget 2018, 9, 27242–27255. [Google Scholar] [CrossRef] [Green Version]
- Debruyne, D.N.; Bhatnagar, N.; Sharma, B.; Luther, W.; Moore, N.F.; Cheung, N.K.; Gray, N.S.; George, R.E. ALK inhibitor resistance in ALK(F1174L)-driven neuroblastoma is associated with AXL activation and induction of EMT. Oncogene 2016, 35, 3681–3689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, J.; van der Wekken, A.J.; Saber, A.; Terpstra, M.M.; Schuuring, E.; Timens, W.; Hiltermann, T.J.N.; Groen, H.J.M.; van den Berg, A.; Kok, K. Mutations in EMT-Related Genes in ALK Positive Crizotinib Resistant Non-Small Cell Lung Cancers. Cancers 2018, 10, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekar, D.; Krishnan, R.; Panagal, M.; Sivakumar, P.; Gopinath, V.; Basam, V. Deciphering the role of microRNA 21 in cancer stem cells (CSCs). Genes Dis. 2016, 3, 277–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.Q.; Ahmed, E.I.; Elareer, N.R.; Junejo, K.; Steinhoff, M.; Uddin, S. Role of miRNA-Regulated Cancer Stem Cells in the Pathogenesis of Human Malignancies. Cells 2019, 8, 840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, Y.Y.; Wright, J.A.; Attema, J.L.; Gregory, P.A.; Bert, A.G.; Smith, E.; Thomas, D.; Lopez, A.F.; Drew, P.A.; Khew-Goodall, Y.; et al. Epigenetic modulation of the miR-200 family is associated with transition to a breast cancer stem-cell-like state. J. Cell Sci. 2013, 126, 2256–2266. [Google Scholar] [CrossRef] [Green Version]
- Burk, U.; Schubert, J.; Wellner, U.; Schmalhofer, O.; Vincan, E.; Spaderna, S.; Brabletz, T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008, 9, 582–589. [Google Scholar] [CrossRef] [Green Version]
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; zur Hausen, A.; et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 2009, 11, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Mateen, S.; Raina, K.; Agarwal, C.; Chan, D.; Agarwal, R. Silibinin synergizes with histone deacetylase and DNA methyltransferase inhibitors in upregulating E-cadherin expression together with inhibition of migration and invasion of human non-small cell lung cancer cells. J. Pharmacol. Exp. Ther. 2013, 345, 206–214. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Zhang, H.; Wang, A.; Ma, Y.; Gan, Y.; Li, G. Silibinin suppresses epithelial-mesenchymal transition in human non-small cell lung cancer cells by restraining RHBDD1. Cell Mol. Biol. Lett. 2020, 25, 36. [Google Scholar] [CrossRef]
- Erler, J.T.; Bennewith, K.L.; Nicolau, M.; Dornhöfer, N.; Kong, C.; Le, Q.T.; Chi, J.T.; Jeffrey, S.S.; Giaccia, A.J. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature 2006, 440, 1222–1226. [Google Scholar] [CrossRef] [PubMed]
- Barker, H.E.; Cox, T.R.; Erler, J.T. The rationale for targeting the LOX family in cancer. Nat. Rev. Cancer 2012, 12, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Cox, T.R.; Gartland, A.; Erler, J.T. Lysyl Oxidase, a Targetable Secreted Molecule Involved in Cancer Metastasis. Cancer Res. 2016, 76, 188–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, K.A.; Lopez, K.M. Lysyl oxidase in cancer inhibition and metastasis. Cancer Lett. 2018, 417, 174–181. [Google Scholar] [CrossRef]
- Hou, X.; Du, H.; Quan, X.; Shi, L.; Zhang, Q.; Wu, Y.; Liu, Y.; Xiao, J.; Li, Y.; Lu, L.; et al. Silibinin Inhibits NSCLC Metastasis by Targeting the EGFR/LOX Pathway. Front. Pharmacol. 2018, 9, 21. [Google Scholar] [CrossRef]
- Kaipa, J.M.; Starkuviene, V.; Erfle, H.; Eils, R.; Gladilin, E. Transcriptome profiling reveals Silibinin dose-dependent response network in non-small lung cancer cells. PeerJ 2020, 8, e10373. [Google Scholar] [CrossRef]
- Pérez-Sánchez, A.; Cuyàs, E.; Ruiz-Torres, V.; Agulló-Chazarra, L.; Verdura, S.; González-Álvarez, I.; Bermejo, M.; Joven, J.; Micol, V.; Bosch-Barrera, J.; et al. Intestinal Permeability Study of Clinically Relevant Formulations of Silibinin in Caco-2 Cell Monolayers. Int. J. Mol. Sci. 2019, 20, 1606. [Google Scholar] [CrossRef] [Green Version]
- Bosch-Barrera, J.; Sais, E.; Cañete, N.; Marruecos, J.; Cuyàs, E.; Izquierdo, A.; Porta, R.; Haro, M.; Brunet, J.; Pedraza, S.; et al. Response of brain metastasis from lung cancer patients to an oral nutraceutical product containing silibinin. Oncotarget 2016, 7, 32006–32014. [Google Scholar] [CrossRef] [Green Version]
- Priego, N.; Zhu, L.; Monteiro, C.; Mulders, M.; Wasilewski, D.; Bindeman, W.; Doglio, L.; Martínez, L.; Martínez-Saez, E.; Ramón, Y.; et al. STAT3 labels a subpopulation of reactive astrocytes required for brain metastasis. Nat. Med. 2018, 24, 1024–1035. [Google Scholar] [CrossRef]
- Sarmiento Soto, M.; Larkin, J.R.; Martin, C.; Khrapitchev, A.A.; Maczka, M.; Economopoulos, V.; Scott, H.; Escartin, C.; Bonvento, G.; Serres, S.; et al. STAT3-Mediated Astrocyte Reactivity Associated with Brain Metastasis Contributes to Neurovascular Dysfunction. Cancer Res. 2020, 80, 5642–5655. [Google Scholar] [CrossRef] [PubMed]
- Verdura, S.; Cuyàs, E.; Llorach-Parés, L.; Pérez-Sánchez, A.; Micol, V.; Nonell-Canals, A.; Joven, J.; Valiente, M.; Sánchez-Martínez, M.; Bosch-Barrera, J.; et al. Silibinin is a direct inhibitor of STAT3. Food Chem. Toxicol. 2018, 116, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E., Jr. Stat3 as an oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef] [Green Version]
- Liddle, F.J.; Alvarez, J.V.; Poli, V.; Frank, D.A. Tyrosine phosphorylation is required for functional activation of disulfide-containing constitutively active STAT mutants. Biochemistry 2006, 45, 5599–5605. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.K.; Dasgupta, A.; Mehla, K.; Gunda, V.; Vernucci, E.; Souchek, J.; Goode, G.; King, R.; Mishra, A.; Rai, I.; et al. Silibinin-mediated metabolic reprogramming attenuates pancreatic cancer-induced cachexia and tumor growth. Oncotarget 2015, 6, 41146–41161. [Google Scholar] [CrossRef] [Green Version]
- Singh, M.; Garg, N.; Venugopal, C.; Hallett, R.; Tokar, T.; McFarlane, N.; Mahendram, S.; Bakhshinyan, D.; Manoranjan, B.; Vora, P.; et al. STAT3 pathway regulates lung-derived brain metastasis initiating cell capacity through miR-21 activation. Oncotarget 2015, 6, 27461–27477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, S.; Fisher, K.H.; Snowden, J.A.; Danson, S.J.; Brown, S.; Zeidler, M.P. Methotrexate Is a JAK/STAT Pathway Inhibitor. PLoS ONE 2015, 10, e0130078. [Google Scholar] [CrossRef]
- Miklossy, G.; Hilliard, T.S.; Turkson, J. Therapeutic modulators of STAT signalling for human diseases. Nat. Rev. Drug Discov. 2013, 12, 611–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shakibaei, M.; Harikumar, K.B.; Aggarwal, B.B. Resveratrol addiction: To die or not to die. Mol. Nutr. Food Res. 2009, 53, 115–128. [Google Scholar] [CrossRef]
- Li, T.; Wang, W.; Chen, H.; Li, T.; Ye, L. Evaluation of anti-leukemia effect of resveratrol by modulating STAT3 signaling. Int. Immunopharmacol. 2010, 10, 18–25. [Google Scholar] [CrossRef]
- Liu, L.J.; Leung, K.H.; Chan, D.S.; Wang, Y.T.; Ma, D.L.; Leung, C.H. Identification of a natural product-like STAT3 dimerization inhibitor by structure-based virtual screening. Cell Death Dis. 2014, 5, e1293. [Google Scholar] [CrossRef] [Green Version]
- Szelag, M.; Sikorski, K.; Czerwoniec, A.; Szatkowska, K.; Wesoly, J.; Bluyssen, H.A. In silico simulations of STAT1 and STAT3 inhibitors predict SH2 domain cross-binding specificity. Eur. J. Pharmacol. 2013, 720, 38–48. [Google Scholar] [CrossRef]
- Tuli, H.S.; Mittal, S.; Aggarwal, D.; Parashar, G.; Parashar, N.C.; Upadhyay, S.K.; Barwal, T.S.; Jain, A.; Kaur, G.; Savla, R.; et al. Path of Silibinin from diet to medicine: A dietary polyphenolic flavonoid having potential anti-cancer therapeutic significance. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2020. [Google Scholar]
- Liakopoulou, C.; Kazazis, C.; Vallianou, N.G. Silimarin and Cancer. Anticancer Agents Med. Chem. 2018, 18, 1970–1974. [Google Scholar] [CrossRef] [PubMed]
- Delmas, D.; Xiao, J.; Vejux, A.; Aires, V. Silymarin and Cancer: A Dual Strategy in Both in Chemoprevention and Chemosensitivity. Molecules 2020, 25, 2009. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, H.M.; Cai, F.; Ko, B.; Yang, C.; Lieu, E.L.; Muhammad, N.; Rhyne, S.; Li, K.; Haloul, M.; et al. The hexosamine biosynthesis pathway is a targetable liability in KRAS/LKB1 mutant lung cancer. Nat. Metab. 2020, 2, 1401–1412. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verdura, S.; Cuyàs, E.; Ruiz-Torres, V.; Micol, V.; Joven, J.; Bosch-Barrera, J.; Menendez, J.A. Lung Cancer Management with Silibinin: A Historical and Translational Perspective. Pharmaceuticals 2021, 14, 559. https://doi.org/10.3390/ph14060559
Verdura S, Cuyàs E, Ruiz-Torres V, Micol V, Joven J, Bosch-Barrera J, Menendez JA. Lung Cancer Management with Silibinin: A Historical and Translational Perspective. Pharmaceuticals. 2021; 14(6):559. https://doi.org/10.3390/ph14060559
Chicago/Turabian StyleVerdura, Sara, Elisabet Cuyàs, Verónica Ruiz-Torres, Vicente Micol, Jorge Joven, Joaquim Bosch-Barrera, and Javier A. Menendez. 2021. "Lung Cancer Management with Silibinin: A Historical and Translational Perspective" Pharmaceuticals 14, no. 6: 559. https://doi.org/10.3390/ph14060559
APA StyleVerdura, S., Cuyàs, E., Ruiz-Torres, V., Micol, V., Joven, J., Bosch-Barrera, J., & Menendez, J. A. (2021). Lung Cancer Management with Silibinin: A Historical and Translational Perspective. Pharmaceuticals, 14(6), 559. https://doi.org/10.3390/ph14060559