
Identification of SARS-CoV-2 E Channel Blockers from a Repurposed Drug Library



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
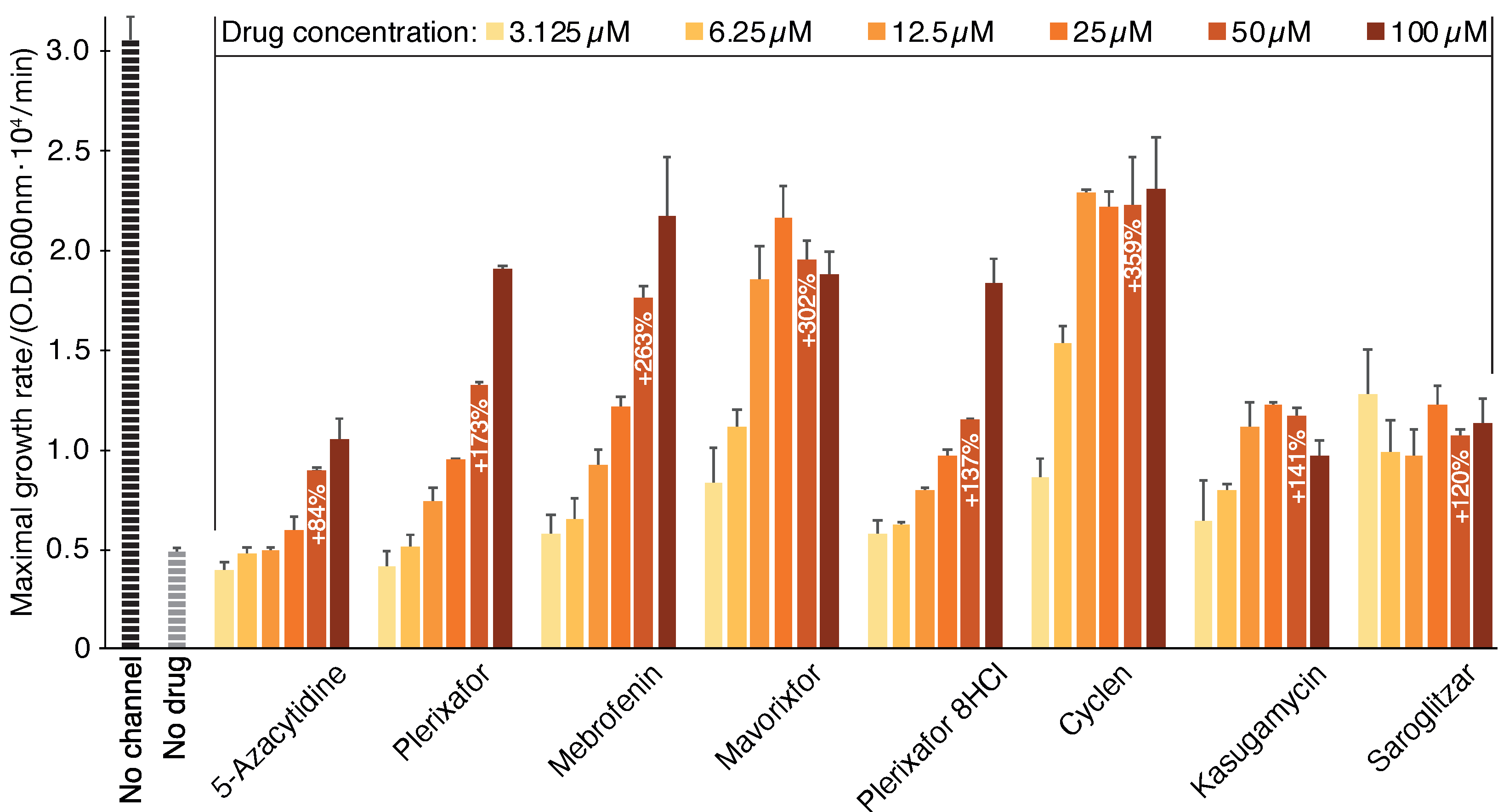
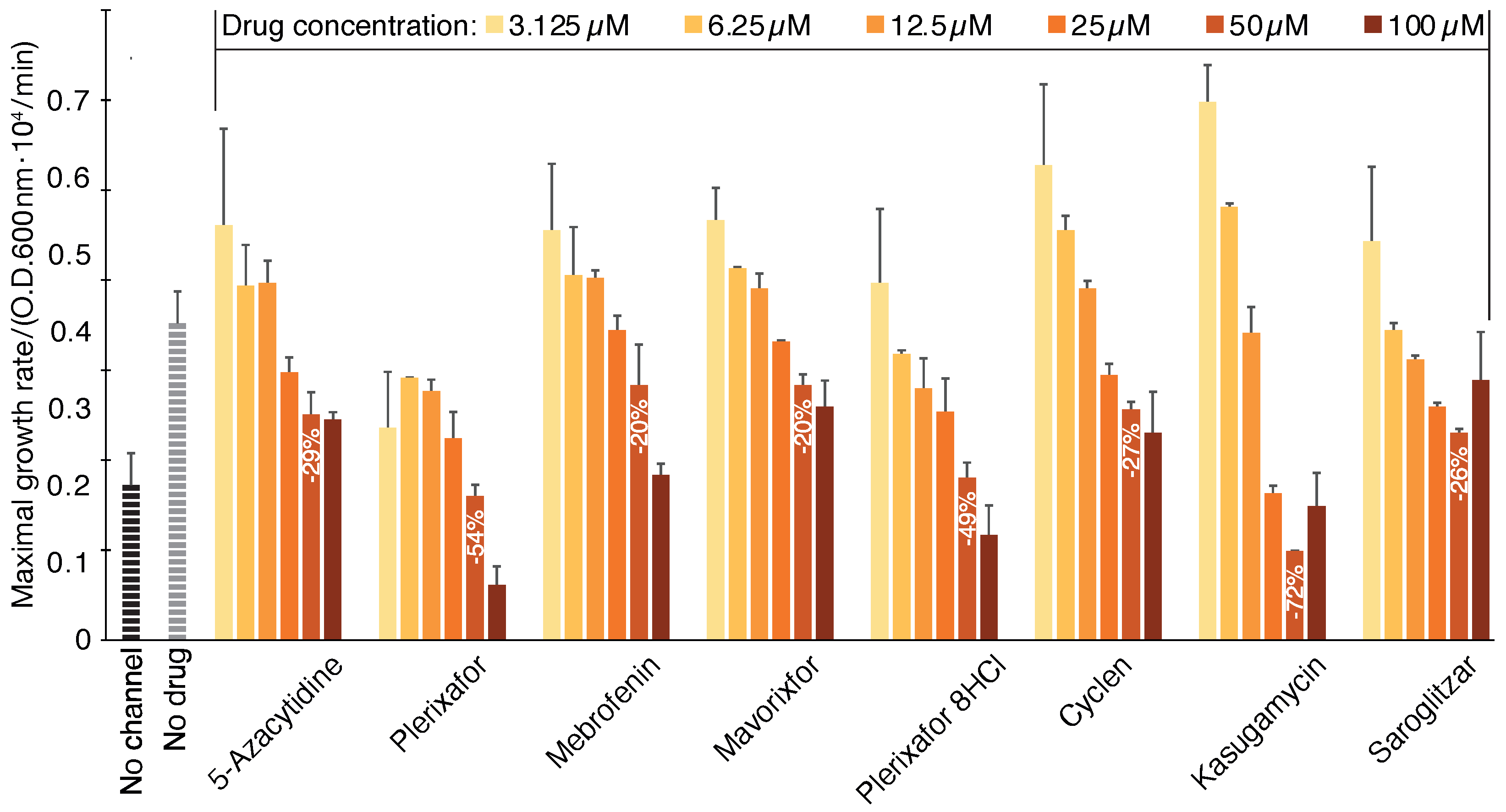
2.1. Negative Assay
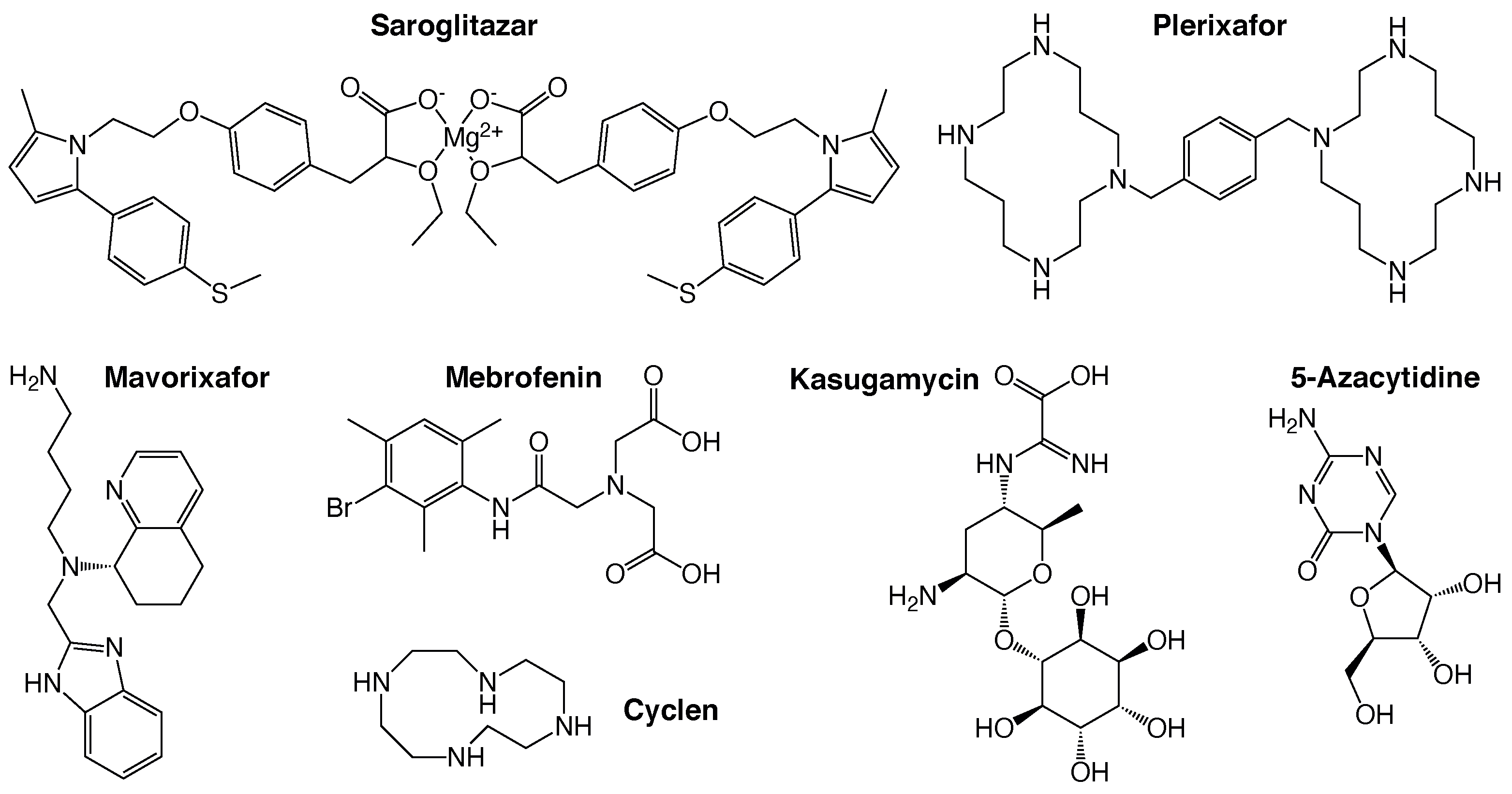
2.2. Positive Assay
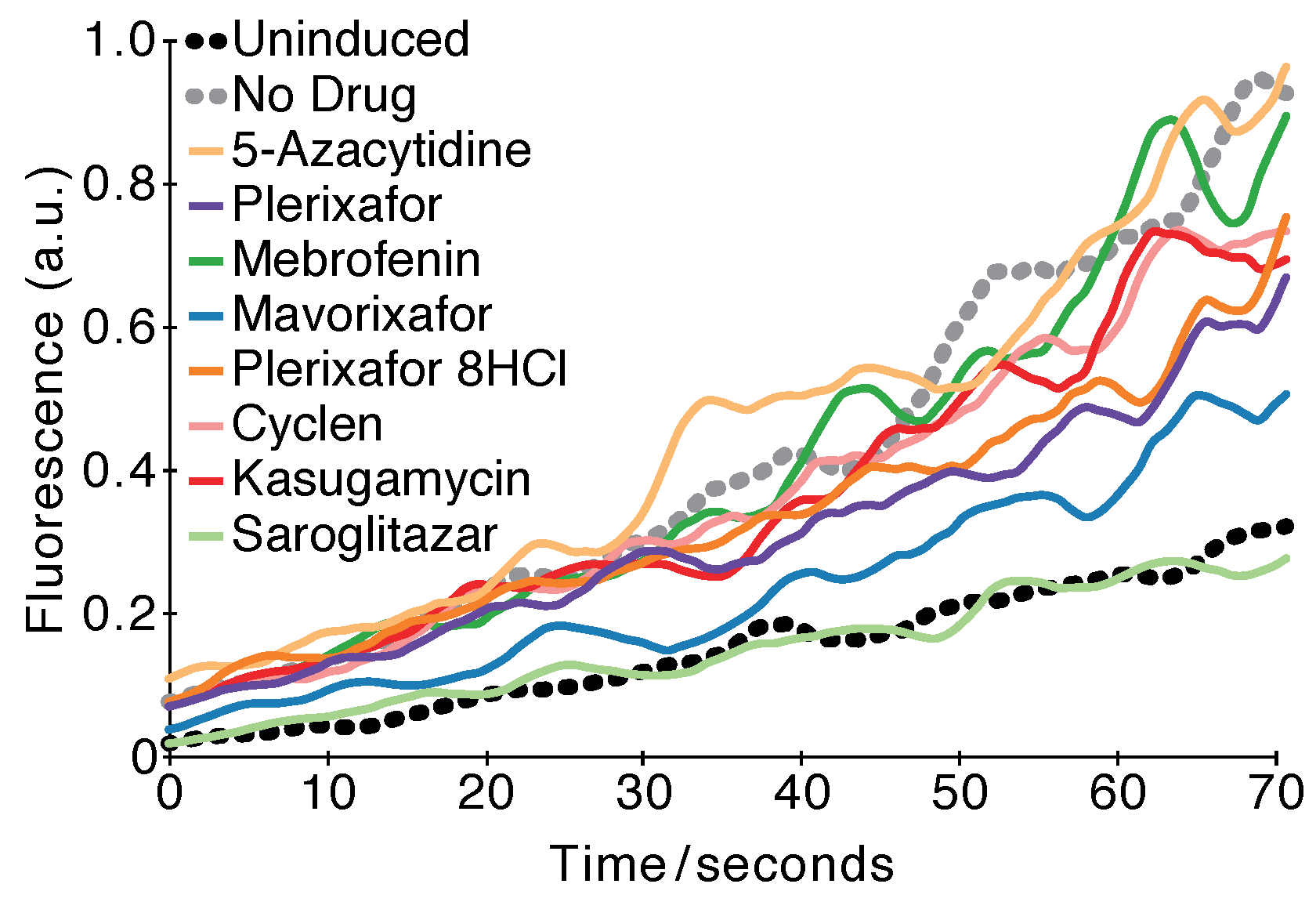
2.3. Fluorescence-Based Test
3. Discussion
4. Materials and Methods
4.1. Channel Assays
4.2. Chemical Screening
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dong, E.; Du, H.; Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ksiazek, T.G.; Erdman, D.; Goldsmith, C.S.; Zaki, S.R.; Peret, T.; Emery, S.; Tong, S.; Urbani, C.; Comer, J.A.; Lim, W.; et al. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1953–1966. [Google Scholar] [CrossRef]
- Rota, P.A.; Oberste, M.S.; Monroe, S.S.; Nix, W.A.; Campagnoli, R.; Icenogle, J.P.; Penaranda, S.; Bankamp, B.; Maher, K.; Chen, M.H.; et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 2003, 300, 1394–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Ho, W.; Huang, Y.; Jin, D.Y.; Li, S.; Liu, S.L.; Liu, X.; Qiu, J.; Sang, Y.; Wang, Q.; et al. SARS-CoV-2 is an appropriate name for the new coronavirus. Lancet 2020, 395, 949–950. [Google Scholar] [CrossRef]
- Schoeman, D.; Fielding, B.C. Coronavirus envelope protein: Current knowledge. Virol. J. 2019, 16, 69. [Google Scholar] [CrossRef] [Green Version]
- DeDiego, M.L.; Alvarez, E.; Almazán, F.; Rejas, M.T.; Lamirande, E.; Roberts, A.; Shieh, W.J.; Zaki, S.R.; Subbarao, K.; Enjuanes, L. A severe acute respiratory syndrome coronavirus that lacks the E gene is attenuated in vitro and in vivo. J. Virol. 2007, 81, 1701–1713. [Google Scholar] [CrossRef] [Green Version]
- Fett, C.; DeDiego, M.L.; Regla-Nava, J.A.; Enjuanes, L.; Perlman, S. Complete protection against severe acute respiratory syndrome coronavirus-mediated lethal respiratory disease in aged mice by immunization with a mouse-adapted virus lacking E protein. J. Virol. 2013, 87, 6551–6559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamirande, E.W.; DeDiego, M.L.; Roberts, A.; Jackson, J.P.; Alvarez, E.; Sheahan, T.; Shieh, W.J.; Zaki, S.R.; Baric, R.; Enjuanes, L.; et al. A live attenuated severe acute respiratory syndrome coronavirus is immunogenic and efficacious in golden Syrian hamsters. J. Virol. 2008, 82, 7721–7724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Netland, J.; DeDiego, M.L.; Zhao, J.; Fett, C.; Álvarez, E.; Nieto-Torres, J.L.; Enjuanes, L.; Perlman, S. Immunization with an attenuated severe acute respiratory syndrome coronavirus deleted in E protein protects against lethal respiratory disease. Virology 2010, 399, 120–128. [Google Scholar] [CrossRef]
- Regla-Nava, J.A.; Nieto-Torres, J.L.; Jimenez-Guardeño, J.M.; Fernandez-Delgado, R.; Fett, C.; Castaño-Rodríguez, C.; Perlman, S.; Enjuanes, L.; DeDiego, M.L. Severe acute respiratory syndrome coronaviruses with mutations in the E protein are attenuated and promising vaccine candidates. J. Virol. 2015, 89, 3870–3887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandala, V.S.; McKay, M.J.; Shcherbakov, A.A.; Dregni, A.J.; Kolocouris, A.; Hong, M. Structure and drug binding of the SARS-CoV-2 envelope protein transmembrane domain in lipid bilayers. Nat. Struct. Mol. Biol. 2020, 27, 1202–1208. [Google Scholar] [CrossRef]
- Wilson, L.; McKinlay, C.; Gage, P.; Ewart, G. SARS coronavirus E protein forms cation-selective ion channels. Virology 2004, 330, 322–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, L.; Gage, P.; Ewart, G. Hexamethylene amiloride blocks E protein ion channels and inhibits coronavirus replication. Virology 2006, 353, 294–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surya, W.; Li, Y.; Verdià-Bàguena, C.; Aguilella, V.M.; Torres, J. MERS coronavirus envelope protein has a single transmembrane domain that forms pentameric ion channels. Virus Res. 2015, 201, 61–66. [Google Scholar] [CrossRef]
- Singh Tomar, P.P.; Arkin, I.T. SARS-CoV-2 E protein is a potential ion channel that can be inhibited by Gliclazide and Memantine. Biochem. Biophys. Res. Commun. 2020, 530, 10–14. [Google Scholar] [CrossRef]
- Zheng, J.; Trudeau, M.C. (Eds.) Handbook of Ion Channels; CRC Press: Boca Raton, FL, USA, 2015. [Google Scholar]
- Scott, C.; Griffin, S. Viroporins: Structure, function and potential as antiviral targets. J. Gen. Virol. 2015, 96, 2000–2027. [Google Scholar] [CrossRef]
- Tonelli, M.; Cichero, E. Fight Against H1N1 Influenza A Virus: Recent Insights Towards the Development of Druggable Compounds. Curr. Med. Chem. 2016, 23, 1802–1817. [Google Scholar] [CrossRef]
- Davies, W.L.; Grunert, R.R.; Haff, R.F.; Mcgahen, J.W.; Neumayer, E.M.; Paulshock, M.; Watts, J.C.; Wood, T.R.; Hermann, E.C.; Hoffmann, C.E. Antiviral activity of 1-adamantanamine (amantadine). Science 1964, 144, 862–863. [Google Scholar] [CrossRef]
- Hay, A.J.; Wolstenholme, A.J.; Skehel, J.J.; Smith, M.H. The molecular basis of the specific anti-influenza action of amantadine. EMBO J. 1985, 4, 3021–3024. [Google Scholar] [CrossRef]
- Pinto, L.H.; Holsinger, L.J.; Lamb, R.A. Influenza Virus M2 Protein Has Ion Channel Activity. Cell 1992, 69, 517–528. [Google Scholar]
- Guan, Y.; Chen, H. Resistance to anti-influenza agents. Lancet 2005, 366, 1139–1140. [Google Scholar] [CrossRef]
- Assa, D.; Alhadeff, R.; Krugliak, M.; Arkin, I.T. Mapping the Resistance Potential of Influenza’s H+ Channel against an Antiviral Blocker. J. Mol. Biol. 2016, 428, 4209–4217. [Google Scholar] [CrossRef] [PubMed]
- Astrahan, P.; Flitman-Tene, R.; Bennett, E.R.; Krugliak, M.; Gilon, C.; Arkin, I.T. Quantitative analysis of influenza M2 channel blockers. Biochim. Biophys. Acta 2011, 1808, 394–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taube, R.; Alhadeff, R.; Assa, D.; Krugliak, M.; Arkin, I.T. Bacteria-based analysis of HIV-1 Vpu channel activity. PLoS ONE 2014, 9, e105387. [Google Scholar] [CrossRef] [PubMed]
- Tomar, P.P.S.; Oren, R.; Krugliak, M.; Arkin, I.T. Potential Viroporin Candidates From Pathogenic Viruses Using Bacteria-Based Bioassays. Viruses 2019, 11, 632. [Google Scholar] [CrossRef] [Green Version]
- Tomar, P.P.S.; Krugliak, M.; Arkin, I.T. Blockers of the SARS-CoV-2 3a Channel Identified by Targeted Drug Repurposing. Viruses 2021, 13, 532. [Google Scholar] [CrossRef]
- Alhadeff, R.; Assa, D.; Astrahan, P.; Krugliak, M.; Arkin, I.T. Computational and experimental analysis of drug binding to the Influenza M2 channel. Biochim. Biophys. Acta 2014, 1838, 1068–1073. [Google Scholar] [CrossRef] [Green Version]
- Stumpe, S.; Bakker, E.P. Requirement of a Large K+-Uptake Capacity and of Extracytoplasmic Protease Activity for Protamine Resistance of Escherichia Coli. Arch. Microbiol. 1997, 167, 126–136. [Google Scholar]
- Miesenböck, G.; De Angelis, D.A.; Rothman, J.E. Visualizing Secretion and Synaptic Transmission With pH-Sensitive Green Fluorescent Proteins. Nature 1998, 394, 192–195. [Google Scholar] [CrossRef] [PubMed]
- Santner, P.; Martins, J.M.d.S.; Laursen, J.S.; Behrendt, L.; Riber, L.; Olsen, C.A.; Arkin, I.T.; Winther, J.R.; Willemoës, M.; Lindorff-Larsen, K. A Robust Proton Flux (pHlux) Assay for Studying the Function and Inhibition of the Influenza A M2 Proton Channel. Biochemistry 2018, 57, 5949–5956. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.Y. Tetrodotoxin, saxitoxin and their significance in the study of excitation phenomena. Pharmacol. Rev. 1966, 18, 997–1049. [Google Scholar]
- Hess, P.; Lansman, J.B.; Tsien, R.W. Different modes of Ca channel gating behaviour favoured by dihydropyridine Ca agonists and antagonists. Nature 1984, 311, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Mitcheson, J.S.; Chen, J.; Lin, M.; Culberson, C.; Sanguinetti, M.C. A structural basis for drug-induced long QT syndrome. Proc. Natl. Acad. Sci. USA 2000, 97, 12329–12333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catterall, W.A.; Striessnig, J. Receptor sites for Ca2+ channel antagonists. Trends Pharmacol. Sci. 1992, 13, 256–262. [Google Scholar] [CrossRef]
- Mitsuya, H.; Weinhold, K.J.; Furman, P.A.; St Clair, M.H.; Lehrman, S.N.; Gallo, R.C.; Bolognesi, D.; Barry, D.W.; Broder, S. 3’-Azido-3’-deoxythymidine (BW A509U): An antiviral agent that inhibits the infectivity and cytopathic effect of human T-lymphotropic virus type III/lymphadenopathy-associated virus in vitro. Proc. Natl. Acad. Sci. USA 1985, 82, 7096–7100. [Google Scholar] [CrossRef] [Green Version]
- Fischl, M.A.; Richman, D.D.; Grieco, M.H.; Gottlieb, M.S.; Volberding, P.A.; Laskin, O.L.; Leedom, J.M.; Groopman, J.E.; Mildvan, D.; Schooley, R.T. The efficacy of azidothymidine (AZT) in the treatment of patients with AIDS and AIDS-related complex. A double-blind, placebo-controlled trial. N. Engl. J. Med. 1987, 317, 185–191. [Google Scholar] [CrossRef]
- Horwitz, J.P.; Chua, J.; Noel, M. Nucleosides. V. The Monomesylates of 1-(2’-Deoxy-β-D-lyxofuranosyl)thymine1,2. J. Org. Chem. 1964, 29, 2076–2078. [Google Scholar] [CrossRef]
- Li, G.; De Clercq, E. Therapeutic options for the 2019 novel coronavirus (2019-nCoV). Nat. Rev. Drug Discov. 2020, 19, 149–150. [Google Scholar] [CrossRef] [Green Version]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of Covid-19-Final Report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J. Covid-19: What now for remdesivir? BMJ 2020, 371, m4457. [Google Scholar] [CrossRef] [PubMed]
- WHO Solidarity Trial Consortium; Pan, H.; Peto, R.; Henao-Restrepo, A.M.; Preziosi, M.P.; Sathiyamoorthy, V.; Abdool Karim, Q.; Alejandria, M.M.; Hernández García, C.; Kieny, M.P.; et al. Repurposed Antiviral Drugs for Covid-19-Interim WHO Solidarity Trial Results. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Riva, L.; Yuan, S.; Yin, X.; Martin-Sancho, L.; Matsunaga, N.; Pache, L.; Burgstaller-Muehlbacher, S.; De Jesus, P.D.; Teriete, P.; Hull, M.V.; et al. Discovery of SARS-CoV-2 antiviral drugs through large-scale compound repurposing. Nature 2020, 586, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Santner, P.; Martins, J.M.d.S.; Kampmeyer, C.; Hartmann-Petersen, R.; Laursen, J.S.; Stein, A.; Olsen, C.A.; Arkin, I.T.; Winther, J.R.; Willemoës, M.; et al. Random Mutagenesis Analysis of the Influenza A M2 Proton Channel Reveals Novel Resistance Mutants. Biochemistry 2018, 57, 5957–5968. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomar, P.P.S.; Krugliak, M.; Arkin, I.T. Identification of SARS-CoV-2 E Channel Blockers from a Repurposed Drug Library. Pharmaceuticals 2021, 14, 604. https://doi.org/10.3390/ph14070604
Tomar PPS, Krugliak M, Arkin IT. Identification of SARS-CoV-2 E Channel Blockers from a Repurposed Drug Library. Pharmaceuticals. 2021; 14(7):604. https://doi.org/10.3390/ph14070604
Chicago/Turabian StyleTomar, Prabhat Pratap Singh, Miriam Krugliak, and Isaiah T. Arkin. 2021. "Identification of SARS-CoV-2 E Channel Blockers from a Repurposed Drug Library" Pharmaceuticals 14, no. 7: 604. https://doi.org/10.3390/ph14070604
APA StyleTomar, P. P. S., Krugliak, M., & Arkin, I. T. (2021). Identification of SARS-CoV-2 E Channel Blockers from a Repurposed Drug Library. Pharmaceuticals, 14(7), 604. https://doi.org/10.3390/ph14070604