
Small Molecule Drugs in Inflammatory Bowel Diseases


Abstract
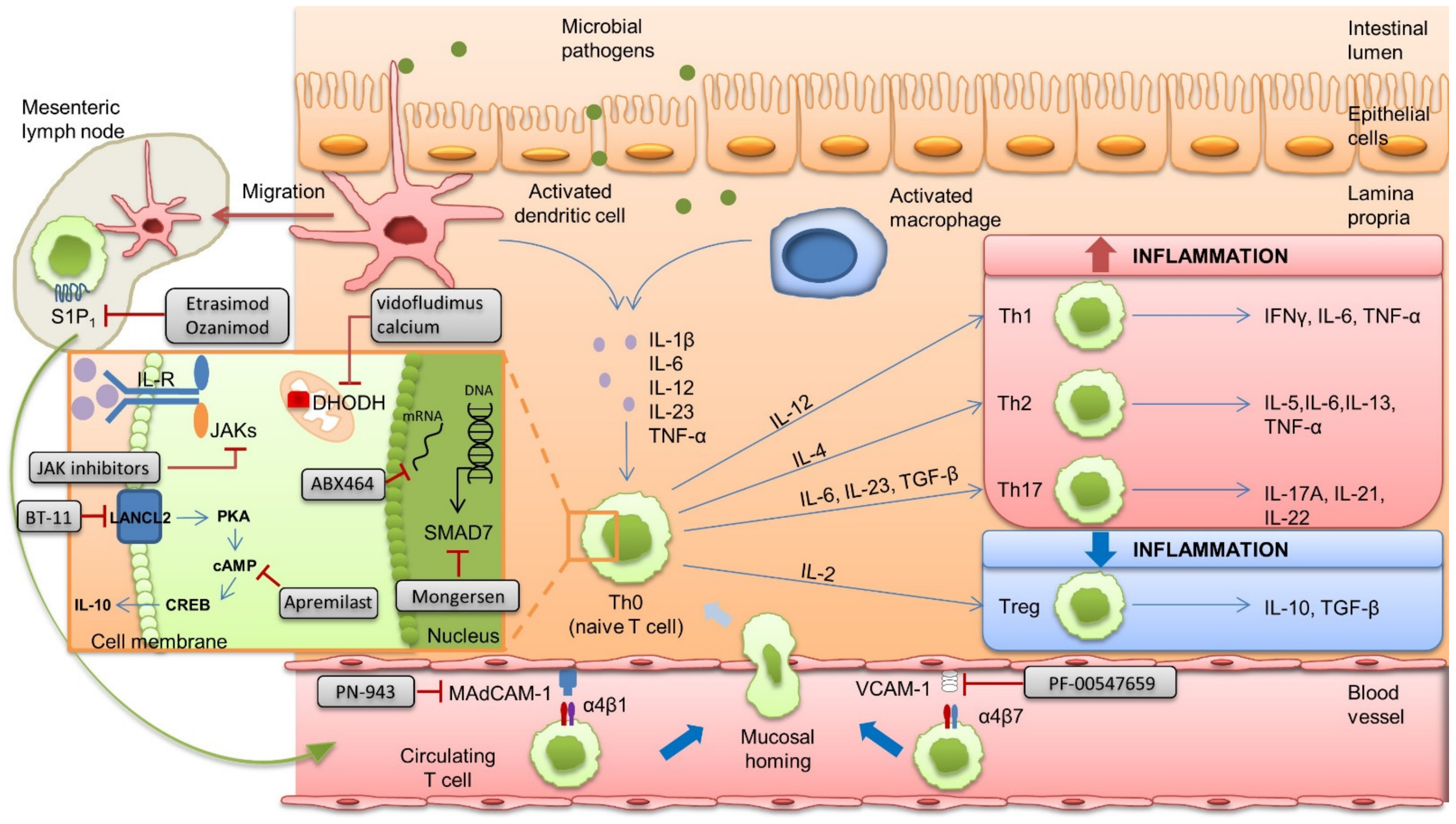
:1. Introduction
2. Materials and Methods
3. Results
3.1. Janus Kinases Inhibitors
3.1.1. Tofacitinib
- Tofacitinib in UC;
- Tofacitinib in CD.
3.1.2. Upadacitinib
- Upadacitinib in UC;
- Upadacitinib in CD.
3.1.3. Filgotinib
- Filgotinib in UC;
- Filgotinib in CD.
3.1.4. Other JAK Inhibitors
3.2. Sphingosine 1-Phosphate Receptor Modulators
- Ozanimod is an orally administered small molecule that selectively targets the sphingosine 1-phosphate receptor subtypes 1 and 5 [96]. Ozanimod was investigated in a phase II clinical trial, TOUCHSTONE, in 197 adults with moderate to severe UC who were randomized to receive either a placebo, ozanimod 0.5 mg, or ozanimod 1 mg once daily, with a dose escalation during the first week (Table 1) [40]. At week 8, ozanimod 1 mg once daily was associated with a slightly higher rate of clinical remission (Mayo Clinic Score (MCS) ≤ 2 with no subscore > 1) than the placebo (16% vs. 6%, p = 0.048). The open-label extension of this study included 170 patients (86%) who were switched to or continued to receive ozanimod 1 mg once daily [97]. During the first year, 28% of the patients discontinued treatment and the discontinuation rates were 15% to 18% annually through years 2–4. The clinical remission rate, including the non-responder imputation, was 54.7% at week 56 and 36.5% at week 200, while the clinical response rate (a decrease in the MCS of ≥3 points and ≥30% and a decrease in the rectal bleeding subscore of ≥1 point or a subscore ≤ 1) was 71.2% and 41.2% at week 56 and 200, respectively [97]. Histologic remission, an endoscopic improvement, endoscopic remission, and mucosal healing at week 56 were achieved in 18.2%, 22.9%, 5.9%, and 2.4% of the patients, respectively. The most common adverse events were hypertension (5.9%), upper respiratory infection (5.9%), lymphopenia (5.3%), and elevated gamma glutamyltransferase (5.3%).
- Etrasimod is an orally administered S1P modulator selectively targeting S1P receptor subtypes 1, 4, and 5 [95]. Etrasimod was investigated in the phase II OASIS randomized, parallel-group induction trial, which included 156 patients with moderate to severe UC [42] (Table 1). At week 12, the clinical improvement (a change from the baseline in the three-component Mayo Clinic Score) was higher in patients treated with 2 mg of etrasimod compared with the placebo (difference, 0.99 points; 90% CI 0.30–1.68) as well as the endoscopic improvement (41.8% vs. 17.8% for the placebo; p = 0.003). In the open-label extension study including 118 patients receiving 2 mg of etrasimod daily up to week 52, 64% of patients had a clinical response, 33% had clinical remission, and 43% had an endoscopic improvement [102]. Patients who achieved a clinical response, clinical remission, or an endoscopic improvement at week 12 maintained the effect of the treatment with 85%, 60%, and 69% of patients with a clinical response, clinical remission, or an endoscopic improvement, respectively, at the end of treatment. Steroid-free clinical remission was observed in 22% of the patients. A total of 60% of the patients experienced treatment emergent adverse events (TEAEs), the most common being worsening of UC (19%) and anemia (11%). When excluding patients with worsening of UC, 51% of the patients (46/91) experienced a TEAE and three patients experienced a serious adverse event [102].
3.3. Other SMDs
3.3.1. Anti-Integrins
3.3.2. Phosphodiesterase 4 Inhibitors
3.3.3. SMAD7 Blockers
3.3.4. Other Small Drugs
4. Conclusions: What Should Be the Future for SMDs
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Baumgart, D.C.; Sandborn, W.J. Inflammatory bowel disease: Clinical aspects and established and evolving therapies. Lancet 2007, 369, 1641–1657. [Google Scholar] [CrossRef]
- Ott, C.; Schölmerich, J. Extraintestinal manifestations and complications in IBD. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 2018, 390, 2769–2778. [Google Scholar] [CrossRef]
- The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 17–30. [CrossRef] [Green Version]
- Na, S.Y.; Moon, W. Perspectives on Current and Novel Treatments for Inflammatory Bowel Disease. Gut. Liver. 2019, 13, 604–616. [Google Scholar] [CrossRef] [Green Version]
- Samanen, J. Chapter 5-Similarities and differences in the discovery and use of biopharmaceuticals and small-molecule chemotherapeutics. In Introduction to Biological and Small Molecule Drug Research and Development; Ganellin, R., Roberts, S., Jefferis, R., Eds.; Elsevier: Oxford, UK, 2013; pp. 161–203. [Google Scholar] [CrossRef]
- Lamb, C.A.; Kennedy, N.A.; Raine, T.; Hendy, P.A.; Smith, P.J.; Limdi, J.K.; Hayee, B.; Lomer, M.C.E.; Parkes, G.C.; Selinger, C.; et al. British Society of Gastroenterology consensus guidelines on the management of inflammatory bowel disease in adults. Gut 2019, 68, s1–s106. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, C.N.; Eliakim, A.; Fedail, S.; Fried, M.; Gearry, R.; Goh, K.L.; Hamid, S.; Khan, A.G.; Khalif, I.; Ng, S.C.; et al. World Gastroenterology Organisation Global Guidelines Inflammatory Bowel Disease: Update August 2015. J. Clin. Gastroenterol. 2016, 50, 803–818. [Google Scholar] [CrossRef] [Green Version]
- Tsui, J.J.; Huynh, H.Q. Is top-down therapy a more effective alternative to conventional step-up therapy for Crohn’s disease? Ann. Gastroenterol. 2018, 31, 413–424. [Google Scholar] [CrossRef]
- Burger, D.; Travis, S. Conventional medical management of inflammatory bowel disease. Gastroenterology 2011, 140, 1827–1837.e1822. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, O.H.; Ainsworth, M.A. Tumor necrosis factor inhibitors for inflammatory bowel disease. N. Engl. J. Med. 2013, 369, 754–762. [Google Scholar] [CrossRef] [Green Version]
- Roda, G.; Jharap, B.; Neeraj, N.; Colombel, J.F. Loss of Response to Anti-TNFs: Definition, Epidemiology, and Management. Clin. Transl. Gastroenterol. 2016, 7, e135. [Google Scholar] [CrossRef]
- Gaujoux, R.; Starosvetsky, E.; Maimon, N.; Vallania, F.; Bar-Yoseph, H.; Pressman, S.; Weisshof, R.; Goren, I.; Rabinowitz, K.; Waterman, M.; et al. Cell-centred meta-analysis reveals baseline predictors of anti-TNFα non-response in biopsy and blood of patients with IBD. Gut 2019, 68, 604–614. [Google Scholar] [CrossRef]
- Vogelaar, L.; Spijker, A.V.; van der Woude, C.J. The impact of biologics on health-related quality of life in patients with inflammatory bowel disease. Clin. Exp. Gastroenterol. 2009, 2, 101–109. [Google Scholar] [CrossRef] [Green Version]
- Van der Have, M.; Oldenburg, B.; Kaptein, A.A.; Jansen, J.M.; Scheffer, R.C.; van Tuyl, B.A.; van der Meulen-de Jong, A.E.; Pierik, M.; Siersema, P.D.; van Oijen, M.G.; et al. Non-adherence to Anti-TNF Therapy is Associated with Illness Perceptions and Clinical Outcomes in Outpatients with Inflammatory Bowel Disease: Results from a Prospective Multicentre Study. J. Crohns. Colitis. 2016, 10, 549–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallick, P.; Taneja, G.; Moorthy, B.; Ghose, R. Regulation of drug-metabolizing enzymes in infectious and inflammatory disease: Implications for biologics-small molecule drug interactions. Expert. Opin. Drug Metab. Toxicol. 2017, 13, 605–616. [Google Scholar] [CrossRef]
- Coskun, M.; Salem, M.; Pedersen, J.; Nielsen, O.H. Involvement of JAK/STAT signaling in the pathogenesis of inflammatory bowel disease. Pharm. Res 2013, 76, 1–8. [Google Scholar] [CrossRef]
- Richmond, C.A.; Rickner, H.; Shah, M.S.; Ediger, T.; Deary, L.; Zhou, F.; Tovaglieri, A.; Carlone, D.L.; Breault, D.T. JAK/STAT-1 Signaling Is Required for Reserve Intestinal Stem Cell Activation during Intestinal Regeneration Following Acute Inflammation. Stem Cell Rep. 2018, 10, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Menet, C.J.; Rompaey, L.V.; Geney, R. Advances in the discovery of selective JAK inhibitors. Prog. Med. Chem. 2013, 52, 153–223. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, D.M.; Kanno, Y.; Villarino, A.; Ward, M.; Gadina, M.; O’Shea, J.J. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat. Rev. Drug Discov. 2017, 16, 843–862. [Google Scholar] [CrossRef] [PubMed]
- Lucaciu, L.A.; Seicean, R.; Seicean, A. Small molecule drugs in the treatment of inflammatory bowel diseases: Which one, when and why?—A systematic review. Eur. J. Gastroenterol. Hepatol. 2020, 32, 669–677. [Google Scholar] [CrossRef]
- Heller, F.; Florian, P.; Bojarski, C.; Richter, J.; Christ, M.; Hillenbrand, B.; Mankertz, J.; Gitter, A.H.; Bürgel, N.; Fromm, M.; et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology 2005, 129, 550–564. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Su, C.; Sands, B.E.; D’Haens, G.R.; Vermeire, S.; Schreiber, S.; Danese, S.; Feagan, B.G.; Reinisch, W.; Niezychowski, W.; et al. Tofacitinib as Induction and Maintenance Therapy for Ulcerative Colitis. N. Engl. J. Med. 2017, 376, 1723–1736. [Google Scholar] [CrossRef]
- Panés, J.; Sandborn, W.J.; Schreiber, S.; Sands, B.E.; Vermeire, S.; D’Haens, G.; Panaccione, R.; Higgins, P.D.R.; Colombel, J.F.; Feagan, B.G.; et al. Tofacitinib for induction and maintenance therapy of Crohn’s disease: Results of two phase IIb randomised placebo-controlled trials. Gut 2017, 66, 1049–1059.e2114. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Ghosh, S.; Panes, J.; Schreiber, S.; D’Haens, G.; Tanida, S.; Siffledeen, J.; Enejosa, J.; Zhou, W.; Othman, A.A.; et al. Efficacy of Upadacitinib in a Randomized Trial of Patients with Active Ulcerative Colitis. Gastroenterology 2020, 158, 2139–2149. [Google Scholar] [CrossRef] [PubMed]
- Sandborn, W.J.; Feagan, B.G.; Loftus, E.V., Jr.; Peyrin-Biroulet, L.; Van Assche, G.; D’Haens, G.; Schreiber, S.; Colombel, J.F.; Lewis, J.D.; Ghosh, S.; et al. Efficacy and Safety of Upadacitinib in a Randomized Trial of Patients with Crohn’s Disease. Gastroenterology 2020, 158, 2123–2138.e2128. [Google Scholar] [CrossRef] [PubMed]
- A Study of the Efficacy and Safety of Upadacitinib (ABT-494) in Participants with Moderately to Severely Active Crohn’s Disease Who Have Inadequately Responded to or Are Intolerant to Biologic Therapy. Available online: https://clinicaltrials.gov/ct2/show/NCT03345836 (accessed on 22 February 2021).
- Filgotinib in the Induction and Maintenance of Remission in Adults with Moderately to Severely Active Ulcerative Colitis (SELECTION1). Available online: https://clinicaltrials.gov/ct2/show/NCT02914522 (accessed on 22 February 2021).
- Vermeire, S.; Schreiber, S.; Petryka, R.; Kuehbacher, T.; Hebuterne, X.; Roblin, X.; Klopocka, M.; Goldis, A.; Wisniewska-Jarosinska, M.; Baranovsky, A.; et al. Clinical remission in patients with moderate-to-severe Crohn’s disease treated with filgotinib (the FITZROY study): Results from a phase 2, double-blind, randomised, placebo-controlled trial. Lancet 2017, 389, 266–275. [Google Scholar] [CrossRef]
- A Phase II Study in Patients with Moderate to Severe Active Ulcerative Colitis. Available online: https://clinicaltrials.gov/ct2/show/NCT03675477 (accessed on 22 February 2021).
- A Phase II Study in Patients with Moderate to Severe Active Crohn’s Disease. Available online: https://clinicaltrials.gov/ct2/show/NCT03677648 (accessed on 22 February 2021).
- Sands, B.E.; Sandborn, W.J.; Feagan, B.G.; Lichtenstein, G.R.; Zhang, H.; Strauss, R.; Szapary, P.; Johanns, J.; Panes, J.; Vermeire, S.; et al. Peficitinib, an Oral Janus Kinase Inhibitor, in Moderate-to-severe Ulcerative Colitis: Results from a Randomised, Phase 2 Study. J. Crohns Colitis 2018, 12, 1158–1169. [Google Scholar] [CrossRef]
- Study to Compare Oral PF-06651600, PF-06700841 and Placebo in Subjects with Moderate to Severe Ulcerative Colitis. Available online: https://clinicaltrials.gov/ct2/show/NCT02958865 (accessed on 22 February 2021).
- Study to Evaluate The Efficacy And Safety Of Oral PF-06651600 And PF-06700841 In Subjects with Moderate To Severe Crohn’s Disease. Available online: https://clinicaltrials.gov/ct2/show/NCT03395184 (accessed on 22 February 2021).
- A Study of the Safety, Efficacy, and Biomarker Response of BMS-986165 in Participants with Moderate to Severe Ulcerative Colitis. Available online: https://clinicaltrials.gov/ct2/show/NCT04613518 (accessed on 22 February 2021).
- An Investigational Study of Experimental Medication BMS-986165 in Participants with Moderate to Severe Crohn’s Disease. Available online: https://www.clinicaltrials.gov/ct2/show/NCT03599622 (accessed on 22 February 2021).
- Efficacy & Safety of TD-1473 in Ulcerative Colitis (RHEA). Available online: https://clinicaltrials.gov/ct2/show/NCT03758443 (accessed on 22 February 2021).
- Efficacy and Safety of TD-1473 in Crohn’s Disease (DIONE). Available online: https://clinicaltrials.gov/ct2/show/NCT03635112 (accessed on 22 February 2021).
- Study of OST-122 in Patients with Moderate to Severe Ulcerative Colitis. Available online: https://clinicaltrials.gov/ct2/show/NCT04353791 (accessed on 22 February 2021).
- Sandborn, W.J.; Feagan, B.G.; Wolf, D.C.; D’Haens, G.; Vermeire, S.; Hanauer, S.B.; Ghosh, S.; Smith, H.; Cravets, M.; Frohna, P.A.; et al. Ozanimod Induction and Maintenance Treatment for Ulcerative Colitis. N. Engl. J. Med. 2016, 374, 1754–1762. [Google Scholar] [CrossRef]
- Feagan, B.G.; Sandborn, W.J.; Danese, S.; Wolf, D.C.; Liu, W.J.; Hua, S.Y.; Minton, N.; Olson, A.; D’Haens, G. Ozanimod induction therapy for patients with moderate to severe Crohn’s disease: A single-arm, phase 2, prospective observer-blinded endpoint study. Lancet Gastroenterol. Hepatol. 2020, 5, 819–828. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Peyrin-Biroulet, L.; Zhang, J.; Chiorean, M.; Vermeire, S.; Lee, S.D.; Kühbacher, T.; Yacyshyn, B.; Cabell, C.H.; Naik, S.U.; et al. Efficacy and Safety of Etrasimod in a Phase 2 Randomized Trial of Patients with Ulcerative Colitis. Gastroenterology 2020, 158, 550–561. [Google Scholar] [CrossRef] [Green Version]
- Vermeire, S.; Sandborn, W.J.; Danese, S.; Hébuterne, X.; Salzberg, B.A.; Klopocka, M.; Tarabar, D.; Vanasek, T.; Greguš, M.; Hellstern, P.A.; et al. Anti-MAdCAM antibody (PF-00547659) for ulcerative colitis (TURANDOT): A phase 2, randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 135–144. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Lee, S.D.; Tarabar, D.; Louis, E.; Klopocka, M.; Klaus, J.; Reinisch, W.; Hébuterne, X.; Park, D.I.; Schreiber, S.; et al. Phase II evaluation of anti-MAdCAM antibody PF-00547659 in the treatment of Crohn’s disease: Report of the OPERA study. Gut 2018, 67, 1824–1835. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, S.; Keshavarzian, A.; Isaacs, K.L.; Schollenberger, J.; Guzman, J.P.; Orlandi, C.; Hanauer, S.B. A randomized, placebo-controlled, phase II study of tetomilast in active ulcerative colitis. Gastroenterology 2007, 132, 76–86. [Google Scholar] [CrossRef]
- Danese, S.; Neurath, M.F.; Kopoń, A.; Zakko, S.F.; Simmons, T.C.; Fogel, R.; Siegel, C.A.; Panaccione, R.; Zhan, X.; Usiskin, K.; et al. Effects of Apremilast, an Oral Inhibitor of Phosphodiesterase 4, in a Randomized Trial of Patients with Active Ulcerative Colitis. Clin. Gastroenterol. Hepatol. 2020, 18, 2526–2534.e2529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteleone, G.; Neurath, M.F.; Ardizzone, S.; Di Sabatino, A.; Fantini, M.C.; Castiglione, F.; Scribano, M.L.; Armuzzi, A.; Caprioli, F.; Sturniolo, G.C.; et al. Mongersen, an oral SMAD7 antisense oligonucleotide, and Crohn’s disease. N. Engl. J. Med. 2015, 372, 1104–1113. [Google Scholar] [CrossRef] [Green Version]
- Phase 2 Dose-finding IMU-838 for Ulcerative Colitis (CALDOSE-1). Available online: https://clinicaltrials.gov/ct2/show/NCT03341962 (accessed on 18 May 2021).
- Safety Evaluation of ABX464 in Patients with Moderate to Severe Active Crohn’s Disease. Available online: https://clinicaltrials.gov/ct2/show/NCT03905109 (accessed on 18 May 2021).
- Vermeire, S.; Hébuterne, X.; Tilg, H.; De Hertogh, G.; Gineste, P.; Steens, J.M. Induction and Long-term Follow-up with ABX464 for Moderate-to-severe Ulcerative Colitis: Results of Phase IIa Trial. Gastroenterology 2021. [Google Scholar] [CrossRef] [PubMed]
- Efficacy and Safety of Oral BT-11 in Mild to Moderate Ulcerative Colitis. Available online: https://clinicaltrials.gov/ct2/show/NCT03861143 (accessed on 18 May 2021).
- Danese, S.; Grisham, M.; Hodge, J.; Telliez, J.B. JAK inhibition using tofacitinib for inflammatory bowel disease treatment: A hub for multiple inflammatory cytokines. Am. J. Physiol Gastrointest Liver Physiol 2016, 310, G155–G162. [Google Scholar] [CrossRef] [Green Version]
- Highlights of prescribing information. XELJANZ. U.S. Food and Drug Administration. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/203214s024,208246s010lbl.pdf (accessed on 19 February 2021).
- Xeljanz. European Medicines Agency. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/xeljanz (accessed on 19 February 2021).
- Hanauer, S.; Panaccione, R.; Danese, S.; Cheifetz, A.; Reinisch, W.; Higgins, P.D.R.; Woodworth, D.A.; Zhang, H.; Friedman, G.S.; Lawendy, N.; et al. Tofacitinib Induction Therapy Reduces Symptoms within 3 Days for Patients with Ulcerative Colitis. Clin. Gastroenterol. Hepatol. 2019, 17, 139–147. [Google Scholar] [CrossRef] [Green Version]
- Sands, B.E.; Armuzzi, A.; Marshall, J.K.; Lindsay, J.O.; Sandborn, W.J.; Danese, S.; Panés, J.; Bressler, B.; Colombel, J.F.; Lawendy, N.; et al. Efficacy and safety of tofacitinib dose de-escalation and dose escalation for patients with ulcerative colitis: Results from OCTAVE Open. Aliment. Pharm. 2020, 51, 271–280. [Google Scholar] [CrossRef]
- A Study of Tofacitinib in Patients with Ulcerative Colitis in Stable Remission. Available online: https://clinicaltrials.gov/ct2/show/NCT03281304 (accessed on 19 February 2021).
- Evaluation of Oral Tofacitinib in Children Aged 2 to 17 Years Old Suffering from Moderate to Severe Ulcerative Colitis. Available online: https://clinicaltrials.gov/ct2/show/NCT04624230 (accessed on 19 February 2021).
- Xeljanz and Healthy Diet. Available online: https://clinicaltrials.gov/ct2/show/NCT04505410 (accessed on 21 February 2021).
- Panés, J.; Su, C.; Bushmakin, A.G.; Cappelleri, J.C.; Mamolo, C.; Healey, P. Randomized trial of tofacitinib in active ulcerative colitis: Analysis of efficacy based on patient-reported outcomes. BMC Gastroenterol. 2015, 15, 14. [Google Scholar] [CrossRef] [Green Version]
- Dowty, M.E.; Lin, J.; Ryder, T.F.; Wang, W.; Walker, G.S.; Vaz, A.; Chan, G.L.; Krishnaswami, S.; Prakash, C. The pharmacokinetics, metabolism, and clearance mechanisms of tofacitinib, a janus kinase inhibitor, in humans. Drug Metab. Dispos. 2014, 42, 759–773. [Google Scholar] [CrossRef] [Green Version]
- Vong, C.; Martin, S.W.; Deng, C.; Xie, R.; Ito, K.; Su, C.; Sandborn, W.J.; Mukherjee, A. Population Pharmacokinetics of Tofacitinib in Patients with Moderate to Severe Ulcerative Colitis. Clin. Pharm. Drug Dev. 2021. [Google Scholar] [CrossRef]
- Sandborn, W.J.; Panés, J.; Sands, B.E.; Reinisch, W.; Su, C.; Lawendy, N.; Koram, N.; Fan, H.; Jones, T.V.; Modesto, I.; et al. Venous thromboembolic events in the tofacitinib ulcerative colitis clinical development programme. Aliment. Pharm. 2019, 50, 1068–1076. [Google Scholar] [CrossRef]
- Curtis, J.R.; Regueiro, M.; Yun, H.; Su, C.; DiBonaventura, M.; Lawendy, N.; Nduaka, C.I.; Koram, N.; Cappelleri, J.C.; Chan, G.; et al. Tofacitinib Treatment Safety in Moderate to Severe Ulcerative Colitis: Comparison of Observational Population Cohort Data from the IBM MarketScan® Administrative Claims Database with Tofacitinib Trial Data. Inflamm. Bowel. Dis. 2020. [Google Scholar] [CrossRef]
- Safety Study Of Tofacitinib Versus Tumor Necrosis Factor (TNF) Inhibitor In Subjects with Rheumatoid Arthritis. Available online: https://clinicaltrials.gov/ct2/show/NCT02092467 (accessed on 21 February 2021).
- Increased Risk of Blood Clots in Lungs and Death with Higher Dose of Xeljanz (tofacitinib) for Rheumatoid Arthritis. European Medicines Agency. Available online: https://www.ema.europa.eu/en/news/increased-risk-blood-clots-lungs-death-higher-dose-xeljanz-tofacitinib-rheumatoid-arthritis (accessed on 21 February 2021).
- Mease, P.; Charles-Schoeman, C.; Cohen, S.; Fallon, L.; Woolcott, J.; Yun, H.; Kremer, J.; Greenberg, J.; Malley, W.; Onofrei, A.; et al. Incidence of venous and arterial thromboembolic events reported in the tofacitinib rheumatoid arthritis, psoriasis and psoriatic arthritis development programmes and from real-world data. Ann. Rheum. Dis. 2020, 79, 1400–1413. [Google Scholar] [CrossRef] [PubMed]
- Initial Safety Trial Results Find Increased Risk of Serious Heart-Related Problems and Cancer with Arthritis and Ulcerative Colitis Medicine Xeljanz, Xeljanz XR (tofacitinib). U.S. Food and Drug Administration. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/initial-safety-trial-results-find-increased-risk-serious-heart-related-problems-and-cancer-arthritis (accessed on 21 February 2021).
- Sandborn, W.J.; Panés, J.; D’Haens, G.R.; Sands, B.E.; Su, C.; Moscariello, M.; Jones, T.; Pedersen, R.; Friedman, G.S.; Lawendy, N.; et al. Safety of Tofacitinib for Treatment of Ulcerative Colitis, Based on 4.4 Years of Data from Global Clinical Trials. Clin. Gastroenterol. Hepatol. 2019, 17, 1541–1550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panaccione, R.; Isaacs, J.D.; Chen, L.A.; Wang, W.; Marren, A.; Kwok, K.; Wang, L.; Chan, G.; Su, C. Characterization of Creatine Kinase Levels in Tofacitinib-Treated Patients with Ulcerative Colitis: Results from Clinical Trials. Dig. Dis. Sci. 2020. [Google Scholar] [CrossRef]
- Panés, J.; D’Haens, G.R.; Higgins, P.D.R.; Mele, L.; Moscariello, M.; Chan, G.; Wang, W.; Niezychowski, W.; Su, C.; Maller, E. Long-term safety and tolerability of oral tofacitinib in patients with Crohn’s disease: Results from a phase 2, open-label, 48-week extension study. Aliment. Pharm. 2019, 49, 265–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klünder, B.; Mohamed, M.F.; Othman, A.A. Population Pharmacokinetics of Upadacitinib in Healthy Subjects and Subjects with Rheumatoid Arthritis: Analyses of Phase I and II Clinical Trials. Clin. Pharm. 2018, 57, 977–988. [Google Scholar] [CrossRef] [Green Version]
- A Study to Evaluate the Safety and Efficacy of Upadacitinib (ABT-494) for Induction and Maintenance Therapy in Participants with Moderately to Severely Active Ulcerative Colitis (UC). Available online: https://clinicaltrials.gov/ct2/show/NCT02819635 (accessed on 21 February 2021).
- A Study of the Efficacy and Safety of Upadacitinib (ABT-494) in Participants with Moderately to Severely Active Ulcerative Colitis (U-Accomplish). Available online: https://clinicaltrials.gov/ct2/show/NCT03653026 (accessed on 21 February 2021).
- A Study to Evaluate the Long-Term Safety and Efficacy of Upadacitinib (ABT-494) in Participants with Ulcerative Colitis (UC). Available online: https://clinicaltrials.gov/ct2/show/NCT03006068 (accessed on 21 February 2021).
- A Study to Evaluate the Long-Term Efficacy, Safety, and Tolerability of Repeated Administration of Upadacitinib (ABT-494) in Participants with Crohn’s Disease. Available online: https://clinicaltrials.gov/ct2/show/NCT02782663 (accessed on 21 February 2021).
- A Study of the Efficacy and Safety of Upadacitnib (ABT-494) in Participants with Moderately to Severely Active Crohn’s Disease Who Have Inadequately Responded to or Are Intolerant to Conventional and/or Biologic Therapies. Available online: https://clinicaltrials.gov/ct2/show/NCT03345849 (accessed on 22 February 2021).
- A Maintenance and Long-Term Extension Study of the Efficacy and Safety of Upadacitinib (ABT-494) in Participants with Crohn’s Disease Who Completed the Studies M14-431 or M14-433. Available online: https://clinicaltrials.gov/ct2/show/NCT03345823 (accessed on 22 February 2021).
- Namour, F.; Diderichsen, P.M.; Cox, E.; Vayssière, B.; Van der Aa, A.; Tasset, C.; Van’t Klooster, G. Pharmacokinetics and Pharmacokinetic/Pharmacodynamic Modeling of Filgotinib (GLPG0634), a Selective JAK1 Inhibitor, in Support of Phase IIB Dose Selection. Clin. Pharm. 2015, 54, 859–874. [Google Scholar] [CrossRef] [Green Version]
- Phase 2b/3 Trial Shows Efficacy of Filgotinib for the Induction and Maintenance of Remission in Moderately and Severely Active Ulcerative Colitis. Gilead. Available online: https://www.gilead.com/news-and-press/press-room/press-releases/2020/10/phase-2b3-trial-shows-efficacy-of-filgotinib-for-the-induction-and-maintenance-of-remission-in-moderately-and-severely-active-ulcerative-colitis (accessed on 12 October 2020).
- Filgotinib in Long-Term Extension Study of Adults with Ulcerative Colitis (SELECTIONLTE). Available online: https://clinicaltrials.gov/ct2/show/NCT02914535 (accessed on 22 February 2021).
- Filgotinib in the Induction and Maintenance of Remission in Adults with Moderately to Severely Active Crohn’s Disease (Diversity1). Available online: https://clinicaltrials.gov/ct2/show/NCT02914561 (accessed on 22 February 2021).
- Filgotinib in Long-Term Extension Study of Adults with Crohn’s Disease (DIVERSITYLTE). Available online: https://clinicaltrials.gov/ct2/show/NCT02914600 (accessed on 22 February 2021).
- Efficacy and Safety of Filgotinib in the Treatment of Small Bowel Crohn’s Disease (SBCD) (Divergence1). Available online: https://clinicaltrials.gov/ct2/show/NCT03046056 (accessed on 22 February 2021).
- Assessment report Jyseleca. European Medicines Agency. Available online: https://www.ema.europa.eu/en/documents/assessment-report/jyseleca-epar-public-assessment-report_en.pdf (accessed on 18 May 2021).
- Gilead Receives Complete Response Letter for Filgotinib for the Treatment of Moderately to Severely Active Rheumatoid Arthritis. Available online: https://www.gilead.com/news-and-press/press-room/press-releases/2020/8/gilead-receives-complete-response-letter-for-filgotinib-for-the-treatment-of-moderately-to-severely-active-rheumatoid-arthritis (accessed on 18 May 2021).
- Study to Evaluate the Testicular Safety of Filgotinib in Adult Males with Moderately to Severely Active Inflammatory Bowel Disease (MANTA). Available online: https://clinicaltrials.gov/ct2/show/NCT03201445 (accessed on 22 February 2021).
- Takeuchi, T.; Tanaka, Y.; Iwasaki, M.; Ishikura, H.; Saeki, S.; Kaneko, Y. Efficacy and safety of the oral Janus kinase inhibitor peficitinib (ASP015K) monotherapy in patients with moderate to severe rheumatoid arthritis in Japan: A 12-week, randomised, double-blind, placebo-controlled phase IIb study. Ann. Rheum. Dis. 2016, 75, 1057–1064. [Google Scholar] [CrossRef]
- Papp, K.; Gordon, K.; Thaçi, D.; Morita, A.; Gooderham, M.; Foley, P.; Girgis, I.G.; Kundu, S.; Banerjee, S. Phase 2 Trial of Selective Tyrosine Kinase 2 Inhibition in Psoriasis. N. Engl. J. Med. 2018, 379, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Bristol Myers Squibb Announces Positive Topline Results from Second Pivotal Phase 3 Psoriasis Study Showing Superiority of Deucravacitinib Compared to Placebo and Otezla® (apremilast). Available online: https://news.bms.com/news/details/2021/Bristol-Myers-Squibb-Announces-Positive-Topline-Results-from-Second-Pivotal-Phase-3-Psoriasis-Study-Showing-Superiority-of-Deucravacitinib-Compared-to-Placebo-and-Otezla-apremilast/default.aspx (accessed on 2 February 2021).
- Safety and Efficacy of BMS-986165 in Participants with Moderate to Severe Ulcerative Colitis. Available online: https://clinicaltrials.gov/ct2/show/NCT03934216 (accessed on 22 February 2021).
- TD-1473 Long-Term Safety (LTS) Ulcerative Colitis (UC) Study. Available online: https://clinicaltrials.gov/ct2/show/NCT03920254 (accessed on 22 February 2021).
- Park, S.J.; Im, D.S. Sphingosine 1-Phosphate Receptor Modulators and Drug Discovery. Biomol. Ther. 2017, 25, 80–90. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Jeldres, T.; Tyler, C.J.; Boyer, J.D.; Karuppuchamy, T.; Bamias, G.; Dulai, P.S.; Boland, B.S.; Sandborn, W.J.; Patel, D.R.; Rivera-Nieves, J. Cell Trafficking Interference in Inflammatory Bowel Disease: Therapeutic Interventions Based on Basic Pathogenesis Concepts. Inflamm. Bowel. Dis. 2019, 25, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Peyrin-Biroulet, L.; Christopher, R.; Behan, D.; Lassen, C. Modulation of sphingosine-1-phosphate in inflammatory bowel disease. Autoimmun. Rev. 2017, 16, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Scott, F.L.; Clemons, B.; Brooks, J.; Brahmachary, E.; Powell, R.; Dedman, H.; Desale, H.G.; Timony, G.A.; Martinborough, E.; Rosen, H.; et al. Ozanimod (RPC1063) is a potent sphingosine-1-phosphate receptor-1 (S1P1) and receptor-5 (S1P5) agonist with autoimmune disease-modifying activity. Br. J. Pharm. 2016, 173, 1778–1792. [Google Scholar] [CrossRef] [Green Version]
- Sandborn, W.J.; Feagan, B.G.; Hanauer, S.; Vermeire, S.; Ghosh, S.; Liu, W.J.; Petersen, A.; Charles, L.; Huang, V.; Usiskin, K.; et al. Long-Term Efficacy and Safety Of Ozanimod In Moderate-To-Severe Ulcerative Colitis: Results From The Open-Label Extension Of The Randomized, Phase 2 Touchstone Study. J. Crohns Colitis 2021. [Google Scholar] [CrossRef]
- Induction Study #1 of Oral Ozanimod as Induction Therapy for Moderately to Severely Active Crohn’s Disease. Available online: https://clinicaltrials.gov/ct2/show/NCT03440372 (accessed on 23 March 2021).
- Induction Study #2 of Oral Ozanimod as Induction Therapy for Moderately to Severely Active Crohn’s Disease. Available online: https://clinicaltrials.gov/ct2/show/NCT03440385 (accessed on 23 March 2021).
- A Placebo-Controlled Study of Oral Ozanimod as Maintenance Therapy for Moderately to Severely Active Crohn’s Disease. Available online: https://clinicaltrials.gov/ct2/show/NCT03464097 (accessed on 22 March 2021).
- An Extension Study of Oral Ozanimod for Moderately to Severely Active Crohn’s Disease. Available online: https://clinicaltrials.gov/ct2/show/NCT03467958 (accessed on 22 March 2021).
- Vermeire, S.; Chiorean, M.; Panés, J.; Peyrin-Biroulet, L.; Zhang, J.; Sands, B.E.; Lazin, K.; Klassen, P.; Naik, S.U.; Cabell, C.H.; et al. Long-term Safety and Efficacy of Etrasimod for Ulcerative Colitis: Results from the Open-label Extension of the OASIS Study. J. Crohns Colitis 2021. [Google Scholar] [CrossRef]
- Park, S.C.; Jeen, Y.T. Anti-integrin therapy for inflammatory bowel disease. World J. Gastroenterol. 2018, 24, 1868–1880. [Google Scholar] [CrossRef] [Green Version]
- Nelson, S.M.; Nguyen, T.M.; McDonald, J.W.; MacDonald, J.K. Natalizumab for induction of remission in Crohn’s disease. Cochrane Database Syst. Rev. 2018, 8, Cd006097. [Google Scholar] [CrossRef]
- Modi, N.B.; Cheng, X.; Mattheakis, L.; Hwang, C.C.; Nawabi, R.; Liu, D.; Gupta, S. Single- and Multiple-Dose Pharmacokinetics and Pharmacodynamics of PN-943, a Gastrointestinal-Restricted Oral Peptide Antagonist of α4β7, in Healthy Volunteers. Clin. Pharm. Drug Dev. 2021. [Google Scholar] [CrossRef]
- Allocca, M.; Gilardi, D.; Fiorino, G.; Furfaro, F.; Argollo, M.; Peyrin-Biroulet, L.; Danese, S. PF-00547659 for the treatment of Crohn’s disease and ulcerative colitis. Expert Opin. Investig. Drugs 2018, 27, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Reinisch, W.; Sandborn, W.J.; Danese, S.; Hébuterne, X.; Kłopocka, M.; Tarabar, D.; Vaňásek, T.; Greguš, M.; Hellstern, P.A.; Kim, J.S.; et al. Long-term safety and efficacy of the anti-MAdCAM-1 monoclonal antibody ontamalimab (SHP647) for the treatment of ulcerative colitis: The open-label study TURANDOT II. J. Crohns Colitis 2021. [Google Scholar] [CrossRef]
- Spadaccini, M.; D’Alessio, S.; Peyrin-Biroulet, L.; Danese, S. PDE4 Inhibition and Inflammatory Bowel Disease: A Novel Therapeutic Avenue. Int. J. Mol. Sci 2017, 18, 1276. [Google Scholar] [CrossRef]
- Monteleone, G.; Boirivant, M.; Pallone, F.; MacDonald, T.T. TGF-beta1 and Smad7 in the regulation of IBD. Mucosal. Immunol. 2008, 1 (Suppl. S1), S50–S53. [Google Scholar] [CrossRef] [PubMed]
- Boirivant, M.; Pallone, F.; Di Giacinto, C.; Fina, D.; Monteleone, I.; Marinaro, M.; Caruso, R.; Colantoni, A.; Palmieri, G.; Sanchez, M.; et al. Inhibition of Smad7 with a specific antisense oligonucleotide facilitates TGF-beta1-mediated suppression of colitis. Gastroenterology 2006, 131, 1786–1798. [Google Scholar] [CrossRef]
- Sands, B.E.; Feagan, B.G.; Sandborn, W.J.; Schreiber, S.; Peyrin-Biroulet, L.; Frédéric Colombel, J.; Rossiter, G.; Usiskin, K.; Ather, S.; Zhan, X.; et al. Mongersen (GED-0301) for Active Crohn’s Disease: Results of a Phase 3 Study. Am. J. Gastroenterol. 2020, 115, 738–745. [Google Scholar] [CrossRef]
- Muehler, A.; Peelen, E.; Kohlhof, H.; Gröppel, M.; Vitt, D. Vidofludimus calcium, a next generation DHODH inhibitor for the Treatment of relapsing-remitting multiple sclerosis. Mult. Scler. Relat. Disord. 2020, 43, 102129. [Google Scholar] [CrossRef]
- Muehler, A.; Kohlhof, H.; Groeppel, M.; Vitt, D. The Selective Oral Immunomodulator Vidofludimus in Patients with Active Rheumatoid Arthritis: Safety Results from the COMPONENT Study. Drugs R D 2019, 19, 351–366. [Google Scholar] [CrossRef] [Green Version]
- Muehler, A.; Kohlhof, H.; Groeppel, M.; Vitt, D. Safety, Tolerability and Pharmacokinetics of Vidofludimus calcium (IMU-838) After Single and Multiple Ascending Oral Doses in Healthy Male Subjects. Eur. J. Drug Metab. Pharm. 2020, 45, 557–573. [Google Scholar] [CrossRef]
- Immunic, Inc. Reports Positive Top-line Data from Phase 2 EMPhASIS Trial of IMU-838 in Patients with Relapsing-Remitting Multiple Sclerosis. Available online: https://www.immunic-therapeutics.com/2020/08/02/immunic-inc-reports-positive-top-line-data-from-phase-2-emphasis-trial-of-imu-838-in-patients-with-relapsing-remitting-multiple-sclerosis/ (accessed on 4 November 2020).
- Vautrin, A.; Manchon, L.; Garcel, A.; Campos, N.; Lapasset, L.; Laaref, A.M.; Bruno, R.; Gislard, M.; Dubois, E.; Scherrer, D.; et al. Both anti-inflammatory and antiviral properties of novel drug candidate ABX464 are mediated by modulation of RNA splicing. Sci. Rep. 2019, 9, 792. [Google Scholar] [CrossRef]
- Efficacy and Safety Study of ABX464 as Maintenance Therapy in Patients with Moderate to Severe Ulcerative Colitis. Available online: https://clinicaltrials.gov/ct2/show/NCT04023396 (accessed on 18 May 2021).
- Leber, A.; Hontecillas, R.; Zoccoli-Rodriguez, V.; Colombel, J.F.; Chauhan, J.; Ehrich, M.; Farinola, N.; Bassaganya-Riera, J. The Safety, Tolerability, and Pharmacokinetics Profile of BT-11, an Oral, Gut-Restricted Lanthionine Synthetase C-Like 2 Agonist Investigational New Drug for Inflammatory Bowel Disease: A Randomized, Double-Blind, Placebo-Controlled Phase I Clinical Trial. Inflamm. Bowel Dis. 2020, 26, 643–652. [Google Scholar] [CrossRef]
- Hirten, R.P.; Iacucci, M.; Shah, S.; Ghosh, S.; Colombel, J.F. Combining Biologics in Inflammatory Bowel Disease and Other Immune Mediated Inflammatory Disorders. Clin. Gastroenterol. Hepatol. 2018, 16, 1374–1384. [Google Scholar] [CrossRef] [Green Version]
- Tian, L.; Xiong, X.; Guo, Q.; Chen, Y.; Wang, L.; Dong, P.; Ma, A. Cost-Effectiveness of Tofacitinib for Patients with Moderate-to-Severe Rheumatoid Arthritis in China. Pharmacoeconomics 2020, 38, 1345–1358. [Google Scholar] [CrossRef]
- Navarro, F.; Martinez-Sesmero, J.M.; Balsa, A.; Peral, C.; Montoro, M.; Valderrama, M.; Gómez, S.; de Andrés-Nogales, F.; Casado, M.A.; Oyagüez, I. Cost-effectiveness analysis of treatment sequences containing tofacitinib for the treatment of rheumatoid arthritis in Spain. Clin. Rheumatol. 2020, 39, 2919–2930. [Google Scholar] [CrossRef] [PubMed]
- Harrington, R.; Al Nokhatha, S.A.; Conway, R. JAK Inhibitors in Rheumatoid Arthritis: An Evidence-Based Review on the Emerging Clinical Data. J. Inflamm Res. 2020, 13, 519–531. [Google Scholar] [CrossRef]
- Mancina, R.M.; Pagnotta, R.; Pagliuso, C.; Albi, V.; Bruno, D.; Garieri, P.; Doldo, P.; Spagnuolo, R. Gastrointestinal Symptoms of and Psychosocial Changes in Inflammatory Bowel Disease: A Nursing-Led Cross-Sectional Study of Patients in Clinical Remission. Medicina 2020, 56, 45. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Type of SMD | Phase, Indication | Design and Intervention | Main Results |
|---|---|---|---|
| JAK inhibitors | |||
| Tofacitinib (pankinase, JAK1, JAK3) | III, UC (OCTAVE PROGRAM) [23] | OCTAVE-1 and OCTAVE-2 trials: an 8-week induction trial in 598 and 541 patients with moderate to severe disease, respectively, naive or biologic-experienced 10 mg/placebo (4:1) | Higher clinical remission in the active group than in the placebo group (OCTAVE-1: 18.5% vs. 8.2%, p = 0.007; OCTAVE-2: 16.6% vs. 3.6%, p < 0.001) |
| A 52-week sustained trial for 593 responders from OCTAVE induction trials; 3 arms: 10 mg/5 mg/placebo (1:1:1) | Higher clinical remission in the 10 mg group vs. the placebo group (40.6% vs. 11.1%, p < 0.001), but not in the 5 mg group (34.3% vs. 11.1%) | ||
| IIb, CD [24] | An 8-week induction trial in 180 patients with moderate to severe disease, naive or biologic-experienced 10 mg/5 mg/placebo (1:1:1) | No significant improvement in remission (43.0%/43.5%/36.7%, all tests vs. placebo NS) | |
| A 48-week OLE trial in 150 patients 10 mg/5 mg (1:1) | Last observation carried forward 85.5% in 10 mg/39.8% in 5 mg | ||
| Upadacitinib (JAK1) | IIb, UC (U-ACHIEVE) [25] | An 8-week induction trial in 250 patients with moderate to severe disease, naive or biologic-experienced 45/30/15/7.5 mg/placebo (1:1:1:1:1) | A higher remission rate in all active groups vs. the placebo group except for the 7.5 mg group (19.6%/13.5%/14.3%/8.5%/0%) |
| II, CD (CELEST) [26] | A 16-week induction trial in 220 patients with moderate to severe disease, naive or biologic-experienced 24 mg qd/24 mg bid/12 mg bid/6 mg bid/3 mg bid/placebo (1:1:1:1:1:1) | Significant clinical remission only for upadacitinib 6 mg bid vs. placebo 14%/22%/11%/27% (p < 0.1)/13%/11% | |
| III, CD (CELEST OLE) [27] | An OLE trial for patients who completed the (52-week) CELEST induction trial | Ongoing, Estimated Primary Completion Date: 2025 | |
| Filgotinib (JAK1) | III, UC [28] | SELECTION-1: a 10-week induction trial in 659 patients with moderate to severe biologic-naive disease and in 689 biologic-experienced patients 200 mg/100 mg/placebo (2:2:1) | Efficacy (clinical remission) of 200 mg filgotinib vs. placebo in both biologic-naive (26.1%vs. 15.3%) biologic-experienced patients (11.5% vs. 4.2%) |
| II, CD [29] | FITZROY: 10-week induction in 172 patients with moderate to severe CD, naive or biologic-experienced 200 mg vs. placebo (3:1) | A higher remission rate in 200 mg filgotinib (47% vs. 23%, p < 0.001) | |
| SHR0302 (JAK1) | II, UC [30] | An 8 to 12-week dose finding and efficacy trial in 164 patients with (expected) moderate to severe disease (3 doses vs. placebo) | Study completed in February 2021; results not published yet |
| II, CD [31] | A 12-week dose finding and efficacy trial in 144 patients with (expected) moderate to severe disease (3 doses vs. placebo) | Ongoing, estimated study completion date: October 2021 | |
| Peficitinib (pankinase inhibitor, JAK3+) | IIb, UC [32] | An 8-week induction trial in 219 patients with moderate to severe disease 150 mg qd/75 mg bid/75 mg qd/25 mg qd/placebo (1:1:1:1:1) | No significantly higher response compared with placebo (27.3%/15.9%/15.9%/15.9%/7.0%) |
| PF-06651600 (JAK3) and PF-06700841 (TYK2/JAK1) | IIb, UC [33] | An 8-week dose ranging trial in 319 patients with (expected) moderate to severe disease, naive or biologic-experienced (3 doses PF-06651600/3 doses PF-06700841/placebo) | Study completed in May 2021; results not published yet |
| IIa, CD [34] | A 12-week efficacy trial in 250 patients with (expected) moderate to severe disease, naive or biologic-experienced PF-06651600 (200 mg for 8 weeks + 50 mg for 4 weeks)/PF-06700841 (60 mg for 12 weeks)/placebo | Ongoing, estimated study completion date: June 2021 | |
| Deucravacitinib (TYK2) | II, UC [35] | A 12-week efficacy trial in 50 patients with (expected) moderate to severe disease, naive or biologic-experienced (2 doses vs. placebo) | Ongoing, estimated study completion date: April 2023 |
| II, CD [36] | A 12-week efficacy trial in 240 patients with (expected) moderate to severe disease, naive or biologic-experienced (2 doses vs. placebo) | Ongoing, estimated study completion date: March 2024 | |
| TD-1473 (gut-selective pankinase inhibitor) | II-III, UC [37] | An 8-week dose ranging phase II trial (3 dose groups or placebo) in 240 patients with moderate to severe disease, naive or biologic-experienced, followed by a phase III 8-week induction trial in 660 patients | Ongoing, estimated study completion date of phase II: July 2021 |
| II, CD [38] | A 12-week efficacy and safety trial in 160 patients with moderate to severe disease; 3 arms (2 doses and placebo) | Ongoing, estimated study completion date: August 2022 | |
| OST-122 (gut-selective JAK3/TYK2/ARK5) | I-II, UC [39] | A 28-day safety trial in 32 patients with moderate to severe disease; 3 arms (low dose, high dose, placebo) | Ongoing, estimated study completion date: April 2022 |
| S-1-P receptor modulators | |||
| Ozanimod S1PR1, S1PR5) | II, UC (TOUCHSTONE) [40] | An 8-week induction trial in 197 patients with moderate to severe disease, naive or biologic-experienced 1 mg/0.5 mg/placebo (1:1:1) | A higher clinical remission rate in the 1 mg group vs. the placebo group (16% vs. 6%, p = 0.048) |
| II, CD (STEPSTONE) [41] | An uncontrolled trial in 69 patients with moderate to severe disease, naive or biologic-experienced 1 mg ozanimod for 11 weeks after a 7-day dose escalation | 39.1% (95% CI 27.6–51.6) achieved clinical remission (CDAI < 150) and 56.5% (95% CI 44.0–68.4) exhibited a clinical response (CDAI decrease from baseline ≥100) | |
| Etrasimod (S1PR1) | II, UC (OASIS) [42] | A 12-week induction trial in 156 patients with moderate to severe disease, naive or biologic-experienced; 3 arms (2 mg/1 mg/placebo) (1:1:1) | A significantly higher response rate (improvement in modified MCS from baseline) in the 2 mg group vs. the placebo group (+0.99, 95% CI 0.30–1.68) |
| Anti-integrins | |||
| Ontamalimab (MAdCAM1) | II, UC (TURANDOT) [43] | A 12-week induction trial in 357 patients with moderate to severe disease, naive or biologic-experienced; 5 arms (225 mg/75 mg/22.5 mg/7.5 mg/placebo) (1:1:1:1:1) | A higher remission rate in all active groups than in the placebo group except for the 225 mg group (5.7%/15.5%/16.7%/11.3%/2.7%) |
| II, CD (OPERA) [44] | A 12-week induction trial in 223 patients with moderate to severe disease, naive or biologic-experienced; 4 arms (225 mg/75 mg/22.5 mg/placebo) (1:1:1:1) | No significant difference in the remission rate at 12 weeks compared with placebo (29.6%/28.5%/26.8%/23.0% | |
| PPD4 inhibitors | |||
| Tetomilast (PDE4) | II, UC [45] | An 8-week induction trial in 186 patients with mildly to moderately active disease, biologic-naive. 3 arms: 50 mg/25 mg/placebo (1:1:1) | No statistical difference in success rates (improvement as defined by a reduction in DAI>3 at week 8) between the active groups and the placebo group (21%/16%/7%) |
| Apremilast (PDE4) | II, UC [46] | A 12-week induction trial in 170 patients with moderate to severe disease, biologic-naive. 3 arms: 40 mg/30 mg/placebo (1:1:1) | Clinical remission was significantly higher than the placebo group for the 30 mg group (31.6% vs. 12.1%), but not for the 40 mg group (21.8%) |
| SMAD7 blockers | |||
| Mongersen | II, CD [47] | A 2-week induction trial in 166 patients with moderate to severe disease, biologic-naive. 4 arms: 160 mg/40 mg/10 mg/placebo (1:1:1:1) | Clinical remission was significantly higher in the active groups compared with the placebo group, except for the 10 mg group (65%/55%/12%/10%) |
| Other SMDs | |||
| Vidofludimus calcium (DHODH) | II, UC (CALDOSE-1) [48] | A 10-week dose finding trial in 240 patients with (expected) moderate to severe disease, naive or biologic-experienced; 4 arms: 10 mg/30 mg/45 mg/placebo | Ongoing, Estimated Primary Completion Date: July 2022 |
| ABX464 (microRNA-124) | II, CD [49] | A 16-week safety and efficacy trial in 30 patients with (expected) moderate to severe disease, naive or biologic-experienced; 2 arms: 50 mg/placebo | Ongoing, Estimated Primary Completion Date: July 2021 |
| IIa, UC [50] | An 8-week induction trial in 29 patients with moderate to severe disease, naive or biologic-experienced; 2 arms: 50 mg/placebo | No significant difference between the active group and the placebo group in clinical remission at 8 weeks (35.0% vs. 11.1%) | |
| BT-11 (LANCL2) | II, UC [51] | A 12-week efficacy and safety trial in 198 patients with (expected) mildly to moderately active disease, naive or biologic-experienced; 3 arms: 1000 mg/500 mg/placebo | Study completed in May 2021; results not published yet |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ben Ghezala, I.; Charkaoui, M.; Michiels, C.; Bardou, M.; Luu, M. Small Molecule Drugs in Inflammatory Bowel Diseases. Pharmaceuticals 2021, 14, 637. https://doi.org/10.3390/ph14070637
Ben Ghezala I, Charkaoui M, Michiels C, Bardou M, Luu M. Small Molecule Drugs in Inflammatory Bowel Diseases. Pharmaceuticals. 2021; 14(7):637. https://doi.org/10.3390/ph14070637
Chicago/Turabian StyleBen Ghezala, Inès, Maëva Charkaoui, Christophe Michiels, Marc Bardou, and Maxime Luu. 2021. "Small Molecule Drugs in Inflammatory Bowel Diseases" Pharmaceuticals 14, no. 7: 637. https://doi.org/10.3390/ph14070637
APA StyleBen Ghezala, I., Charkaoui, M., Michiels, C., Bardou, M., & Luu, M. (2021). Small Molecule Drugs in Inflammatory Bowel Diseases. Pharmaceuticals, 14(7), 637. https://doi.org/10.3390/ph14070637