
Hacking Pancreatic Cancer: Present and Future of Personalized Medicine

 ,
,  ,
,  , and
, and
Abstract
:1. Introduction
2. Biomolecular Landscape of Pancreatic Cancer
3. DNA Damage Response and Repair Genes (DDR)
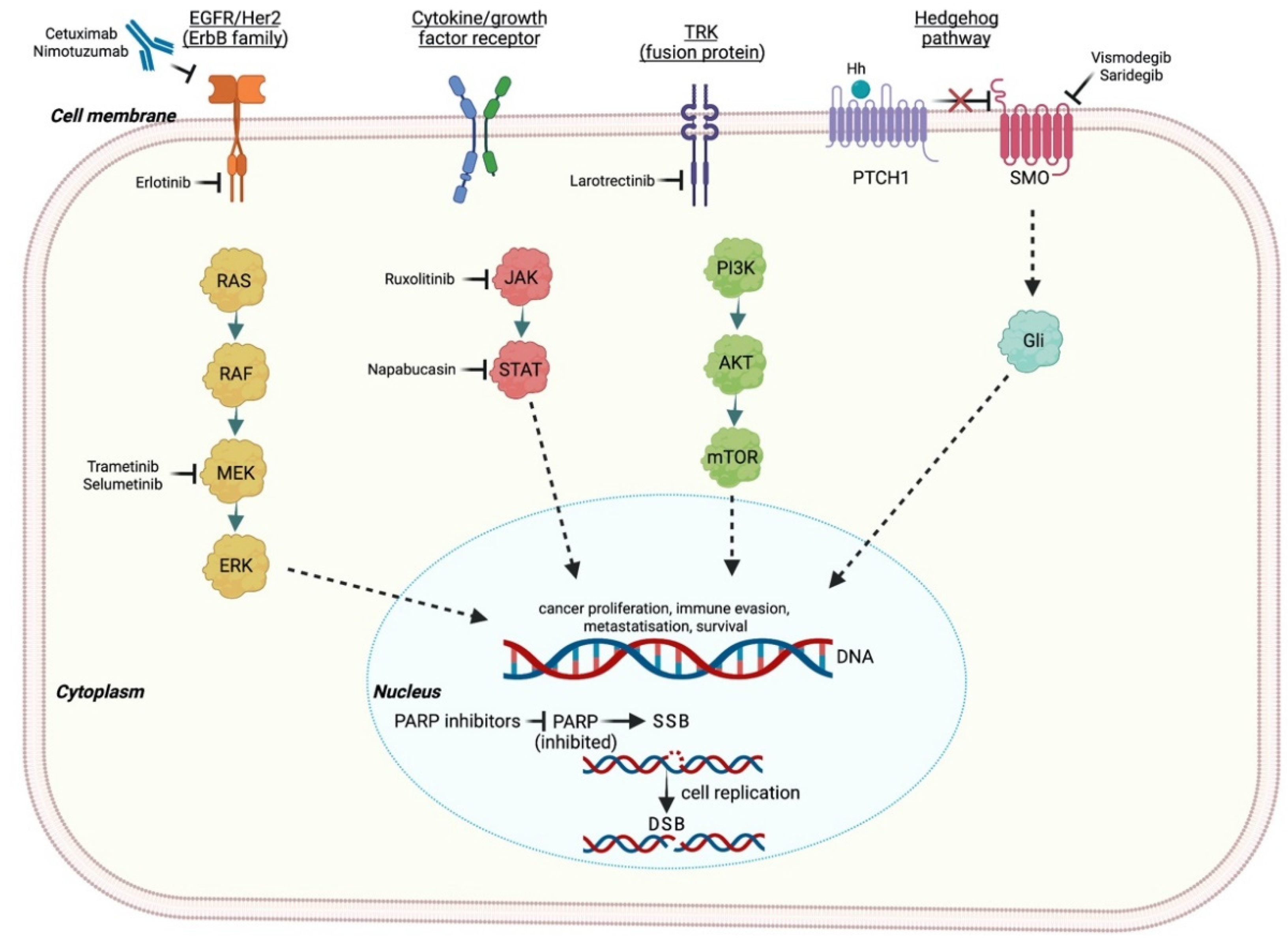
3.1. PARP Inhibitors
3.1.1. Olaparib
3.1.2. Veliparib
3.1.3. Rucaparib
3.1.4. Talazoparib
3.2. Further DDR Targeting Agents
4. MEK Inhibitors
5. EGFR Inhibitors
6. KRAS Targeting Agents
7. Targeting Tumor Microenvironment: The First Yet Harder Obstacle to Overcome
Hedgehog Pathway
8. Microenvironment Targeting Agents
9. JAK/STAT Inhibitors
10. NTRK Inhibitors
11. Targeting Cancer Metabolism
Hydroxychloroquine
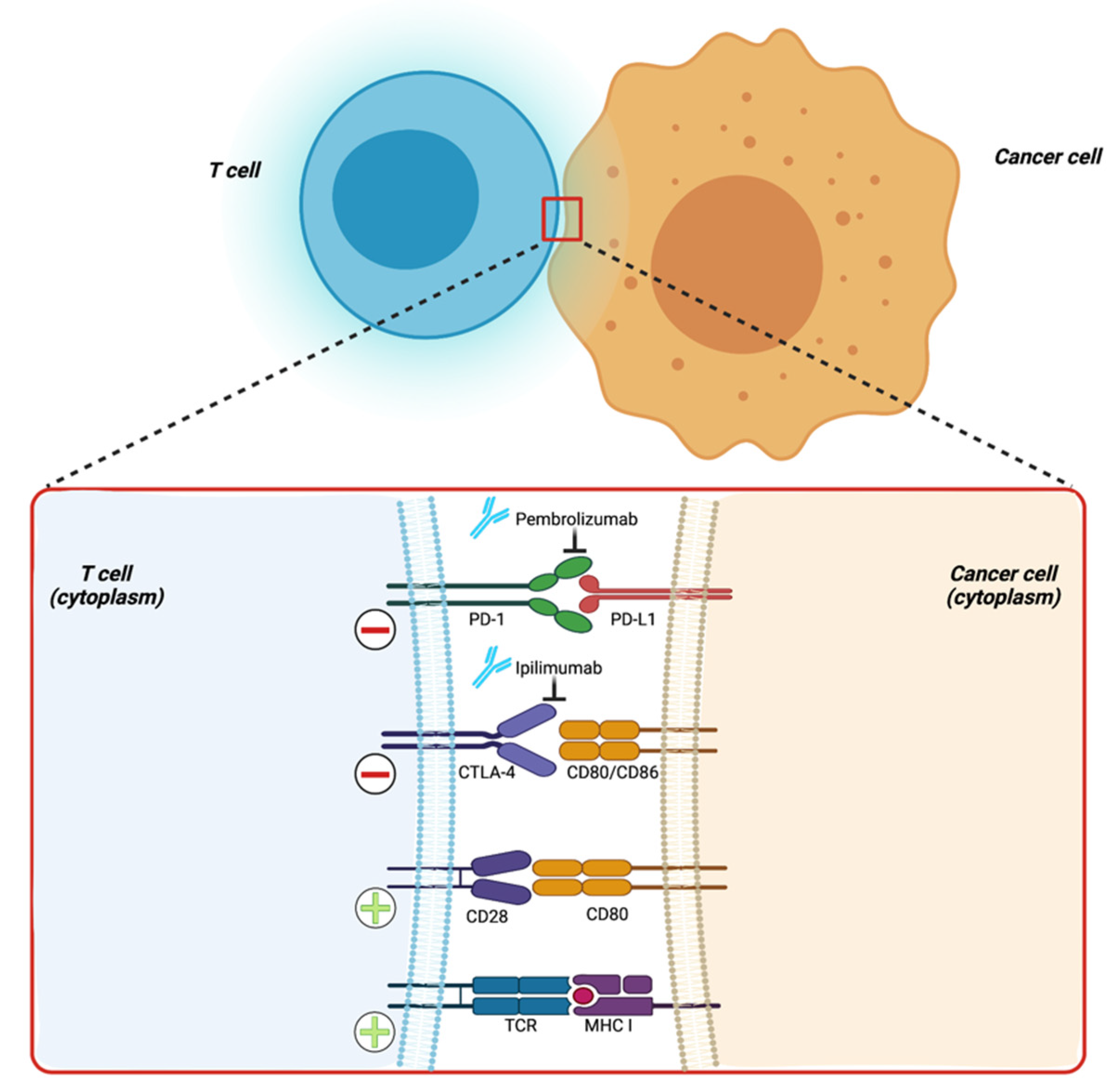
12. Immunotherapy
13. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McGuigan, A.; Kelly, P.; Turkington, R.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol. 2018, 24, 4846–4861. [Google Scholar] [CrossRef]
- Vincent, A.; Herman, J.; Schulick, R.; Hruban, R.H.; Goggins, M. Pancreatic cancer. Lancet 2011, 378, 607–620. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [Green Version]
- Pishvaian, M.J.; Bender, R.J.; Halverson, D.; Rahib, L.; Hendifar, A.E.; Mikhail, S.; Chung, V.; Picozzi, V.J.; Sohal, D.; Blais, E.M.; et al. Molecular Profiling of Patients with Pancreatic Cancer: Initial Results from the Know Your Tumor Initiative. Clin. Cancer Res. 2018, 24, 5018–5027. [Google Scholar] [CrossRef] [Green Version]
- Aung, K.L.; Fischer, S.E.; Denroche, R.E.; Jang, G.-H.; Dodd, A.; Creighton, S.; Southwood, B.; Liang, S.-B.; Chadwick, D.; Zhang, A.; et al. Genomics-Driven Precision Medicine for Advanced Pancreatic Cancer: Early Results from the COMPASS Trial. Clin. Cancer Res. 2017, 24, 1344–1354. [Google Scholar] [CrossRef] [Green Version]
- Pishvaian, M.J.; Blais, E.M.; Brody, J.R.; Lyons, E.; DeArbeloa, P.; Hendifar, A.; Mikhail, S.; Chung, V.; Sahai, V.; Sohal, D.P.S.; et al. Overall survival in patients with pancreatic cancer receiving matched therapies following molecular profiling: A retrospective analysis of the Know Your Tumor registry trial. Lancet Oncol. 2020, 21, 508–518. [Google Scholar] [CrossRef]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: Integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.-P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.; et al. Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair–Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Andre, T.; Shiu, K.-K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.J.A.; Smith, D.M.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab versus chemotherapy for microsatellite instability-high/mismatch repair deficient metastatic colorectal cancer: The phase 3 KEYNOTE-177 Study. J. Clin. Oncol. 2020, 38, LBA4. [Google Scholar] [CrossRef]
- Raphael, B.J.; Hruban, R.H.; Aguirre, A.J.; Moffitt, R.; Yeh, J.J.; Stewart, C.; Robertson, A.G.; Cherniack, A.D.; Gupta, M.; Getz, G.; et al. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203.e13. [Google Scholar] [CrossRef] [Green Version]
- Al-Hader, A.; Al-Rohil, R.N.; Han, H.; Von Hoff, D. Pancreatic acinar cell carcinoma: A review on molecular profiling of patient tumors. World J. Gastroenterol. 2017, 23, 7945–7951. [Google Scholar] [CrossRef]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Hoeijmakers, J.H.J. Genome maintenance mechanisms for preventing cancer. Nat. Cell Biol. 2001, 411, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Lal, G.; Liu, G.; Schmocker, B.; Kaurah, P.; Ozcelik, H.; Narod, A.S.; Redston, M.; Gallinger, S. Inherited predisposition to pancreatic adenocarcinoma: Role of family history and germ-line p16, BRCA1, and BRCA2 mutations. Cancer Res. 2000, 60, 409–416. [Google Scholar]
- Roberts, N.; Jiao, Y.; Yu, J.; Kopelovich, L.; Petersen, G.M.; Bondy, M.L.; Gallinger, S.; Schwartz, A.G.; Syngal, S.; Cote, M.L.; et al. ATM Mutations in Patients with Hereditary Pancreatic Cancer. Cancer Discov. 2011, 2, 41–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, S.; Hruban, R.H.; Kamiyama, M.; Borges, M.; Zhang, X.; Parsons, D.W.; Lin, J.C.-H.; Palmisano, E.; Brune, K.; Jaffee, E.M.; et al. Exomic Sequencing Identifies PALB2 as a Pancreatic Cancer Susceptibility Gene. Science 2009, 324, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguirre, A.J.; Nowak, J.A.; Camarda, N.; Moffitt, R.A.; Ghazani, A.A.; Hazar-Rethinam, M.; Raghavan, S.; Kim, J.; Brais, L.K.; Ragon, D.; et al. Real-time Genomic Characterization of Advanced Pancreatic Cancer to Enable Precision Medicine. Cancer Discov. 2018, 8, 1096–1111. [Google Scholar] [CrossRef] [Green Version]
- Heeke, A.L.; Pishvaian, M.J.; Lynce, F.; Xiu, J.; Brody, J.R.; Chen, W.-J.; Baker, T.M.; Marshall, J.L.; Isaacs, C. Prevalence of Homologous Recombination–Related Gene Mutations Across Multiple Cancer Types. JCO Precis. Oncol. 2018, 2018, 1–13. [Google Scholar] [CrossRef]
- Russell, R.; Perkhofer, L.; Liebau, S.; Lin, Q.; Lechel, A.; Feld, F.M. Loss of ATM accelerates pancreatic cancer formation and epithelial-mesenchymal tran- sition. Nat. Commun. 2015, 6, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamberti, G.; Andrini, E.; Sisi, M.; Di Federico, A.; Ricciuti, B. Targeting DNA damage response and repair genes to en-hance anticancer immunotherapy: Rationale and clinical implication. Future Oncol. 2020, 16, 1751–1766. [Google Scholar]
- O’Neil, N.; Bailey, M.L.; Hieter, P. Synthetic lethality and cancer. Nat. Rev. Genet. 2017, 18, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A.R.; Nussenzweig, A.R.C.A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Scott, C.L.; Swisher, E.M.; Kaufmann, S. Poly (ADP-Ribose) Polymerase Inhibitors: Recent Advances and Future Development. J. Clin. Oncol. 2015, 33, 1397–1406. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmaña, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib Monotherapy in Patients With Advanced Cancer and a Germline BRCA1/2 Mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Bendell, J.; O’Reilly, E.M.; Middleton, M.R.; Chau, I.; Hochster, H.; Fielding, A.; Burke, W.; Burris, I.H. Phase I study of olaparib plus gemcitabine in patients with advanced solid tumours and comparison with gemcitabine alone in patients with locally advanced/metastatic pancreatic cancer. Ann. Oncol. 2015, 26, 804–811. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Myzak, M.C.; Johnson, B.A.; De Jesus-Acosta, A.; Le, D.T.; Jaffee, E.; Azad, N.S.; Donehower, R.C.; Zheng, L.; Oberstein, P.E.; et al. Olaparib in combination with irinotecan, cisplatin, and mitomycin C in patients with advanced pancreatic cancer. Oncotarget 2017, 8, 44073–44081. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 4459–4471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pommier, Y.; O’Connor, M.J.; De Bono, J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med. 2016, 8, 362ps17. [Google Scholar] [CrossRef]
- Lowery, M.A.; Kelsen, D.P.; Capanu, M.; Smith, S.C.; Lee, J.W.; Stadler, Z.K.; Moore, M.J.; Kindler, H.L.; Golan, T.; Segal, A.; et al. Phase II trial of veliparib in patients with previously treated BRCA-mutated pancreas ductal adenocarcinoma. Eur. J. Cancer 2018, 89, 19–26. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, E.M.; Lee, J.W.; Zalupski, M.; Capanu, M.; Park, J.; Golan, T.; Tahover, E.; Lowery, M.; Chou, J.F.; Sahai, V.; et al. Randomized, Multicenter, Phase II Trial of Gemcitabine and Cisplatin With or Without Veliparib in Patients With Pancreas Adenocarcinoma and a Germline BRCA/PALB2 Mutation. J. Clin. Oncol. 2020, 38, 1378–1388. [Google Scholar] [CrossRef] [PubMed]
- Chiorean, E.G.; A Guthrie, K.; Philip, P.A.; Swisher, E.M.; Jalikis, F.; Pishvaian, M.J.; Berlin, J.; Noel, M.S.; Suga, J.M.; Garrido-Laguna, I.; et al. Randomized phase II study of second-line modified FOLFIRI with PARP inhibitor ABT-888 (Veliparib) (NSC-737664) versus FOLFIRI in metastatic pancreatic cancer (mPC): SWOG S1513. J. Clin. Oncol. 2019, 37, 4014. [Google Scholar] [CrossRef]
- Shroff, R.T.; Hendifar, A.; McWilliams, R.R.; Geva, R.; Epelbaum, R.; Rolfe, L.; Goble, S.; Lin, K.K.; Biankin, A.V.; Giordano, H.; et al. Rucaparib Monotherapy in Patients with Pancreatic Cancer and a Known Deleterious BRCA Mutation. JCO Precis. Oncol. 2018, 2, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Binder, K.A.R.; Mick, R.; O’Hara, M.; Teitelbaum, U.; Karasic, T.; Schneider, C.; O’Dwyer, P.J.; Carpenter, E.; Pantel, A.; Makvandi, M.; et al. Abstract CT234: A Phase II, single arm study of maintenance rucaparib in patients with platinum-sensitive advanced pancreatic cancer and a pathogenic germline or somatic mutation in BRCA1, BRCA2 or PALB2. Cancers 2019, 11, 1980. [Google Scholar]
- De Bono, J.; Ramanathan, R.K.; Mina, L.; Chugh, R.; Glaspy, J.; Rafii, S.; Kaye, S.; Sachdev, J.; Heymach, J.; Smith, D.; et al. Phase I, Dose-Escalation, Two-Part Trial of the PARP Inhibitor Talazoparib in Patients with Advanced Germline BRCA1/2 Mutations and Selected Sporadic Cancers. Cancer Discov. 2017, 7, 620–629. [Google Scholar] [CrossRef] [Green Version]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.-K.; Hsu, J.-M.; Hsu, J.L.; Yu, W.-H.; Du, Y.; Lee, H.-H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef] [Green Version]
- Robillard, L.; Nguyen, M.; Loehr, A.; Orsulic, S.; Kristeleit, R.S.; Lin, K.; Raponi, M.; Harding, T.C.; Simmons, A.D. Abstract 3650: Preclinical evaluation of the PARP inhibitor rucaparib in combination with PD-1 and PD-L1 inhibition in a syngeneic BRCA1 mutant ovarian cancer model. Immunology 2017, 77, 3650. [Google Scholar]
- Sun, C.; Fang, Y.; Yin, J.; Chen, J.; Ju, Z.; Zhang, D.; Chen, X.; Vellano, C.P.; Jeong, K.J.; Ng, P.K.-S.; et al. Rational combination therapy with PARP and MEK inhibitors capitalizes on therapeutic liabilities in RAS mutant cancers. Sci. Transl. Med. 2017, 9, eaal5148. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, C.C.; Mateo, J.; Walsh, M.F.; DeSarkar, N.; Abida, W.; Beltran, H. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N. Engl. J. Med. 2016, 375, 443–453. [Google Scholar] [CrossRef]
- Armstrong, S.A.; Schultz, C.W.; Azimi-Sadjadi, A.; Brody, J.R.; Pishvaian, M.J. ATM Dysfunction in Pancreatic Adenocarcinoma and Associated Therapeutic Implications. Mol. Cancer Ther. 2019, 18, 1899–1908. [Google Scholar] [CrossRef] [Green Version]
- Choi, M.; Kipps, T.; Kurzrock, R. ATM Mutations in Cancer: Therapeutic Implications. Mol. Cancer Ther. 2016, 15, 1781–1791. [Google Scholar] [CrossRef] [Green Version]
- Perkhofer, L.; Schmitt, A.; Carrasco, M.C.R.; Ihle, M.; Hampp, S.; Ruess, D.A.; Hessmann, E.; Russell, R.; Lechel, A.; Azoitei, N.; et al. ATM Deficiency Generating Genomic Instability Sensitizes Pancreatic Ductal Adenocarcinoma Cells to Therapy-Induced DNA Damage. Cancer Res. 2017, 77, 5576–5590. [Google Scholar] [CrossRef] [Green Version]
- Wallez, Y.; Dunlop, C.R.; Johnson, I.T.; Koh, S.-B.; Fornari, C.; Yates, J.W.; Fernández, S.B.D.Q.; Lau, A.; Richards, F.M.; Jodrell, D.I. The ATR Inhibitor AZD6738 Synergizes with Gemcitabine In Vitro and In Vivo to Induce Pancreatic Ductal Adenocarcinoma Regression. Mol. Cancer Ther. 2018, 17, 1670–1682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, M.; Zhao, T.; Ma, L.; Guo, Y. CHK1 inhibition sensitizes pancreatic cancer cells to gemcitabine via promoting CDK-dependent DNA damage and ribonucleotide reductase downregulation. Oncol. Rep. 2017, 39, 1322–1330. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, M.J.; Raleigh, J.M.; Verkade, H.; Nurse, P. Chk1 is a wee1 kinase in the G2 DNA damage checkpoint inhibiting cdc2 by Y15 phosphorylation. EMBO J. 1997, 16, 545–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- RajeshKumar, N.; De Oliveira, E.; Ottenhof, N.; Watters, J.; Brooks, D.; DeMuth, T.; Shumway, S.D.; Mizuarai, S.; Hirai, H.; Maitra, A.; et al. MK-1775, a Potent Wee1 Inhibitor, Synergizes with Gemcitabine to Achieve Tumor Regressions, Selectively in p53-Deficient Pancreatic Cancer Xenografts. Clin. Cancer Res. 2011, 17, 2799–2806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lal, S.; Zarei, M.; Chand, S.N.; Dylgjeri, E.; Mambelli-Lisboa, N.C.; Pishvaian, M.J.; Yeo, C.J.; Winter, J.M.; Brody, J.R. WEE1 inhibition in pancreatic cancer cells is dependent on DNA repair status in a context dependent manner. Sci. Rep. 2016, 6, 33323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leijen, S.; Van Geel, R.M.; Pavlick, A.C.; Tibes, R.; Rosen, L.; Razak, A.R.A.; Lam, R.; Demuth, T.; Rose, S.; Lee, M.A.; et al. Phase I Study Evaluating WEE1 Inhibitor AZD1775 As Monotherapy and in Combination With Gemcitabine, Cisplatin, or Carboplatin in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2016, 34, 4371–4380. [Google Scholar] [CrossRef] [PubMed]
- Beijnen, J.H.; Schellens, J.H. Abrogation of the G2 Checkpoint by Inhibition of Wee-1 Kinase Results in Sensitization of p53-Deficient Tumor Cells to DNA-Damaging Agents. Curr. Clin. Pharmacol. 2010, 5, 186–191. [Google Scholar] [CrossRef]
- Cuneo, K.C.; Morgan, M.A.; Sahai, V.; Schipper, M.J.; Parsels, L.A.; Parsels, J.D.; Devasia, T.; Al-Hawaray, M.; Cho, C.S.; Nathan, H.; et al. Dose Escalation Trial of the Wee1 Inhibitor Adavosertib (AZD1775) in Combination With Gemcitabine and Radiation for Patients With Locally Advanced Pancreatic Cancer. J. Clin. Oncol. 2019, 37, 2643–2650. [Google Scholar] [CrossRef] [PubMed]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Infante, J.R.; Fecher, A.L.; Falchook, G.S.; Nallapareddy, S.; Gordon, M.S.; Becerra, C.; DeMarini, D.J.; Cox, D.S.; Xu, Y.; Morris, S.R.; et al. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: A phase 1 dose-escalation trial. Lancet Oncol. 2012, 13, 773–781. [Google Scholar] [CrossRef]
- Infante, J.R.; Somer, B.G.; Park, J.O.; Li, C.-P.; Scheulen, M.E.; Kasubhai, S.M.; Oh, D.-Y.; Liu, Y.; Redhu, S.; Steplewski, K.; et al. A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Eur. J. Cancer 2014, 50, 2072–2081. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Hidalgo, M.; Canon, J.; Macarulla, T.; Bazin, I.; Poddubskaya, E.; Manojlovic, N.; Radenkovic, D.; Verslype, C.; Raymond, E.; et al. Phase I/II trial of pimasertib plus gemcitabine in patients with metastatic pancreatic cancer. Int. J. Cancer 2018, 143, 2053–2064. [Google Scholar] [CrossRef] [PubMed]
- Mirzoeva, O.K.; Collisson, E.A.; Schaefer, P.M.; Hann, B.; Hom, Y.K.; Ko, A.H.; Korn, W.M. Subtype-Specific MEK-PI3 Kinase Feedback as a Therapeutic Target in Pancreatic Adenocarcinoma. Mol. Cancer Ther. 2013, 12, 2213–2225. [Google Scholar] [CrossRef] [Green Version]
- Ko, A.H.; Bekaii-Saab, T.; Van Ziffle, J.; Mirzoeva, O.M.; Joseph, N.M.; Talasaz, A.; Kuhn, P.; Tempero, M.A.; Collisson, E.A.; Kelley, R.K.; et al. A Multicenter, Open-Label Phase II Clinical Trial of Combined MEK plus EGFR Inhibition for Chemotherapy-Refractory Advanced Pancreatic Adenocarcinoma. Clin. Cancer Res. 2016, 22, 61–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, V.; McDonough, S.; Philip, P.A.; Cardin, D.; Wang-Gillam, A.; Hui, L.; Tejani, M.A.; Seery, T.E.; Dy, I.A.; Al Baghdadi, T.; et al. Effect of Selumetinib and MK-2206 vs Oxaliplatin and Fluorouracil in Patients With Metastatic Pancreatic Cancer After Prior Therapy. JAMA Oncol. 2017, 3, 516–522. [Google Scholar] [CrossRef]
- Uegaki, K.; Nio, Y.; Inoue, Y.; Minari, Y.; Sato, Y.; Song, M.M.; Dong, M.; Tamura, K.; Uegaki, K.; Nio, Y.; et al. Clinicopathological significance of epidermal growth factor and its receptor in human pancreatic cancer. Anticancer. Res. 1998, 17, 3841–3847. [Google Scholar]
- Moore, M.J.; Goldstein, D.; Hamm, J.; Figer, A.; Hecht, J.R.; Gallinger, S.; Au, H.J.; Murawa, P.; Walde, D.; Wolff, R.A.; et al. Erlotinib Plus Gemcitabine Compared With Gemcitabine Alone in Patients With Advanced Pancreatic Cancer: A Phase III Trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2007, 25, 1960–1966. [Google Scholar] [CrossRef]
- Da Cunha Santos, G.; Dhani, N.; Tu, D.; Chin, K.; Ludkovski, O.; Kamel-Reid, S.; Squire, J.; Parulekar, W.; Moore, M.J.; Tsao, M.S. Molecular predictors of outcome in a phase 3 study of gemcitabine and erlotinib therapy in patients with advanced pancreatic cancer: National Cancer Institute of Canada Clinical Trials Group Study PA. Cancer 2010, 116, 5599–5607. [Google Scholar] [CrossRef] [PubMed]
- Hammel, P.; Huguet, F.F.; Van Laethem, J.-L.; Goldstein, D.D.; Glimelius, B.; Artru, P.P.; Borbath, I.; Bouché, O.; Shannon, J.J.; André, T.; et al. Effect of Chemoradiotherapy vs Chemotherapy on Survival in Patients With Locally Advanced Pancreatic Cancer Controlled After 4 Months of Gemcitabine With or Without Erlotinib. JAMA 2016, 315, 1844–1853. [Google Scholar] [CrossRef]
- Wang, J.P.; Wu, C.-Y.; Yeh, Y.-C.; Shyr, Y.-M.; Wu, Y.-Y.; Kuo, C.-Y.; Hung, Y.-P.; Chen, M.-H.; Lee, W.-P.; Luo, J.-C.; et al. Erlotinib is effective in pancreatic cancer with epidermal growth factor receptor mutations: A randomized, open-label, prospective trial. Oncotarget 2015, 6, 18162–18173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, N.I.; Prewett, M.; Zuklys, K.; Rockwell, P.; Mendelsohn, J. Biological efficacy of a chimeric antibody to the epidermal growth factor receptor in a human tumor xenograft model. Clin. Cancer Res. 1995, 1, 1311–1318. [Google Scholar]
- Forster, T.; Huettner, F.J.; Springfeld, C.; Loehr, M.; Kalkum, E.; Hackbusch, M.; Hackert, T.; Diener, M.K.; Probst, P. Cetuximab in Pancreatic Cancer Therapy: A Systematic Review and Meta-Analysis. Oncology 2020, 98, 53–60. [Google Scholar] [CrossRef]
- Su, D.; Jiao, S.; Wang, L.-J.; Shi, W.-W.; Long, Y.-Y.; Li, J.; Bai, L. Efficacy of nimotuzumab plus gemcitabine usage as first-line treatment in patients with advanced pancreatic cancer. Tumor Biol. 2013, 35, 2313–2318. [Google Scholar] [CrossRef]
- Schultheis, B.; Reuter, D.; Ebert, M.P.; Siveke, J.; Kerkhoff, A.; Berdel, W.E.; Hofheinz, R.; Behringer, D.M.; Schmidt, W.E.; Goker, E.; et al. Gemcitabine combined with the monoclonal antibody nimotuzumab is an active first-line regimen inKRAS wildtype patients with locally advanced or metastatic pancreatic cancer: A multicenter, randomized phase IIb study. Ann. Oncol. 2017, 28, 2429–2435. [Google Scholar] [CrossRef] [PubMed]
- di Magliano, M.P.; Logsdon, C.D. Roles for KRAS in pancreatic tumor development and progression. Gastroenterology 2013, 144, 1220–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryant, K.L.; Mancias, J.D.; Kimmelman, A.C.; Der, C.J. KRAS: Feeding pancreatic cancer proliferation. Trends Biochem. Sci. 2014, 39, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Molina-Arcas, M.; Samani, A.; Downward, J. Drugging the Undruggable: Advances on RAS Targeting in Cancer. Genes 2021, 12, 899. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Van De Velde, H.; Karasek, P.; Oettle, H.; Vervenne, W.; Szawlowski, A.; Schoffski, P.; Post, S.; Verslype, C.; Neumann, H.; et al. Phase III Trial of Gemcitabine Plus Tipifarnib Compared with Gemcitabine Plus Placebo in Advanced Pancreatic Cancer. J. Clin. Oncol. 2004, 22, 1430–1438. [Google Scholar] [CrossRef]
- Laheru, D.; Shah, P.; RajeshKumar, N.V.; McAllister, F.; Taylor, G.; Goldsweig, H.; Le, D.T.; Donehower, R.C.; Jimeno, A.; Linden, S.; et al. Integrated preclinical and clinical development of S-trans, trans-farnesylthiosalicylic acid (FTS, Salirasib) in pancreatic cancer. Investig. New Drugs 2012, 30, 2391–2399. [Google Scholar] [CrossRef] [Green Version]
- Furuse, J.; Kurata, T.; Okano, N.; Fujisaka, Y.; Naruge, D.; Shimizu, T.; Kitamura, H.; Iwasa, T.; Nagashima, F.; Nakagawa, K. An early clinical trial of Salirasib, an oral RAS inhibitor, in Japanese patients with relapsed/refractory solid tumors. Cancer Chemother. Pharmacol. 2018, 82, 511–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrow, B.; Albo, D.; Berger, D.H. The Role of the Tumor Microenvironment in the Progression of Pancreatic Cancer. J. Surg. Res. 2008, 149, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Fearon, D.T. The Carcinoma-Associated Fibroblast Expressing Fibroblast Activation Protein and Escape from Immune Surveillance. Cancer Immunol. Res. 2014, 2, 187–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marigo, V.; Tabin, C.J. Regulation of patched by sonic hedgehog in the developing neural tube. Proc. Natl. Acad. Sci. USA 1996, 93, 9346–9351. [Google Scholar] [CrossRef] [Green Version]
- Holman, K.W.; Jones, R.J.; Marian, A.; Cundiff, S.T.; Ye, J. Intensity-related dynamics of femtosecond frequency combs. Opt. Lett. 2003, 28, 851–853. [Google Scholar] [CrossRef] [PubMed]
- Niyaz, M.; Khan, M.S.; Wani, R.A.; Shah, O.J.; Mudassar, S. Sonic Hedgehog Protein is Frequently Up-Regulated in Pancreatic Cancer Compared to Colorectal Cancer. Pathol. Oncol. Res. 2018, 26, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Takebe, N.; Miele, L.; Harris, P.J.; Jeong, W.; Bando, H.; Kahn, M.G.; Yang, S.X.; Ivy, S.P. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: Clinical update. Nat. Rev. Clin. Oncol. 2015, 12, 445–464. [Google Scholar] [CrossRef]
- Tang, D.; Wang, D.; Yuan, Z.; Xue, X.; Zhang, Y.; An, Y.; Chen, J.; Tu, M.; Lu, Z.; Wei, J.; et al. Persistent activation of pancreatic stellate cells creates a microenvironment favorable for the malignant behavior of pancreatic ductal adenocarcinoma. Int. J. Cancer 2012, 132, 993–1003. [Google Scholar] [CrossRef]
- Bailey, J.M.; Swanson, B.J.; Hamada, T.; Eggers, J.P.; Singh, P.K.; Caffery, T.; Ouellette, M.M.; Hollingsworth, M.A. Sonic Hedgehog Promotes Desmoplasia in Pancreatic Cancer. Clin. Cancer Res. 2008, 14, 5995–6004. [Google Scholar] [CrossRef] [Green Version]
- Catenacci, D.V.T.; Junttila, M.R.; Karrison, T.; Bahary, N.; Horiba, M.N.; Nattam, S.R.; Marsh, R.; Wallace, J.; Kozloff, M.; Rajdev, L.; et al. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients With Metastatic Pancreatic Cancer. J. Clin. Oncol. 2015, 33, 4284–4292. [Google Scholar] [CrossRef] [PubMed]
- Ko, A.H.; LoConte, N.; Tempero, M.A.; Walker, E.; Kelley, R.K.; Lewis, S.; Chang, W.-C.; Kantoff, E.; Vannier, M.W.; Catenacci, D.V.; et al. A Phase I Study of FOLFIRINOX Plus IPI-926, a Hedgehog Pathway Inhibitor, for Advanced Pancreatic Adenocarcinoma. Pancreas 2016, 45, 370–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madden, J.I. Infinity reports update from Phase 2 study of Saridegib plus Gemcitabine in patients with metastatic pancreatic cancer. Infin. Pharm. 2012, 2012–2014. [Google Scholar]
- Hingorani, S.; Zheng, L.; Bullock, A.J.; Seery, T.E.; Harris, W.P.; Sigal, D.S.; Braiteh, F.; Ritch, P.S.; Zalupski, M.M.; Bahary, N.; et al. HALO 202: Randomized Phase II Study of PEGPH20 Plus Nab-Paclitaxel/Gemcitabine Versus Nab-Paclitaxel/Gemcitabine in Patients With Untreated, Metastatic Pancreatic Ductal Adenocarcinoma. J. Clin. Oncol. 2018, 36, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, R.K.; McDonough, S.L.; Philip, P.A.; Hingorani, S.; Lacy, J.; Kortmansky, J.S.; Thumar, J.; Chiorean, E.G.; Shields, A.F.; Behl, D.; et al. Phase IB/II Randomized Study of FOLFIRINOX Plus Pegylated Recombinant Human Hyaluronidase Versus FOLFIRINOX Alone in Patients With Metastatic Pancreatic Adenocarcinoma: SWOG S1313. J. Clin. Oncol. 2019, 37, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. TGFβ in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [Green Version]
- Melisi, D.; Garcia-Carbonero, R.; Macarulla, T.; Pezet, D.; Deplanque, G.; Fuchs, M.; Trojan, J.; Oettle, H.; Kozloff, M.; Cleverly, A.; et al. Galunisertib plus gemcitabine vs. gemcitabine for first-line treatment of patients with unresectable pancreatic cancer. Br. J. Cancer 2018, 119, 1208–1214. [Google Scholar] [CrossRef] [Green Version]
- Thomas, S.J.; A Snowden, J.; Zeidler, M.; Danson, S. The role of JAK/STAT signalling in the pathogenesis, prognosis and treatment of solid tumours. Br. J. Cancer 2015, 113, 365–371. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Talukder, A.; Savage, N.M.; Singh, N.; Liu, K. JAK-STAT-mediated chronic inflammation impairs cytotoxic T lymphocyte activation to decrease anti-PD-1 immunotherapy efficacy in pancreatic cancer. OncoImmunology 2017, 6, e1291106. [Google Scholar] [CrossRef] [Green Version]
- Groner, B.; von Manstein, V. Jak Stat signaling and cancer: Opportunities, benefits and side effects of targeted inhibition. Mol. Cell. Endocrinol. 2017, 451, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Bekaii-Saab, T.S.; Starodub, A.; El-Rayes, B.F.; Shahda, S.; O’Neil, B.H.; Noonan, A.M.; Shaib, W.L.; Hanna, W.T.; Mikhail, S.; Neki, A.S.; et al. Phase 1b/2 trial of cancer stemness inhibitor napabucasin (NAPA) + nab-paclitaxel (nPTX) and gemcitabine (Gem) in metastatic pancreatic adenocarcinoma (mPDAC). J. Clin. Oncol. 2018, 36, 4110. [Google Scholar] [CrossRef]
- Sonbol, M.; Ahn, D.H.; Goldstein, D.; Okusaka, T.; Tabernero, J.; Macarulla, T.; Reni, M.; Li, C.-P.; O’Neil, B.; Van Cutsem, E.; et al. CanStem111P trial: A Phase III study of napabucasin plus nab-paclitaxel with gemcitabine. Future Oncol. 2019, 15, 1295–1302. [Google Scholar] [CrossRef]
- Bekaii-Saab, T.; Okusaka, T.; Goldstein, D.; O’Neill, B.; Tabernero, J.; Li, C.; Reni, M.; Oh, C.; Borodyansky, L.; Van Cutsem, E. CanStem111P trial: A Phase 3 Study of napabucasin (NAPA) plus nab-paclitaxel (nPTX) with gemcitabine (Gem) in adult patients with metastatic pancreatic adenocarcinoma (mPDAC)—Trial in progress. Ann. Oncol. 2018, 29 (Suppl. 5), V51–V52. [Google Scholar] [CrossRef]
- Hurwitz, H.; Van Cutsem, E.; Bendell, J.; Hidalgo, M.; Li, C.-P.; Salvo, M.G.; Macarulla, T.; Sahai, V.; Sama, A.; Greeno, E.; et al. Ruxolitinib + capecitabine in advanced/metastatic pancreatic cancer after disease progression/intolerance to first-line therapy: JANUS 1 and 2 randomized phase III studies. Investig. New Drugs 2018, 36, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Shahda, S.; Beck, T.; Uppal, N.; Cohen, S.J.; Donehower, R.; Gabayan, A.E.; Assad, A.; Switzky, J.; Zhen, H.; et al. A Phase Ib/II Study of the JAK1 Inhibitor, Itacitinib, plus nab -Paclitaxel and Gemcitabine in Advanced Solid Tumors. Oncologist 2019, 24, 14. [Google Scholar] [CrossRef]
- Khotskaya, Y.B.; Holla, V.R.; Farago, A.F.; Shaw, K.R.M.; Meric-Bernstam, F.; Hong, D.S. Targeting TRK family proteins in cancer. Pharmacol. Ther. 2017, 173, 58–66. [Google Scholar] [CrossRef]
- Nakagawara, A. Trk receptor tyrosine kinases: A bridge between cancer and neural development. Cancer Lett. 2001, 169, 107–114. [Google Scholar] [CrossRef]
- Zito Marino, F.; Pagliuca, F.; Ronchi, A.; Cozzolino, I.; Montella, M.; Berretta, M.; Errico, M.E.; Donofrio, V.; Bianco, R.; Franco, R. NTRK Fusions, from the Diagnostic Algorithm to Innovative Treatment in the Era of Precision Medicine. Int. J. Mol. Sci. 2020, 21, 3718. [Google Scholar] [CrossRef]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; Dubois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib inTRKFusion–Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.J. Larotrectinib: First Global Approval. Drugs 2019, 79, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Galenkamp, K.M.O.; Sosicka, P.; Jung, M. Golgi Acidification by NHE7 Regulates Cytosolic pH Homeostasis in Pancreatic Cancer Cells. Cancer Discov. 2020, 10, 822–835. [Google Scholar] [CrossRef] [Green Version]
- Halbrook, C.J.; Lyssiotis, C.A. Employing Metabolism to Improve the Diagnosis and Treatment of Pancreatic Cancer. Cancer Cell 2017, 31, 5–19. [Google Scholar] [CrossRef] [Green Version]
- Pardee, T.S.; Luther, S.; Buyse, M.; Powell, B.L.; Cortes, J. Devimistat in combination with high dose cytarabine and mitoxantrone compared with high dose cytarabine and mitoxantrone in older patients with relapsed/refractory acute myeloid leukemia: ARMADA 2000 Phase III study. Future Oncol. 2019, 15, 3197–3208. [Google Scholar] [CrossRef]
- Alistar, A.; Morris, B.B.; Desnoyer, R.; Klepin, H.D.; Hosseinzadeh, K.; Clark, C.; Cameron, A.; Leyendecker, J.; D’Agostino, R.; Topaloglu, U.; et al. Safety and tolerability of the first-in-class agent CPI-613 in combination with modified FOLFIRINOX in patients with metastatic pancreatic cancer: A single-centre, open-label, dose-escalation, phase 1 trial. Lancet Oncol. 2017, 18, 770–778. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Wolpin, B.M.; Rubinson, D.A.; Wang, X.; Chan, J.A.; Cleary, J.M.; Enzinger, P.C.; Fuchs, C.S.; McCleary, N.J.; Meyerhardt, J.A.; Ng, K.; et al. Phase II and Pharmacodynamic Study of Autophagy Inhibition Using Hydroxychloroquine in Patients With Metastatic Pancreatic Adenocarcinoma. Oncology 2014, 19, 637–638. [Google Scholar] [CrossRef] [Green Version]
- Karasic, T.B.; O’Hara, M.H.; Loaiza-Bonilla, A.; Reiss, K.A.; Teitelbaum, U.R.; Borazanci, E.; De Jesus-Acosta, A.; Redlinger, C.; Burrell, J.A.; Laheru, D.A.; et al. Effect of Gemcitabine and nab-Paclitaxel With or Without Hydroxychloroquine on Patients With Advanced Pancreatic Cancer. JAMA Oncol. 2019, 5, 993–998. [Google Scholar] [CrossRef] [PubMed]
- Zeh, H.J.; Bahary, N.; Boone, B.A.; Singhi, A.D.; Miller-Ocuin, J.L.; Normolle, D.P.; Zureikat, A.H.; Hogg, M.E.; Bartlett, D.L.; Lee, K.K.; et al. A Randomized Phase II Preoperative Study of Autophagy Inhibition with High-Dose Hydroxychloroquine and Gemcitabine/Nab-Paclitaxel in Pancreatic Cancer Patients. Clin. Cancer Res. 2020, 26, 3126–3134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinsey, C.G.; Camolotto, S.A.; Boespflug, A.; Guillen, K.P.; Foth, M.; Truong, A.; Schuman, S.S.; Shea, J.E.; Seipp, M.T.; Yap, J.; et al. Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med. 2019, 25, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Upadhrasta, S.; Zheng, L. Strategies in Developing Immunotherapy for Pancreatic Cancer: Recognizing and Correcting Multiple Immune "Defects" in the Tumor Microenvironment. J. Clin. Med. 2019, 8, 1472. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Royal, R.E.; Levy, C.; Turner, K.; Mathur, A.; Hughes, M.; Kammula, U.S.; Sherry, R.M.; Topalian, S.L.; Yang, J.C.; Lowy, I.; et al. Phase 2 Trial of Single Agent Ipilimumab (Anti-CTLA-4) for Locally Advanced or Metastatic Pancreatic Adenocarcinoma. J. Immunother. 2010, 33, 828–833. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, E.M.; Oh, D.Y.; Dhani, N. Durvalumab With or Without Tremelimumab for Patients with Metastatic Pancreatic Ductal Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 1431–1438. [Google Scholar] [CrossRef]
- Weiss, G.J.; Blaydorn, L.; Beck, J. Correction to: Phase Ib/II study of gemcitabine, nab-paclitaxel, and pembrolizumab in metastatic pancreatic adenocarcinoma. Investig. New Drugs 2018, 36, 96–102. [Google Scholar] [CrossRef]
- Renouf, D.J.; Knox, J.J.; Kavan, P. The Canadian Cancer Trials Group PA.7 trial: Results of a randomized phase II study of gemcitabine (GEM) and nab-paclitaxel (Nab-P) vs GEM, nab-P, durvalumab (D) and tremelimumab (T) as first line therapy in metastatic pancreatic ductal adenocarcinoma (mPDAC). Ann. Oncol. 2018, 31, 1142–1215. [Google Scholar]
- Jaffee, E.M.; Hruban, R.H.; Biedrzycki, B.; Laheru, D.; Schepers, K.; Sauter, P.R.; Goemann, M.; Coleman, J.; Grochow, L.; Donehower, R.C.; et al. Novel Allogeneic Granulocyte-Macrophage Colony-Stimulating Factor–Secreting Tumor Vaccine for Pancreatic Cancer: A Phase I Trial of Safety and Immune Activation. J. Clin. Oncol. 2001, 19, 145–156. [Google Scholar] [CrossRef]
- Lutz, E.R.; Wu, A.A.; Bigelow, E.; Sharma, R.; Mo, G.; Soares, K.; Solt, S.; Dorman, A.; Wamwea, A.; Yager, A.; et al. Immunotherapy Converts Nonimmunogenic Pancreatic Tumors into Immunogenic Foci of Immune Regulation. Cancer Immunol. Res. 2014, 2, 616–631. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Lutz, E.; Uram, J.N.; Sugar, E.A.; Onners, B.; Solt, S.; Zheng, L.; Diaz, L.; Donehower, R.C.; Jaffee, E.; et al. Evaluation of Ipilimumab in Combination With Allogeneic Pancreatic Tumor Cells Transfected With a GM-CSF Gene in Previously Treated Pancreatic Cancer. J. Immunother. 2013, 36, 382–389. [Google Scholar] [CrossRef] [Green Version]
- Hardacre, J.M.; Mulcahy, M.; Small, W.; Talamonti, M.; Obel, J.; Krishnamurthi, S.; Rocha-Lima, C.S.; Safran, H.; Lenz, H.-J.; Chiorean, E.G. Addition of Algenpantucel-L Immunotherapy to Standard Adjuvant Therapy for Pancreatic Cancer: A Phase 2 Study. J. Gastrointest. Surg. 2013, 17, 94–101. [Google Scholar] [CrossRef]
- Hewitt, D.B.; Nissen, N.; Hatoum, H.; Musher, B.; Seng, J.; Coveler, A.L.; Al-Rajabi, R.; Yeo, C.J.; Leiby, B.; Banks, J.; et al. A Phase 3 Randomized Clinical Trial of Chemotherapy With or Without Algenpantucel-L (HyperAcute-Pancreas) Immunotherapy in Subjects with Borderline Resectable or Locally Advanced Unresectable Pancreatic Cancer. Ann. Surg. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bahary, N.; Garrido-Laguna, I.; Cinar, P.; O’Rourke, M.A.; Somer, B.G.; Nyak-Kapoor, A.; Lee, J.S.; Munn, D.; Kennedy, E.P.; Vahanian, N.N.; et al. Phase 2 trial of the indoleamine 2,3-dioxygenase pathway (IDO) inhibitor indoximod plus gemcitabine/nab-paclitaxel for the treatment of metastatic pancreas cancer: Interim analysis. J. Clin. Oncol. 2016, 34, 3020. [Google Scholar] [CrossRef]
- Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Mayer, A.; Deshpande, A.D.; Carpenter, D.; Mitchem, J.; Plambeck-Suess, S.M.; Worley, L.A.; Goetz, B.D.; et al. Inflammatory Monocyte Mobilization Decreases Patient Survival in Pancreatic Cancer: A Role for Targeting the CCL2/CCR2 Axis. Clin. Cancer Res. 2013, 19, 3404–3415. [Google Scholar] [CrossRef] [Green Version]
- Nywening, T.M.; Wang-Gillam, A.; Sanford, E.D.; Belt, A.B.; Panni, R.Z.; Cusworth, B.M.; Toriola, A.; Nieman, R.K.; Worley, A.L.; Yano, M.; et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: A single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016, 17, 651–662. [Google Scholar] [CrossRef] [Green Version]
- Noel, M.; O’Reilly, E.M.; Wolpin, B.M.; Ryan, D.P.; Bullock, A.J.; Britten, C.D.; Linehan, D.C.; Belt, B.A.; Gamelin, E.C.; Ganguly, B.; et al. Phase 1b study of a small molecule antagonist of human chemokine (C-C motif) receptor 2 (PF-04136309) in combination with nab-paclitaxel/gemcitabine in first-line treatment of metastatic pancreatic ductal adenocarcinoma. Investig. New Drugs 2020, 38, 800–811. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Target | Tumor | Setting | Treatment Arms | Phase | Primary Outcome | N of Patients | clinicaltrial.gov Identifier |
|---|---|---|---|---|---|---|---|
| PARP | PC | Advanced, pretreated, without germline BRCA1/2 mutations but with BRCAness phenotype | olaparib | II | ORR | 34 | NCT02677038 |
| PARP | AST | Advanced, pretreated | (1) AZD6738 | II | ORR | 68 | NCT03682289 |
| ATR | (2) AZD6738 + olaparib | ||||||
| PARP | AST | Advanced, pretreated | cediranib + olaparib | II | ORR | 126 | NCT02498613 |
| VEGF | |||||||
| PARP | PC | Advanced, with BRCA1/2 or PALB2 mutation | (1) veliparib + gemcitabine hydrochloride + cisplatin | II | ORR Dose-finding | 107 | NCT01585805 |
| (2) gemcitabine hydrochloride + cisplatin | |||||||
| (3) veliparib | |||||||
| PARP | PC | Metastatic, untreated, with HRD | rucaparib + nal-IRI, leucovorin, fluorouracil | II | ORR | 110 | NCT03337087 |
| DLTs | |||||||
| PARP | AST | Advanced, pretreated, with HRD | rucaparib | II | ORR | 220 | NCT04171700 |
| PARP | PC | Advanced, pretreated, with BRCA1/2, PALB2, CHEK2 or ATM mutation | niraparib | II | PFS | 32 | NCT03601923 |
| PARP | PC | Advanced, pretreated | niraparib + dostarlimab + RT | II | DCR | 25 | NCT04409002 |
| PARP | PC | Advanced, pretreated, with DDR genes alteration | niraparib | II | ORR | 18 | NCT03553004 |
| PARP | PC | Advanced, following platinum-based CT without PD | (1) niraparib + nivolumab | Ib/II | PFS | 84 | NCT03404960 |
| (2) niraparib + ipilimumab | |||||||
| PARP | PC | Resected, after completion of (neo)adjuvant CT (+/− RT), with BRCA1/2 or PALB2 mutation | (1) olaparib (2) placebo | II | RFS | 152 | NCT04858334 |
| PARP | PC | metastatic, following platinum-based CT without PD, with BRCA1/2 mutation | (1) olaparib + pembrolizumab (2) olaparib | II | PFS | 88 | NCT04548752 |
| PD-1 | |||||||
| PARP, PD-1 | PC | Metastatic, pretreated, with BRCA1/2, PALB2, BARD1, RAD51c/d mutation | niraparib + dostarlimab | II | DCR | 20 | NCT04493060 |
| PARP, PD-1 | PC | metastatic, untreated, following low-dose CT with gemcitabine, nab-paclitaxel, capecitabine, cisplatin, and irinotecan (GAX-CI) | olaparib + pembrolizumab | II | PFS | 38 | NCT04753879 |
| PARP | PC | Advanced, untreated, with BRCA1/2 or PALB2 mutation | (1) fluzoparib + mFOLFIRINOX followed by fluzoparib maintenance | Ib/II | DLTs MTD ORR | 66 | NCT04228601 |
| (2) placebo + mFOLFIRINOX followed by placebo maintenance | |||||||
| WEE1 | PC | Metastatic, untreated | (1) adavosertib (MK-1775) + nab-paclitaxel + gemcitabine | I/II | MTD PFS | 133 | NCT02194829 |
| (2) placebo + nab-paclitaxel + gemcitabine | |||||||
| RAD51 | AST | Advanced, any line | CYT-0851 | I/II | DLTs ORR | 165 | NCT03997968 |
| Target | Tumor | Setting | Treatment Arms | Phase | Primary Outcome | N of Patients | clinicaltrial.gov Identifier |
|---|---|---|---|---|---|---|---|
| FAK PD-1 | PC | Resectable | (1) perioperative CT followed by pembrolizumab + defactinib (2) perioperative CT followed by pembrolizumab | II | pCR rate | 36 | NCT03727880 |
| FAK | PC | Locally advanced | (1) CT followed by SBRT + defactinib (2) CT followed by SBRT alone | II | PFS | 42 | NCT04331041 |
| MEK FAK | PC | Advanced, pretreated | GSK2256098 + trametinib | II | ORR | 16 | NCT02428270 |
| MEK BCL-2 | AST | Advanced, pretreated, with KRAS or NRAS mutation | trametinib + navitoclax | Ib/II | ORR PFS Safety | 130 | NCT02079740 |
| MEK BRAF | PC | Advanced, pretreated, with BRAF V600E mutation | binimetinib + encorafenib | II | ORR | 29 | NCT04390243 |
| ERK | PC | Metastatic, pretreated | (1) LY3214996 + hydroxychloroquine (2) LY3214996 | II | DCR, Safety | 52 | NCT04386057 |
| ALK5 | PC | Metastatic, pretreated | TEW-7197 + FOLFOX | Ib/II | PFS | 36 | NCT03666832 |
| EGFR | PC | Advanced, pretreated | CT followed by anti-CD3 x anti-EGFR bispecific antibody armed activated T cells | I/II | OS Safety | 22 | NCT03269526 |
| EGFR HDAC | PC | advanced, first-line | CG200745 PPA + gemcitabine + erlotinib | I/II | ORR | 24 | NCT02737228 |
| EGFR | PC | Resected, adjuvant | (1) CT (2) gemcitabine + erlotinib | II/III | OS | 545 | NCT01013649 |
| CTGF | PC | Locally advanced, neoadjuvant | (1) pamrevlumab + gemcitabine + nab-paclitaxel (2) placebo + gemcitabine + nab-paclitaxel | III | OS Proportion of R0 or R1 resection | 256 | NCT03941093 |
| KRAS | PC | Advanced, pretreated | cyclophosphamide + fludarabine + T cell therapy (+ anti-PD-1) | I/II | ORR Safety | 30 | NCT04146298 |
| KRAS | AST | Advanced, pretreated, with KRAS G12C mutation | adagrasib | I/II | ORR Safety Plasma concentration | 391 | NCT03785249 |
| KRAS NRAS | AST | Detectable ctDNA despite prior therapy, with KRAS/NRAS mutation | ELI-002 | I/II | MTD RFS Safety | 159 | NCT04853017 |
| HER2 | AST | Advanced, pretreated, with HER2 expression or amplification | A166 | I/II | MTD ORR | 82 | NCT03602079 |
| HDAC PD-1 | PC CC | Advanced pretreated | entinostat + nivolumab | II | ORR | 44 | NCT03250273 |
| XPO1 | PC | Metastatic, untreated | (1) selinexor + gemcitabine/nab-paclitaxel gemcitabine/nab-paclitaxel | I/II | MTD OS Safety | 56 | NCT02178436 |
| NTRK ROS1 ALK | AST | Advanced, with NTRK/ROS1/ALK gene rearrangement | entrectinib | II | ORR | 300 | NCT02568267 |
| NTRK | AST | Advanced, pretreated, with NTRAK gene rearrangement | larotrectinib | II | ORR | 203 | NCT02576431 |
| Metabolism | PC | Metastatic, untreated | (1) devimistat + FOLFIRINOX(2) FOLFIRINOX | III | ORR PFS | 500 | NCT03504423 |
| Target | Tumor | Setting | Treatment Arms | Phase | Primary Outcome | N of Patients | clinicaltrial.gov Identifier |
|---|---|---|---|---|---|---|---|
| PD-L1 TGF-βRII | PC | Advanced, pretreated | gemcitabine + nab-paclitaxel + SHR-1701 | Ib/II | ORR RP2D | 54 | NCT04624217 |
| PD-L1 CTLA-4 | PC | Locally advanced | minimally invasive surgical microwave ablation + durvalumab + tremelimumab + gemcitabine | II | PFS | 20 | NCT04156087 |
| PD-1 CTLA-4 | PC | Metastatic | nivolumab + ipilimumab + radiation | II | ORR | 30 | NCT04361162 |
| PD-1 | PC | Metastatic | (1) FOLFIRINOX (2) FOLFIRINOX + Anti-PD-1 antibody | III | OS | 110 | NCT03977272 |
| PD-1 | PC | Locally advanced | (1) FOLFIRINOX (2) FOLFIRINOX + anti-PD-1 antibody | III | PFS | 830 | NCT03983057 |
| PD-1 | PC | Metastatic, untreated | gemcitabine + S-1 + nivolumab | II | ORR | 38 | NCT04377048 |
| CSF1R PD-1 | PC | Advanced, pretreated | (1) gemcitabine/nab-paclitaxel or 5-FU/leucovorin/irinotecan liposome (2) cabiralizumab + nivolumab (3) gemcitabine + nab-paclitaxel + cabiralizumab + nivolumab (4) cabiralizumab + nivolumab + FOLFOX | II | PFS | 179 | NCT03336216 |
| PD-1 | PC | Metastatic | (1) FOLFIRINOX/mFOLFIRINOX + anti-PD-1 (2) FOLFIRINOX/mFOLFIRINOX | III | OS | 110 | NCT03977272 |
| PD-L1 CTLA4 | PC | Advanced | (1) 2nd line PD-L1/CTLA4 inhibitor(2) 1st line PD-L1/CTLA4 inhibitor + gemcitabine/nab-paclitaxel (3) 1st line PD-L1/CTLA4 inhibitor + FOLFIRINOX | I/II | ORR | 60 | NCT04324307 |
| CXCR4 PD-1 | PC | Metastatic pretreated | plerixafor + cemiplimab | II | ORR | 21 | NCT04177810 |
| PD-L1 ICOS | AST | Advanced, pretreated | (1) KY1044 (2) KY1044 + atezolizumab | I/II | ORR Safety | 412 | NCT03829501 |
| ETBR PD-1 | AST | Advanced pretreated | ENB-003 + pembrolizumab | I/II | ORR Safety | 130 | NCT04205227 |
| PD-1 | AST | Advanced, pretreated | (1) pembrolizumab + lenvatinib (2) lenvatinib | II | ORR Safety | 760 | NCT03797326 |
| CD11b PD-1 | AST | Advanced | (1) GB1275 GB1275 + anti PD-1 | I/II | ORR Safety | 242 | NCT04060342 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Federico, A.; Tateo, V.; Parisi, C.; Formica, F.; Carloni, R.; Frega, G.; Rizzo, A.; Ricci, D.; Di Marco, M.; Palloni, A.; et al. Hacking Pancreatic Cancer: Present and Future of Personalized Medicine. Pharmaceuticals 2021, 14, 677. https://doi.org/10.3390/ph14070677
Di Federico A, Tateo V, Parisi C, Formica F, Carloni R, Frega G, Rizzo A, Ricci D, Di Marco M, Palloni A, et al. Hacking Pancreatic Cancer: Present and Future of Personalized Medicine. Pharmaceuticals. 2021; 14(7):677. https://doi.org/10.3390/ph14070677
Chicago/Turabian StyleDi Federico, Alessandro, Valentina Tateo, Claudia Parisi, Francesca Formica, Riccardo Carloni, Giorgio Frega, Alessandro Rizzo, Dalia Ricci, Mariacristina Di Marco, Andrea Palloni, and et al. 2021. "Hacking Pancreatic Cancer: Present and Future of Personalized Medicine" Pharmaceuticals 14, no. 7: 677. https://doi.org/10.3390/ph14070677
APA StyleDi Federico, A., Tateo, V., Parisi, C., Formica, F., Carloni, R., Frega, G., Rizzo, A., Ricci, D., Di Marco, M., Palloni, A., & Brandi, G. (2021). Hacking Pancreatic Cancer: Present and Future of Personalized Medicine. Pharmaceuticals, 14(7), 677. https://doi.org/10.3390/ph14070677