
Development of Machine Learning Models for Accurately Predicting and Ranking the Activity of Lead Molecules to Inhibit PRC2 Dependent Cancer


 ,
,  ,
,
Abstract
:1. Introduction
2. Results
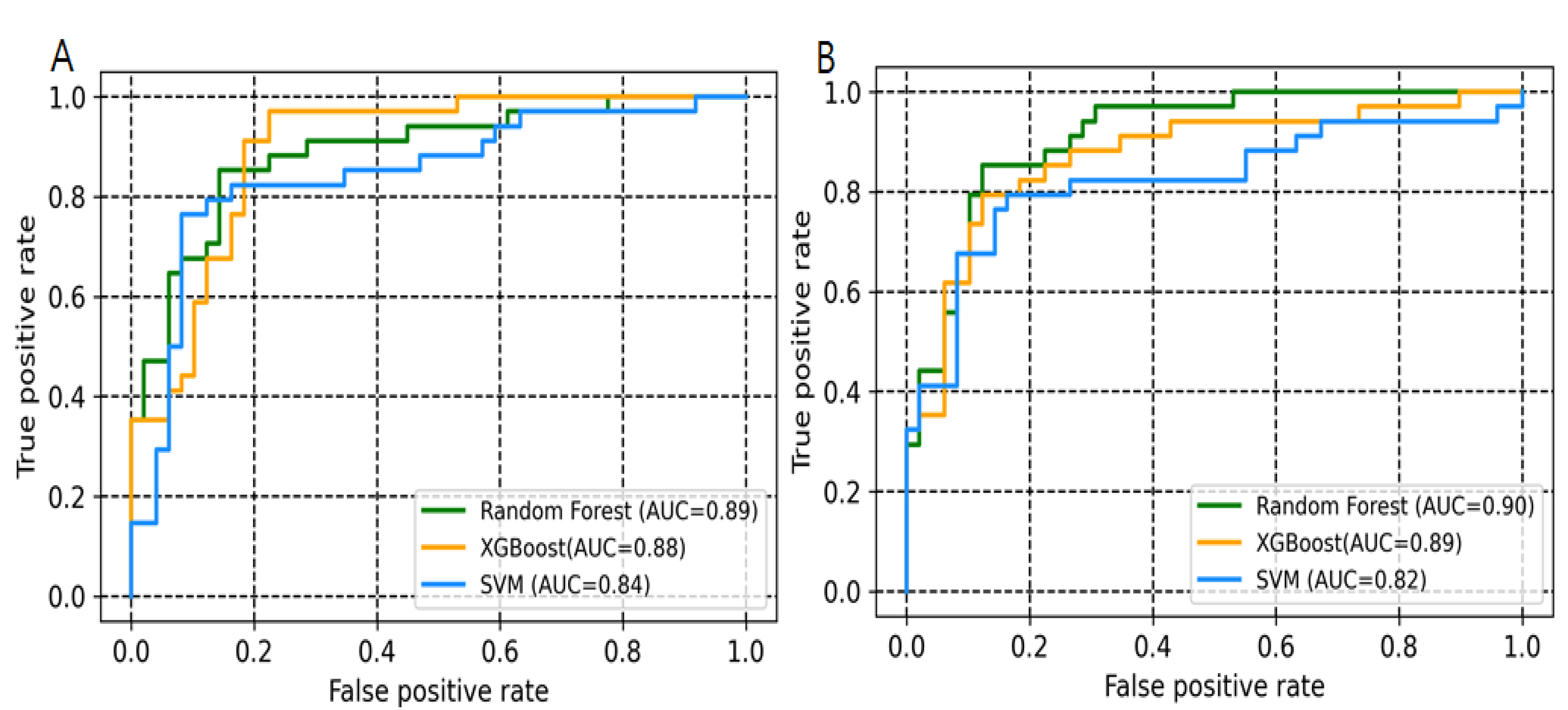
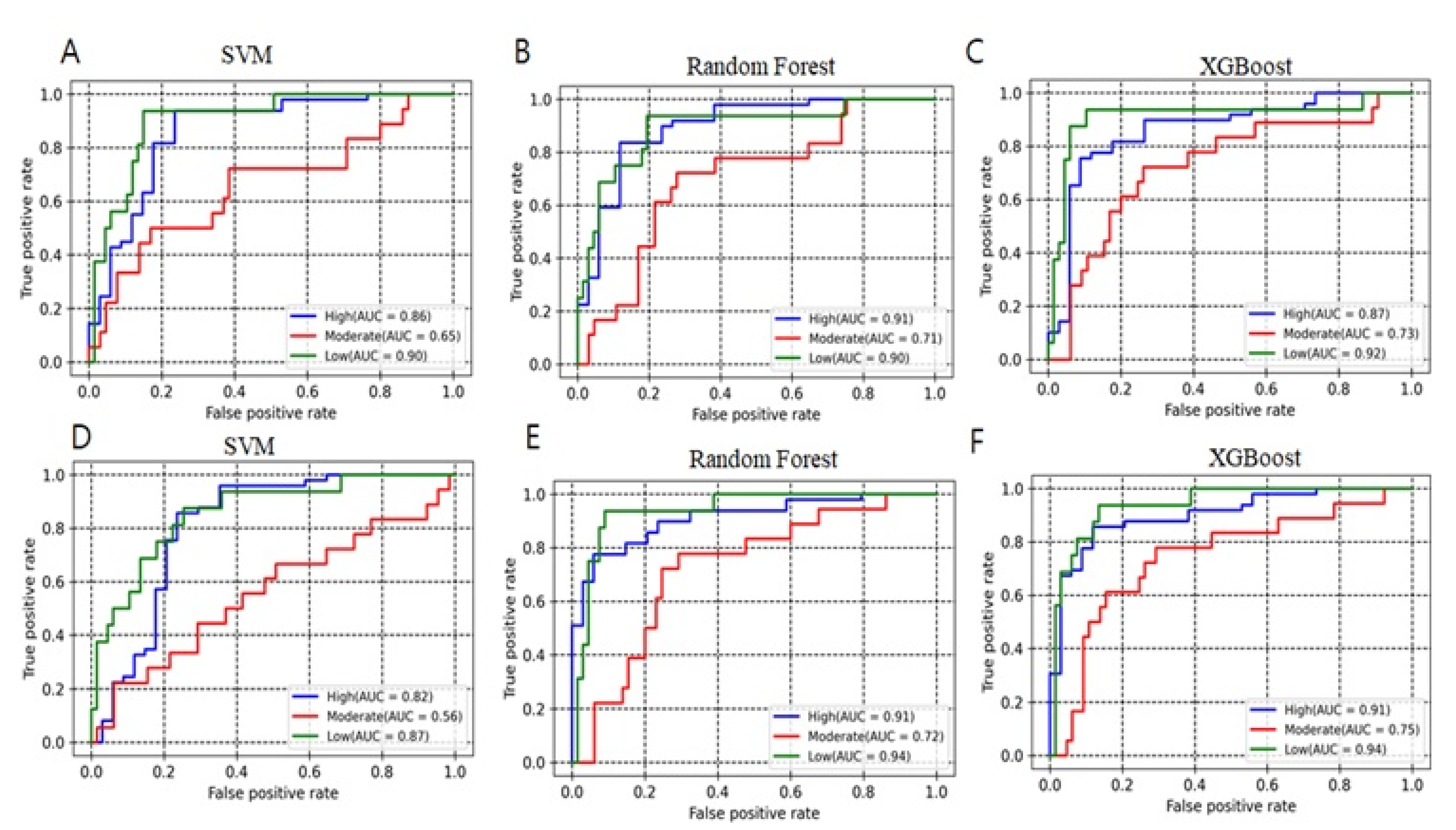
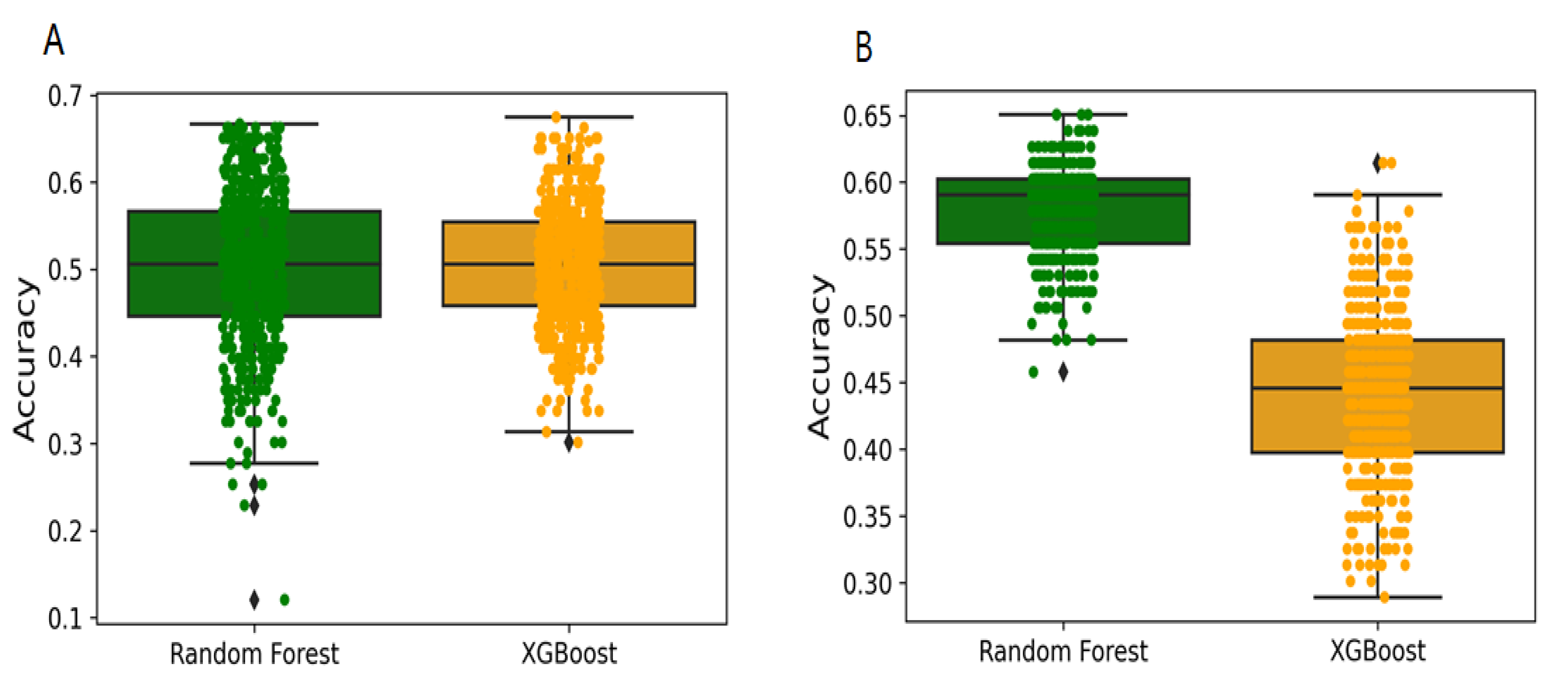
2.1. Model Development and Evaluation
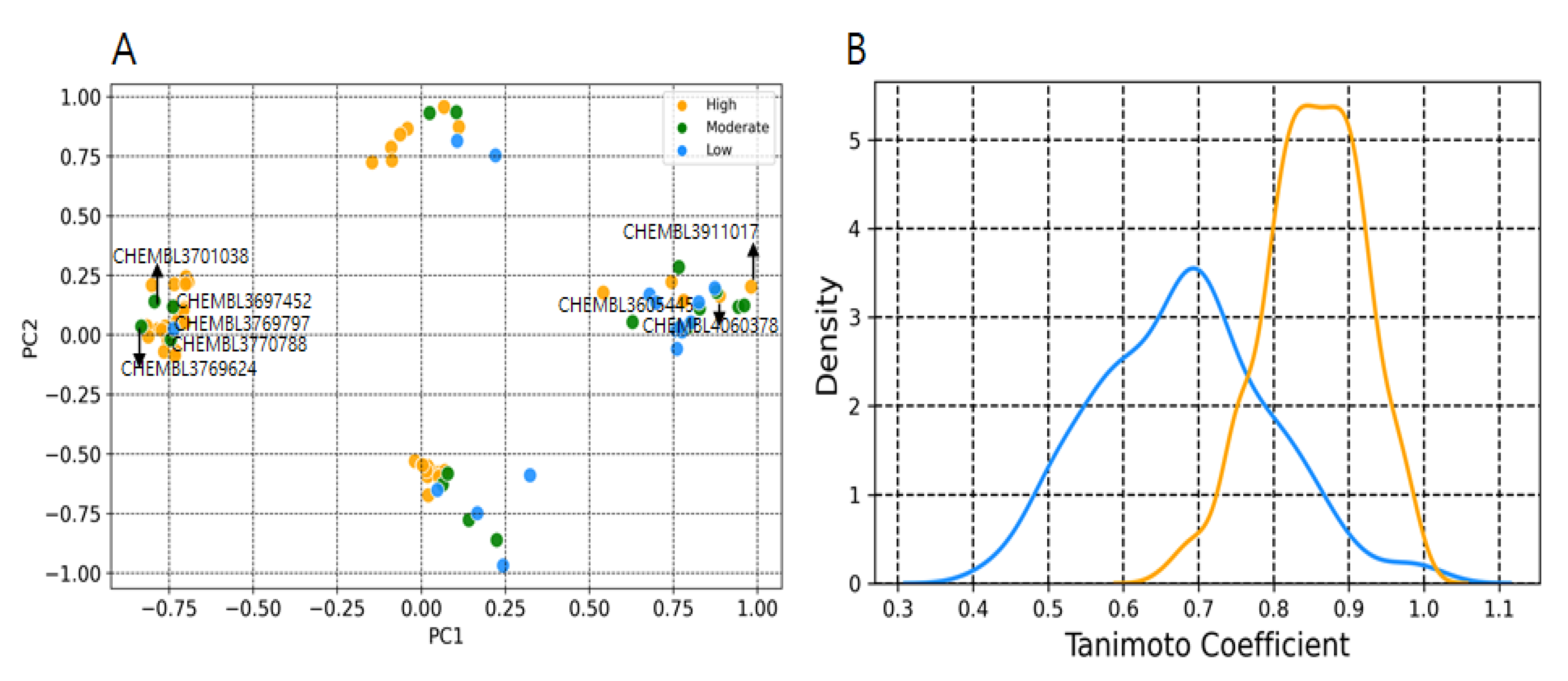
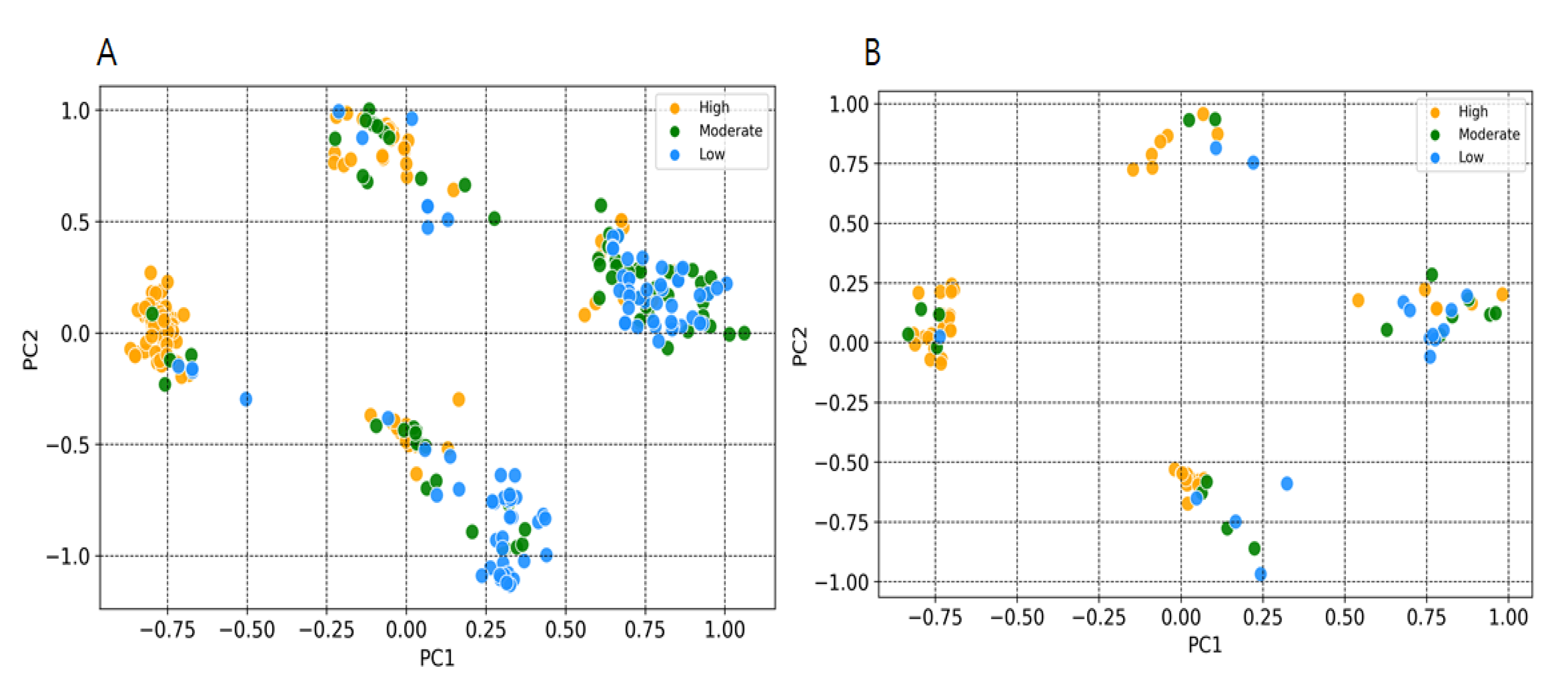
2.2. Y-Randomization and Applicability Domain
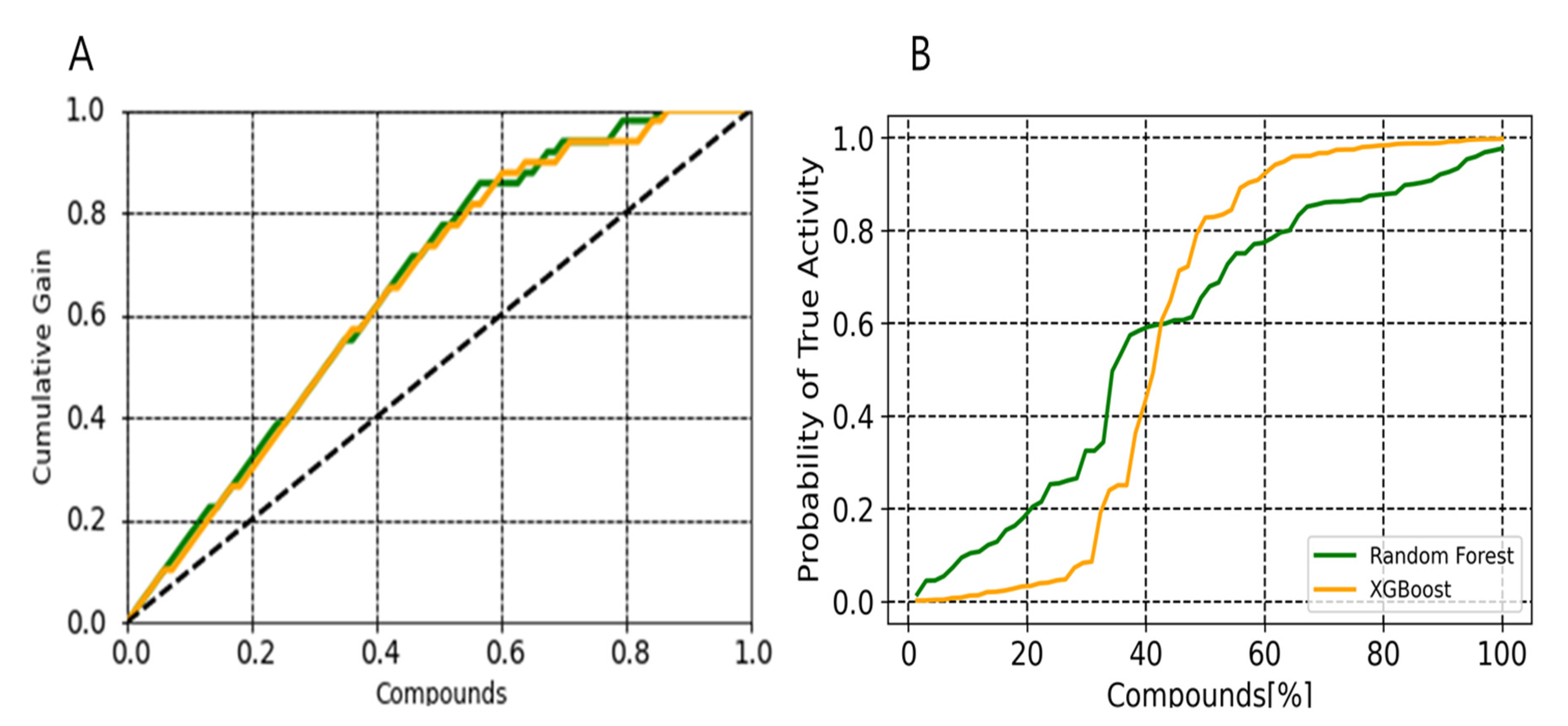
2.3. Probabilistic Distribution
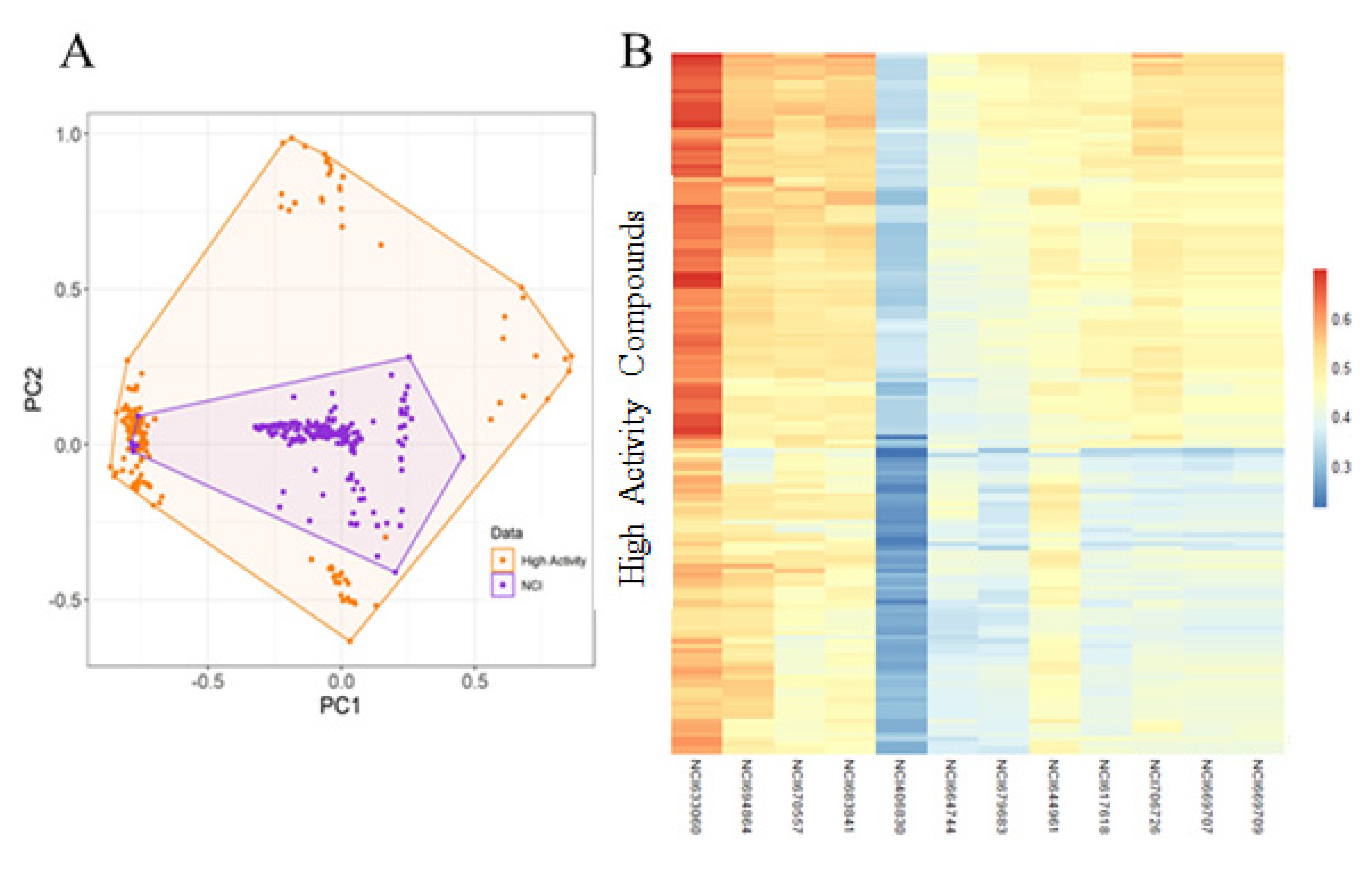
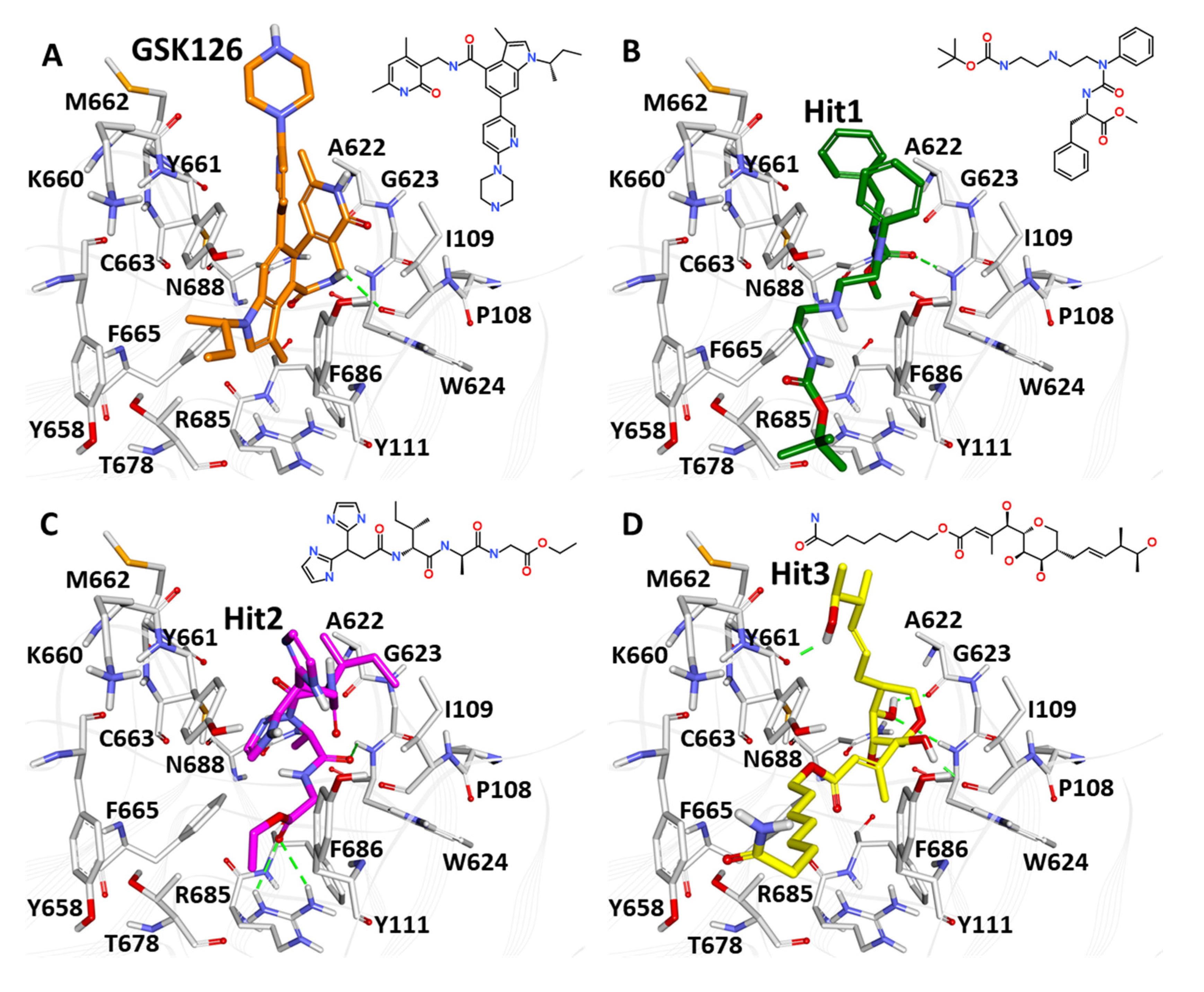
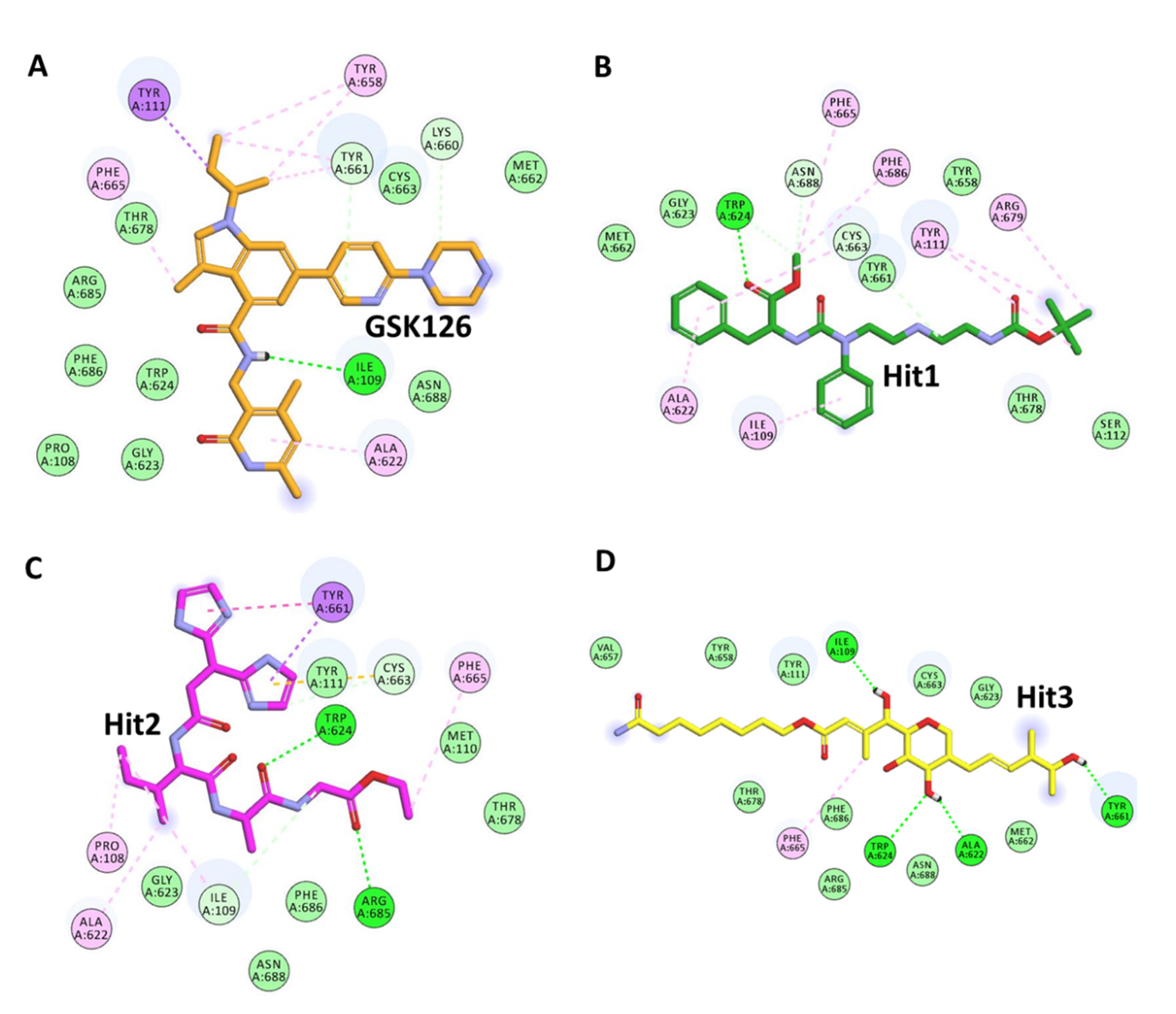
2.4. Virtual Screening and Molecular Docking
3. Discussion
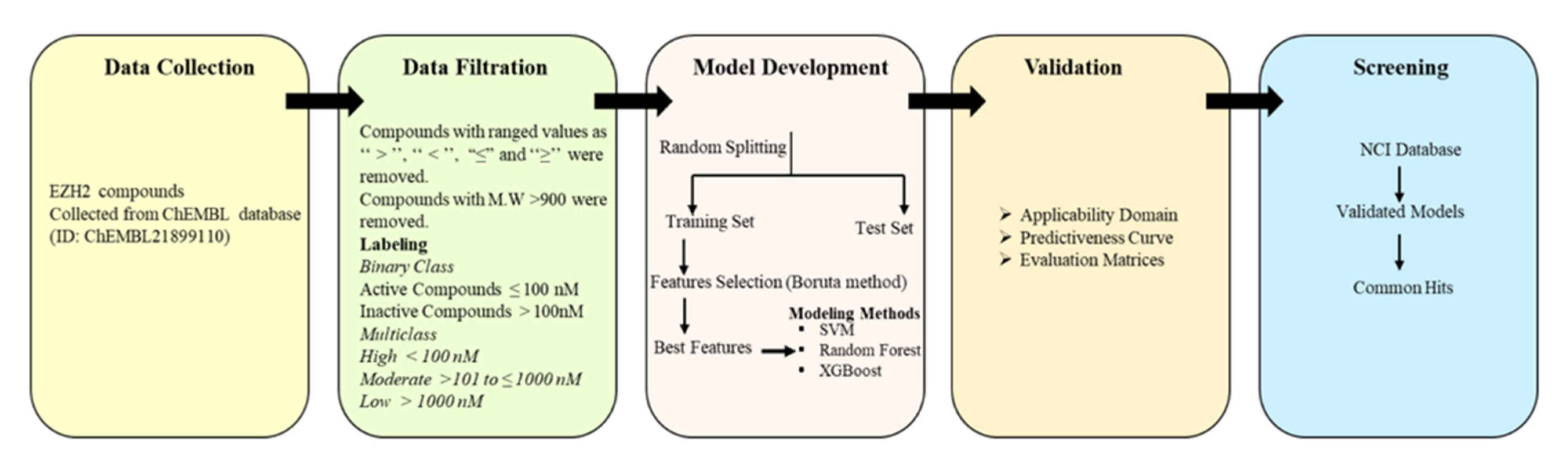
4. Materials and Methods
4.1. Descriptor Calculation and Selection
4.2. Model Building
4.3. Model Validation
4.4. Predictiveness Curve
4.5. Applicability Domain
4.6. Y-Randomization
4.7. Similarity Calculations
4.8. Library Screening and Molecular Docking
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lund, A.H.; Van Lohuizen, M. Epigenetics and cancer. Genes Dev. 2004, 18, 2315–2335. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2009, 31, 27–36. [Google Scholar] [CrossRef]
- Laugesen, A.; Højfeldt, J.W.; Helin, K. Molecular mechanisms directing PRC2 recruitment and H3K27 methylation. Mol. Cell 2019, 74, 8–18. [Google Scholar] [CrossRef] [Green Version]
- Dockerill, M.; Gregson, C.; O’ Donovan, D.H. Targeting PRC2 for the treatment of cancer: An updated patent review (2016–2020). Expert Opin. Ther. Pat. 2021, 31, 119–135. [Google Scholar] [CrossRef]
- Brooun, A.; Gajiwala, K.S.; Deng, Y.L.; Liu, W.; Bolaños, B.; Bingham, P.; He, Y.A.; Diehl, W.; Grable, N.; Kung, P.P.; et al. Polycomb repressive complex 2 structure with inhibitor reveals a mechanism of activation and drug resistance. Nat. Commun. 2016, 7, 11384. [Google Scholar] [CrossRef]
- Duan, R.; Du, W.; Guo, W. EZH2: A novel target for cancer treatment. J. Hematol. Oncol. 2020, 13, 104. [Google Scholar] [CrossRef] [PubMed]
- Min, J.; Zhang, Y.; Xu, R.M. Structural basis for specific binding of polycomb chromodomain to histone H3 methylated at Lys 27. Genes Dev. 2003, 17, 1823–1828. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Wang, X.X.; Zhuang, Y.W.; Jiang, Y.; Melcher, K.; Xu, H.E. Structure of the PRC2 complex and application to drug discovery. Acta Pharmacol. Sin. 2017, 38, 963–976. [Google Scholar] [CrossRef] [PubMed]
- McCabe, M.T.; Graves, A.P.; Ganji, G.; Diaz, E.; Halsey, W.S.; Jiang, Y.; Smitheman, K.N.; Ott, H.M.; Pappalardi, M.B.; Allen, K.E.; et al. Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27). Proc. Natl. Acad. Sci. USA 2012, 109, 2989–2994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, T.; Nerle, S.; Pritchard, J.; Zhao, B.; Rivera, V.M.; Garner, A.; Gonzalvez, F. Acquisition of a single EZH2 D1 domain mutation confers acquired resistance to EZH2-targeted inhibitors. Oncotarget 2015, 6, 32646–32655. [Google Scholar] [CrossRef] [Green Version]
- Danishuddin; Subbarao, N.; Faheem, M.; Khan, S.N. Polycomb repressive complex 2 inhibitors: Emerging epigenetic modulators. Drug Discov. Today 2019, 24, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Gulati, N.; Béguelin, W.; Giulino-Roth, L. Enhancer of zeste homolog 2 (EZH2) inhibitors. Leuk. Lymphoma 2018, 59, 1574–1585. [Google Scholar] [CrossRef] [PubMed]
- Velcheti, V.; Wong, K.K.; Saunthararajah, Y. EZH2 inhibitors: Take it EZy, it is all about context. Cancer Discov. 2019, 9, 472–475. [Google Scholar] [CrossRef] [Green Version]
- Lue, J.K.; Amengual, J.E. Emerging EZH2 Inhibitors and Their Application in Lymphoma. Curr. Hematol. Malig. Rep. 2018, 13, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Bird, G.H.; Neff, T.; Guo, G.; Kerenyi, M.A.; Walensky, L.D.; Orkin, S.H. Targeted disruption of the EZH2-EED complex inhibits EZH2-dependent cancer. Nat. Chem. Biol. 2013, 9, 643–650. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.; Konze, K.D.; Jin, J.; Wang, G.G. Targeting EZH2 and PRC2 dependence as novel anticancer therapy. Exp. Hematol. 2015, 43, 698–712. [Google Scholar] [CrossRef] [Green Version]
- Knutson, S.K.; Wigle, T.J.; Warholic, N.M.; Sneeringer, C.J.; Allain, C.J.; Klaus, C.R.; Sacks, J.D.; Raimondi, A.; Majer, C.R.; Song, J.; et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat. Chem. Biol. 2012, 8, 890–896. [Google Scholar] [CrossRef] [PubMed]
- Garapaty-Rao, S.; Nasveschuk, C.; Gagnon, A.; Chan, E.Y.; Sandy, P.; Busby, J.; Balasubramanian, S.; Campbell, R.; Zhao, F.; Bergeron, L.; et al. Identification of EZH2 and EZH1 small molecule inhibitors with selective impact on diffuse large B cell lymphoma cell growth. Chem. Biol. 2013, 20, 1329–1339. [Google Scholar] [CrossRef] [Green Version]
- McCabe, M.T.; Ott, H.M.; Ganji, G.; Korenchuk, S.; Thompson, C.; Van Aller, G.S.; Liu, Y.; Graves, A.P.; Diaz, E.; LaFrance, L.V.; et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012, 492, 108–112. [Google Scholar] [CrossRef]
- Yu, T.; Wang, Y.; Hu, Q.; Wu, W.N.; Wu, Y.; Wei, W.; Han, D.; You, Y.; Lin, N.; Liu, N. The EZH2 inhibitor GSK343 suppresses cancer stem-like phenotypes and reverses mesenchymal transition in glioma cells. Oncotarget 2017, 8, 98348–98359. [Google Scholar] [CrossRef] [Green Version]
- Qi, W.; Chan, H.M.; Teng, L.; Li, L.; Chuai, S.; Zhang, R.; Zeng, J.; Li, M.; Fan, H.; Lin, Y.; et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc. Natl. Acad. Sci. USA 2012, 109, 21360–21365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konze, K.D.; Ma, A.; Li, F.; Barsyte-Lovejoy, D.; Parton, T.; MacNevin, C.J.; Liu, F.; Gao, C.; Huang, X.P.; Kuznetsova, E.; et al. An orally bioavailable chemical probe of the lysine methyltransferases EZH2 and EZH1. ACS Chem. Biol. 2013, 8, 1324–1334. [Google Scholar] [CrossRef] [PubMed]
- Bisserier, M.; Wajapeyee, N. Mechanisms of resistance to ezh2 inhibitors in diffuse large b-cell lymphomas. Blood 2018, 131, 2125–2137. [Google Scholar] [CrossRef]
- Khanna, A.; Côté, A.; Arora, S.; Moine, L.; Gehling, V.S.; Brenneman, J.; Cantone, N.; Stuckey, J.I.; Apte, S.; Ramakrishnan, A.; et al. Design, synthesis, and pharmacological evaluation of second generation EZH2 inhibitors with Long residence time. ACS Med. Chem. Lett. 2020, 11, 1205–1212. [Google Scholar] [CrossRef]
- Wu, Y.; Hu, J.; Ding, H.; Chen, L.; Zhang, Y.; Liu, R.; Xu, P.; Du, D.; Lu, W.; Liu, J.; et al. Identification of novel EZH2 inhibitors through pharmacophore-based virtual screening and biological assays. Bioorg. Med. Chem. Lett. 2016, 26, 3813–3817. [Google Scholar] [CrossRef] [PubMed]
- Misawa, K.; Yamaotsu, N.; Hirono, S. Identification of novel EED-EZH2 PPI inhibitors using an in silico fragment mapping method. J. Comput. Aided. Mol. Des. 2021, 35, 601–611. [Google Scholar] [CrossRef]
- Zhu, K.; Du, D.; Yang, R.; Tao, H.; Zhang, H. Identification and assessments of novel and potent small-molecule inhibitors of EED-EZH2 interaction of polycomb repressive complex 2 by computational methods and biological evaluations. Chem. Pharm. Bull. 2020, 68, 58–63. [Google Scholar] [CrossRef] [Green Version]
- Stuckey, J.I.; Cantone, N.R.; Côté, A.; Arora, S.; Vivat, V.; Ramakrishnan, A.; Mertz, J.A.; Khanna, A.; Brenneman, J.; Gehling, V.S.; et al. Identification and characterization of second-generation EZH2 inhibitors with extended residence times and improved biological activity. J. Biol. Chem. 2021, 296. [Google Scholar] [CrossRef]
- Kursa, M.B.; Jankowski, A.; Rudnicki, W.R. Boruta—A system for feature selection. Fundam. Inform. 2010, 101, 271–285. [Google Scholar] [CrossRef]
- Lipiński, P.F.J.; Szurmak, P. SCRAMBLE’N’GAMBLE: A tool for fast and facile generation of random data for statistical evaluation of QSAR models. Chem. Pap. 2017, 71, 2217–2232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, K.M.; Sung, J.M.; Kim, W.J.; An, S.K.; Namkoong, K.; Lee, E.; Chang, H.J. Population-based dementia prediction model using Korean public health examination data: A cohort study. PLoS ONE 2019, 14, e0211957. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.B. Classifiers and their Metrics Quantified. Mol. Inform. 2018, 37, 1700127. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Du, D.H.; Wang, J.; Cai, X.Q.; Deng, A.X.; Nosjean, O.; Boutin, J.A.; Renard, P.; Yang, D.H.; Luo, C.; et al. Identification of catalytic and non-catalytic activity inhibitors against PRC2-EZH2 complex through multiple high-throughput screening campaigns. Chem. Biol. Drug Des. 2020, 96, 1024–1051. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Gao, S.; Li, J.; Liu, D.; Sheng, C.; Yao, C.; Jiang, W.; Wu, J.; Chen, S.; Huang, W. Wedelolactone disrupts the interaction of EZH2-EED complex and inhibits PRC2-dependent cancer. Oncotarget 2015, 6, 13049–13059. [Google Scholar] [CrossRef] [Green Version]
- Hajian-Tilaki, K. Receiver operating characteristic (ROC) curve analysis for medical diagnostic test evaluation. Casp. J. Intern. Med. 2013, 4, 627–635. [Google Scholar]
- Arthur, D.E.; Uzairu, A.; Mamza, P.; Abechi, S.E.; Shallangwa, G. Insilico modelling of quantitative structure–activity relationship of pGI50 anticancer compounds on K-562 cell line. Cogent Chem. 2018, 4, 1432520. [Google Scholar] [CrossRef]
- Ogura, K.; Sato, T.; Yuki, H.; Honma, T. Support Vector Machine model for hERG inhibitory activities based on the integrated hERG database using descriptor selection by NSGA-II. Sci. Rep. 2019, 9, 12220. [Google Scholar] [CrossRef]
- Cai, C.; Guo, P.; Zhou, Y.; Zhou, J.; Wang, Q.; Zhang, F.; Fang, J.; Cheng, F. Deep Learning-Based Prediction of Drug-Induced Cardiotoxicity. J. Chem. Inf. Model. 2019, 59, 1073–1084. [Google Scholar] [CrossRef]
- Mervin, L.H.; Afzal, A.M.; Drakakis, G.; Lewis, R.; Engkvist, O.; Bender, A. Target prediction utilising negative bioactivity data covering large chemical space. J. Cheminform. 2015, 7, 51. [Google Scholar] [CrossRef] [Green Version]
- Nidhi; Glick, M.; Davies, J.W.; Jenkins, J.L. Prediction of biological targets for compounds using multiple-category bayesian models trained on chemogenomics databases. J. Chem. Inf. Model. 2006, 46, 1124–1133. [Google Scholar] [CrossRef]
- Yang, Z.Y.; Dong, J.; Yang, Z.J.; Lu, A.P.; Hou, T.J.; Cao, D.S. Structural analysis and identification of false positive hits in Luciferase-based assays. J. Chem. Inf. Model. 2020, 60, 2031–2043. [Google Scholar] [CrossRef]
- Armutlu, P.; Ozdemir, M.E.; Uney-Yuksektepe, F.; Halil, I.H.; Turkay, M. Classification of drug molecules considering their IC50 values using mixed-integer linear programming based hyper-boxes method. BMC Bioinform. 2008, 9, 411. [Google Scholar] [CrossRef] [Green Version]
- Ponzoni, I.; Sebastián-Pérez, V.; Martínez, M.J.; Roca, C.; De la Cruz Pérez, C.; Cravero, F.; Vazquez, G.E.; Páez, J.A.; Díaz, M.F.; Campillo, N.E. QSAR classification models for predicting the activity of inhibitors of Beta-Secretase (BACE1) associated with Alzheimer’s disease. Sci. Rep. 2019, 9, 9102. [Google Scholar] [CrossRef] [Green Version]
- Rücker, C.; Rücker, G.; Meringer, M. Y-randomization and its variants in QSPR/QSAR. J. Chem. Inf. Model. 2007, 47, 2345–2357. [Google Scholar] [CrossRef]
- Ruiz, I.L.; Gómez-Nieto, M.Á. Study of the applicability domain of the QSAR classification models by means of the rivality and modelability indexes. Molecules 2018, 23, 2756. [Google Scholar] [CrossRef] [Green Version]
- Jaworska, J.; Nikolova-Jeliazkova, N.; Aldenberg, T. QSAR applicability domain estimation by projection of the training set in descriptor space: A review. ATLA Altern. Lab. Anim. 2005, 33, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Sullivan Pepe, M.; Feng, Z. Evaluating the predictiveness of a continuous marker. Biometrics 2007, 63, 1181–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pepe, M.S.; Feng, Z.; Huang, Y.; Longton, G.; Prentice, R.; Thompson, I.M.; Zheng, Y. Integrating the predictiveness of a marker with its performance as a classifier. Am. J. Epidemiol. 2008, 167, 362–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sachs, M.C.; Zhou, X.H. Partial summary measures of the predictiveness curve. Biom. J. 2013, 55, 589–602. [Google Scholar] [CrossRef]
- Rabal, O.; Castellar, A.; Oyarzabal, J. Novel pharmacological maps of protein lysine methyltransferases: Key for target deorphanization. J. Cheminform. 2018, 10, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef] [PubMed]
- Danishuddin; Khan, A.U. Descriptors and their selection methods in QSAR analysis: Paradigm for drug design. Drug Discov. Today 2016, 21, 1291–1302. [Google Scholar] [CrossRef] [PubMed]
- Mercader, A.G.; Duchowicz, P.R.; Fernández, F.M.; Castro, E.A. Modified and enhanced replacement method for the selection of molecular descriptors in QSAR and QSPR theories. Chemom. Intell. Lab. Syst. 2008, 92, 138–144. [Google Scholar] [CrossRef]
- Yap, C.W. PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef] [PubMed]
- TensorFlow Lite. Available online: https://www.tensorflow.org/lite/guide (accessed on 18 May 2021).
- Warmuth, M.K.; Liao, J.; Rätsch, G.; Mathieson, M.; Putta, S.; Lemmen, C. Active Learning with Support Vector Machines in the Drug Discovery Process. J. Chem. Inf. Comput. Sci. 2003, 43, 667–673. [Google Scholar] [CrossRef] [Green Version]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016. [Google Scholar]
- Grandini, M.; Bagli, E.; Visani, G. Metrics for multi-class classification: An overview. arXiv 2020, arXiv:2008.05756. [Google Scholar]
- El-Ansary, A.; Bjørklund, G.; Chirumbolo, S.; Alnakhli, O.M. Predictive value of selected biomarkers related to metabolism and oxidative stress in children with autism spectrum disorder. Metab. Brain Dis. 2017, 32, 1209–1221. [Google Scholar] [CrossRef] [PubMed]
- Rakhimbekova, A.; Madzhidov, T.I.; Nugmanov, R.I.; Gimadiev, T.R.; Baskin, I.I.; Varnek, A. Comprehensive analysis of applicability domains of QSPR models for chemical reactions. Int. J. Mol. Sci. 2020, 21, 5542. [Google Scholar] [CrossRef]
- Sushko, I.; Novotarskyi, S.; Körner, R.; Pandey, A.K.; Cherkasov, A.; Li, J.; Gramatica, P.; Hansen, K.; Schroeter, T.; Müller, K.R.; et al. Applicability domains for classification problems: Benchmarking of distance to models for ames mutagenicity set. J. Chem. Inf. Model. 2010, 50, 2094–2111. [Google Scholar] [CrossRef]
- Bento, A.P.; Hersey, A.; Félix, E.; Landrum, G.; Gaulton, A.; Atkinson, F.; Bellis, L.J.; De Veij, M.; Leach, A.R. An open source chemical structure curation pipeline using RDKit. J. Cheminform. 2020, 12, 51. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Descriptor | Number | Name |
|---|---|---|
| Autocorrelation | 6 | AATS7v, AATS7i, AATS7s, ATSC3v, ATSC8e, GATS6s |
| Burden modified eigenvalues | 4 | SpMin3_Bhm, SpMax1_Bhv, SpMin2_Bhv, SpMax2_Bhs |
| Atom type electrotopological state | 2 | maxHBa, maxHBint5 |
| Molecular distance edge | 1 | MDEC-33 |
| Rotatable bonds count | 1 | RotBFrac |
| Topological charge | 1 | JGI7 |
| Physicochemical | 1 | AMR |
| MACCSFP105 | 1 | A$A($A)$A |
| MACCSFP114 | 1 | CH3CH2A |
| Descriptors Set | Methods | Accuracy | Precision | Recall | F1 | AUC |
|---|---|---|---|---|---|---|
| ALL Descriptors | SVM | 0.82 | 0.85 | 0.84 | 0.85 | 0.84 |
| XGB | 0.78 | 0.75 | 0.70 | 0.78 | 0.88 | |
| RF | 0.79 | 0.77 | 0.78 | 0.79 | 0.89 | |
| Selected Descriptors | SVM | 0.77 | 0.76 | 0.75 | 0.76 | 0.82 |
| XGB | 0.82 | 0.77 | 0.79 | 0.81 | 0.89 | |
| RF | 0.80 | 0.82 | 0.80 | 0.81 | 0.90 |
| Descriptors Set | Methods | Metrices | AUC | |||||
|---|---|---|---|---|---|---|---|---|
| Accuracy | Precision (Macro) | Recall (Macro) | FI (Macro) | High | Moderate | Low | ||
| ALL Descriptors | SVM | 0.70 | 0.44 | 0.60 | 0.60 | 0.86 | 0.65 | 0.90 |
| RF | 0.72 | 0.63 | 0.64 | 0.64 | 0.91 | 0.71 | 0.90 | |
| XGB | 0.73 | 0.65 | 0.67 | 0.64 | 0.87 | 0.73 | 0.92 | |
| Selected Descriptors | SVM | 0.68 | 0.41 | 0.56 | 0.56 | 0.82 | 0.56 | 0.87 |
| RF | 0.73 | 0.63 | 0.62 | 0.62 | 0.91 | 0.72 | 0.94 | |
| XGB | 0.75 | 0.67 | 0.67 | 0.67 | 0.91 | 0.75 | 0.94 | |
| Compound ID | Classifier Probability | CDOCKER Interaction Energy (Kcal/mol) | |
|---|---|---|---|
| Random Forest | XGBoost | ||
| GSK-126 | - | - | −46.55 |
| NCI694864 | 0.634 | 0.947 | −53.68 |
| NCI670557 | 0.661 | 0.784 | −47.70 |
| NCI706726 | 0.622 | 0.905 | −47.34 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Danishuddin; Kumar, V.; Parate, S.; Bahuguna, A.; Lee, G.; Kim, M.O.; Lee, K.W. Development of Machine Learning Models for Accurately Predicting and Ranking the Activity of Lead Molecules to Inhibit PRC2 Dependent Cancer. Pharmaceuticals 2021, 14, 699. https://doi.org/10.3390/ph14070699
Danishuddin, Kumar V, Parate S, Bahuguna A, Lee G, Kim MO, Lee KW. Development of Machine Learning Models for Accurately Predicting and Ranking the Activity of Lead Molecules to Inhibit PRC2 Dependent Cancer. Pharmaceuticals. 2021; 14(7):699. https://doi.org/10.3390/ph14070699
Chicago/Turabian StyleDanishuddin, Vikas Kumar, Shraddha Parate, Ashutosh Bahuguna, Gihwan Lee, Myeong Ok Kim, and Keun Woo Lee. 2021. "Development of Machine Learning Models for Accurately Predicting and Ranking the Activity of Lead Molecules to Inhibit PRC2 Dependent Cancer" Pharmaceuticals 14, no. 7: 699. https://doi.org/10.3390/ph14070699
APA StyleDanishuddin, Kumar, V., Parate, S., Bahuguna, A., Lee, G., Kim, M. O., & Lee, K. W. (2021). Development of Machine Learning Models for Accurately Predicting and Ranking the Activity of Lead Molecules to Inhibit PRC2 Dependent Cancer. Pharmaceuticals, 14(7), 699. https://doi.org/10.3390/ph14070699