
Current Therapies in Clinical Trials of Parkinson’s Disease: A 2021 Update



Abstract
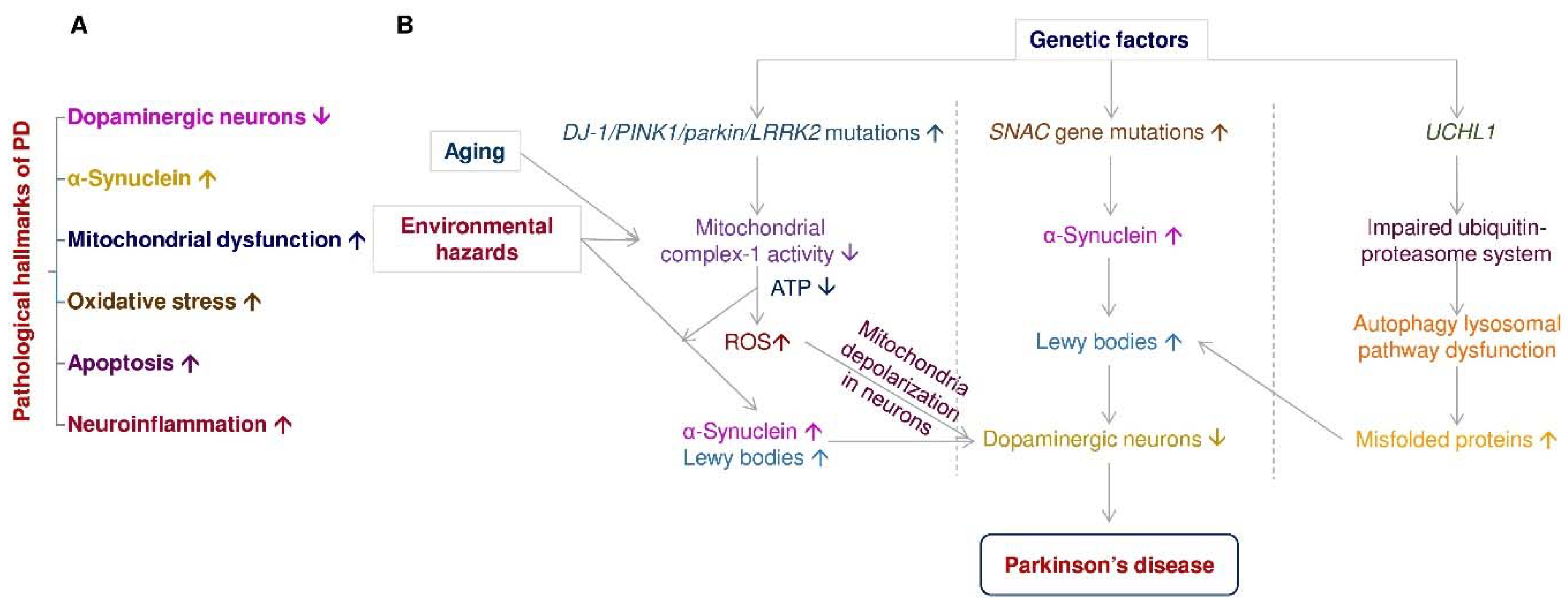
:1. Introduction
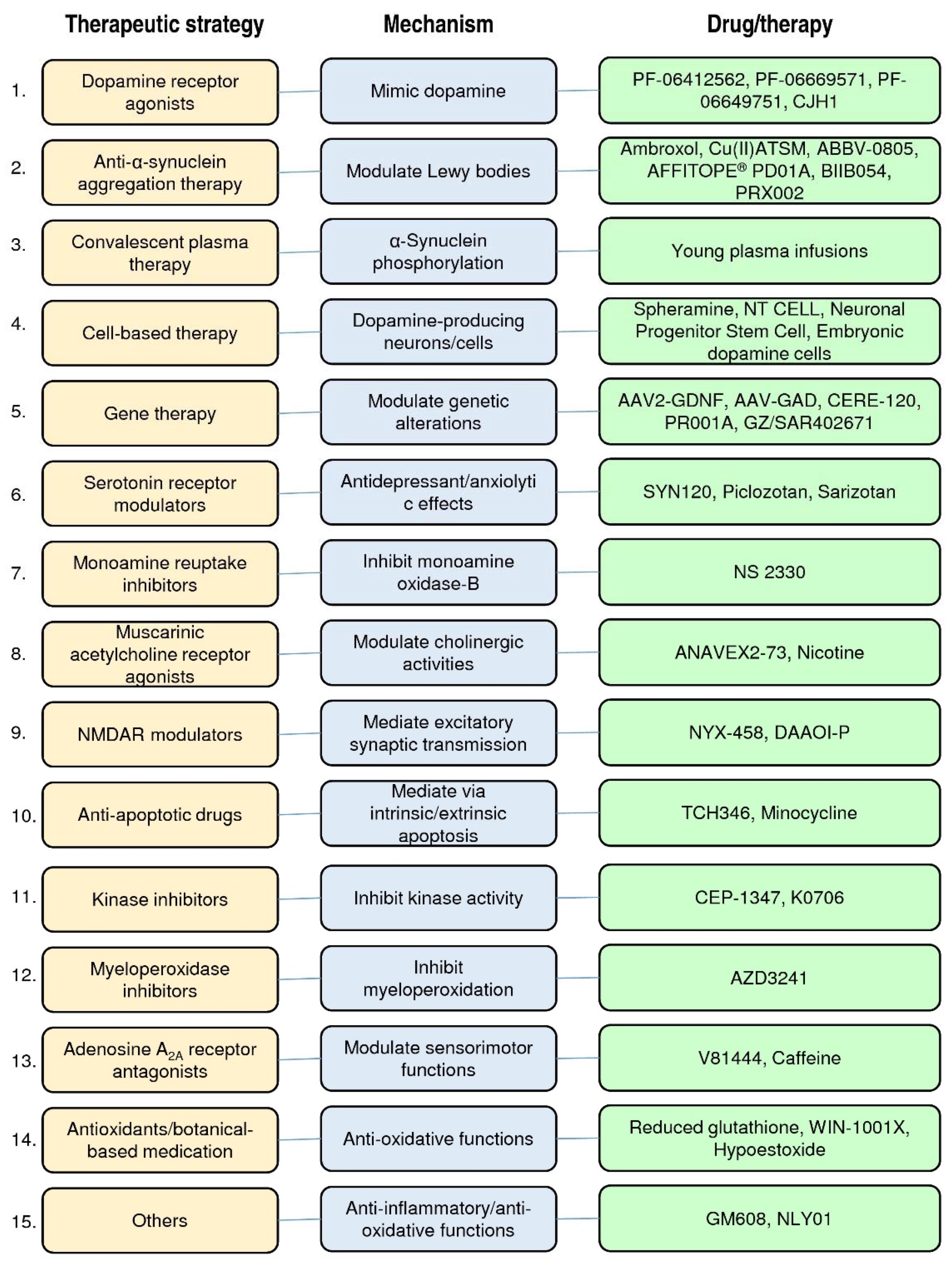
2. PD Therapeutic Strategies in Clinical Trials
2.1. Dopamine Receptor Agonists
2.2. Anti-α-Synuclein Aggregation Therapy
2.3. Convalescent Plasma Therapy
2.4. Cell-Based Therapy
2.5. Gene Therapy
2.6. Serotonin Receptor Agonists or Antagonists
2.7. Monoamine Reuptake Inhibitors
2.8. Muscarinic and Nicotinic Acetylcholine Receptor Agonists
2.9. N-Methyl-d-Aspartate Receptor (NMDAR) Modulators
2.10. Anti-Apoptotic Drugs
2.11. Kinase Inhibitors
2.12. Myeloperoxidase Inhibitors
2.13. Adenosine A2A Receptor Antagonists
2.14. Antioxidants and Botanical-Based Medication
2.15. Others
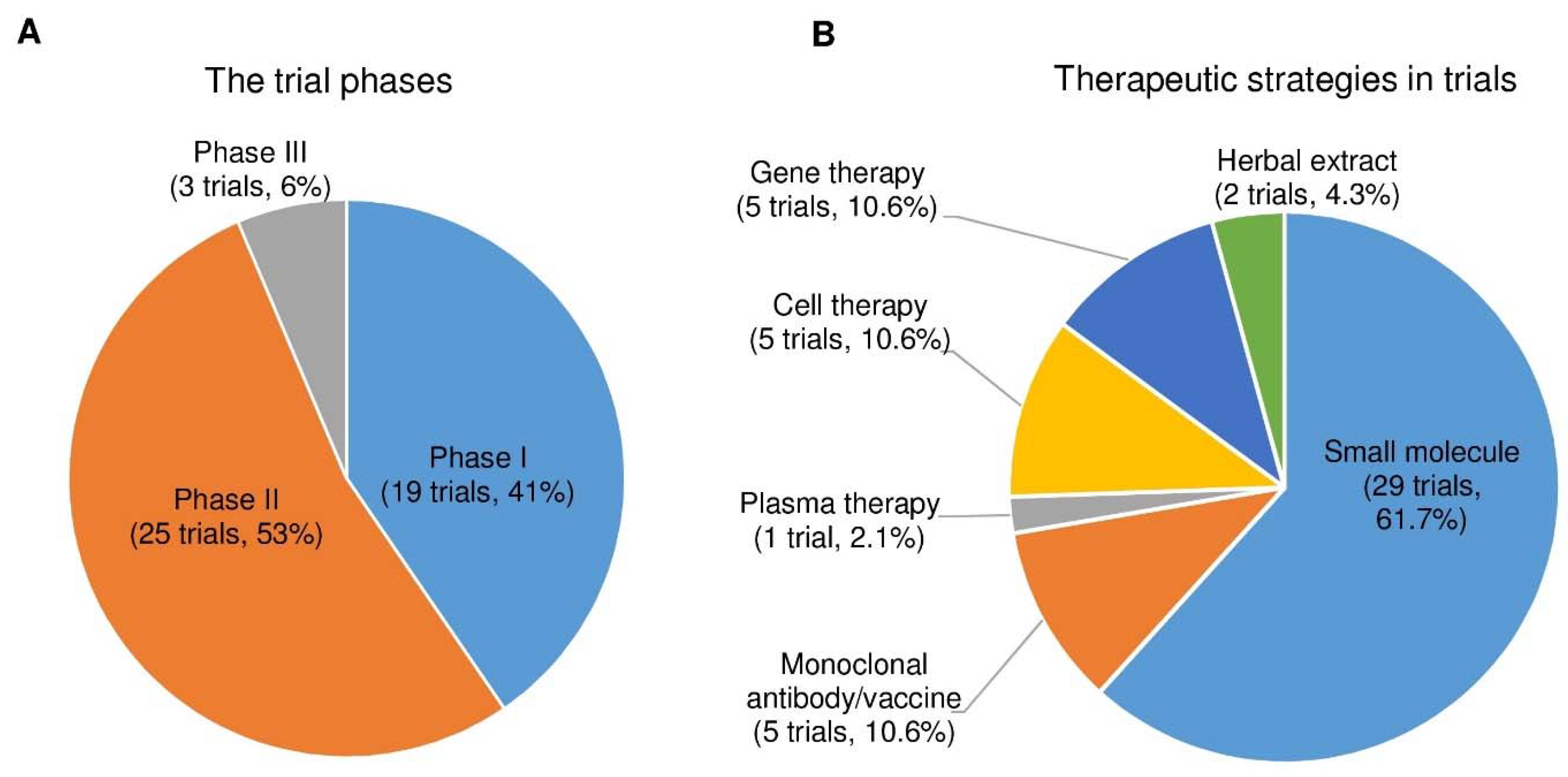
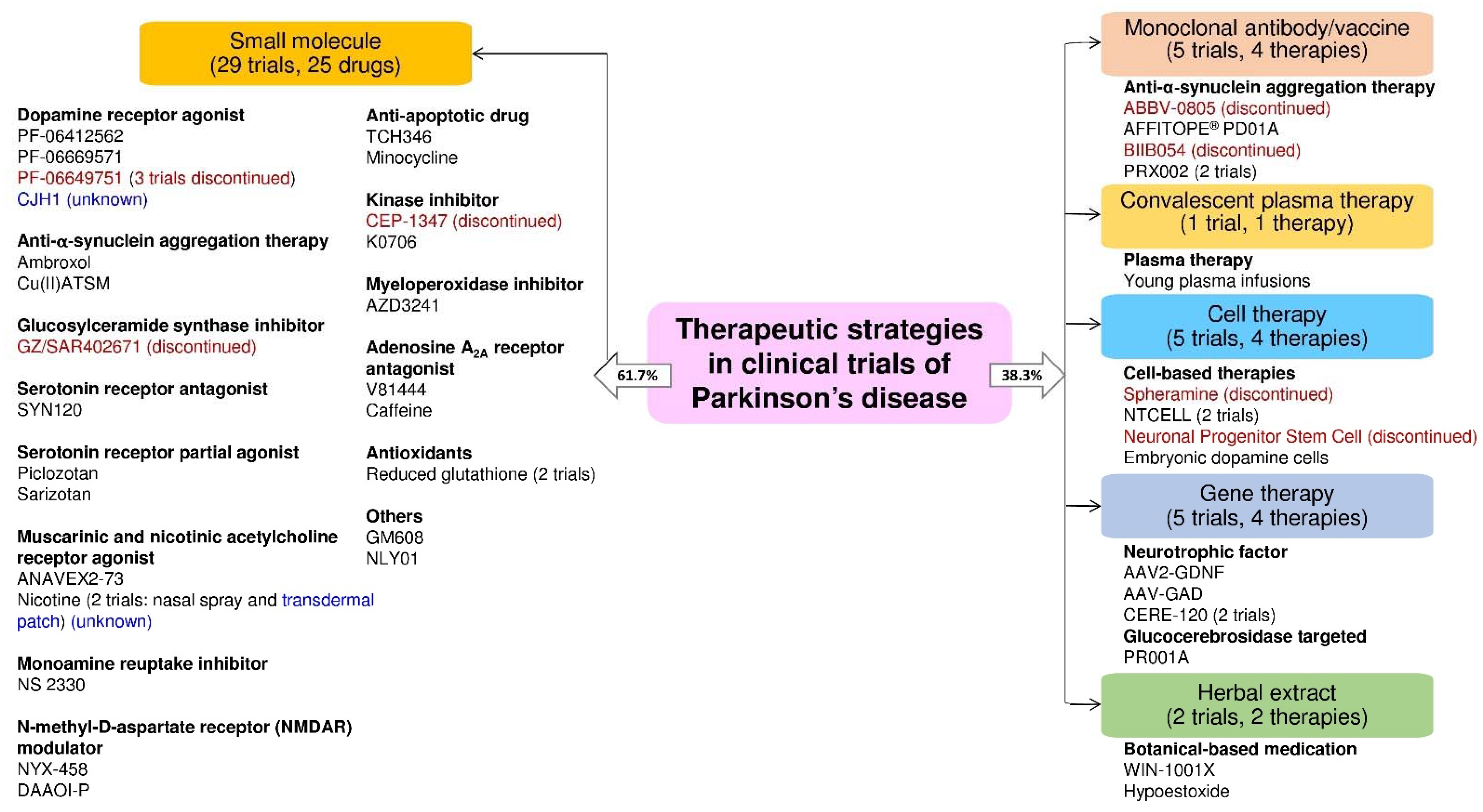
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Marino, S.; Ciurleo, R.; Di Lorenzo, G.; Barresi, M.; De Salvo, S.; Giacoppo, S.; Bramanti, A.; Lanzafame, P.; Bramanti, P. Magnetic resonance imaging markers for early diagnosis of Parkinson’s disease. Neural Regen. Res. 2012, 7, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef] [Green Version]
- Rabinstein, A.A.; Shulman, L.M. Management of behavioral and psychiatric problems in Parkinson’s disease. Parkinsonism Relat. Disord. 2000, 7, 41–50. [Google Scholar] [CrossRef]
- Horstink, D.M.W.I.M. Parkinson’s disease and parkinsonism in the elderly. Brain 2000, 123, 2569–2571. [Google Scholar] [CrossRef]
- Boxall, J. Early onset Parkinson’s disease. Part 2: Physician’s viewpoint. Can. Fam. Phys. 1994, 40, 513–515. [Google Scholar] [PubMed]
- Foltynie, T.; Sawcer, S.; Brayne, C.; Barker, R.A. The genetic basis of Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2002, 73, 363–370. [Google Scholar] [CrossRef] [Green Version]
- Maroteaux, L.; Campanelli, J.T.; Scheller, R.H. Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 1988, 8, 2804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maguire-Zeiss, K.A. α-Synuclein: A therapeutic target for Parkinson’s disease? Pharm. Res. 2008, 58, 271–280. [Google Scholar] [CrossRef] [Green Version]
- Abbott, A. Levodopa: The story so far. Nature 2010, 466, S6–S7. [Google Scholar] [CrossRef]
- Lesser, R.P.; Fahn, S.; Snider, S.R.; Cote, L.J.; Isgreen, W.P.; Barrett, R.E. Analysis of the clinical problems in parkinsonism and the complications of long-term levodopa therapy. Neurology 1979, 29, 1253–1260. [Google Scholar] [CrossRef]
- Guebila, M.B.; Thiele, I. Model-based dietary optimization for late-stage, levodopa-treated, Parkinson’s disease patients. NPJ Syst. Biol. Appl. 2016, 2, 16013. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, A.; Goodgold, A.; Jonas, S.; Leibowitz, M. Comparison of dopa decarboxylase inhibitor (carbidopa) combined with levodopa and levodopa alone in Parkinson’s disease. Neurology 1975, 25, 911. [Google Scholar] [CrossRef]
- Kaakkola, S.; Männistö, P.T.; Nissinen, E.; Vuorela, A.; Mäntylä, R. The effect of an increased ratio of carbidopa to levodopa on the pharmacokinetics of levodopa. Acta Neurol. Scand. 1985, 72, 385–391. [Google Scholar] [CrossRef]
- Dodd, M.L.; Klos, K.J.; Bower, J.H.; Geda, Y.E.; Josephs, K.A.; Ahlskog, J.E. Pathological gambling caused by drugs used to treat parkinson disease. Arch. Neurol. 2005, 62, 1377–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Lim, E.-C.; Nadkarni, N.V.; Tan, E.-K.; Kumar, P.M. The impact of levodopa therapy-induced complications on quality of life in Parkinson’s disease patients in Singapore. Sci. Rep. 2019, 9, 9248. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Singh, S.; Sharma, V.; Singh, V.P.; Deshmukh, R. Neurobiology of l-DOPA induced dyskinesia and the novel therapeutic strategies. Biomed. Pharmacother. 2015, 70, 283–293. [Google Scholar] [CrossRef]
- AlDakheel, A.; Kalia, L.V.; Lang, A.E. Pathogenesis-targeted, disease-modifying therapies in Parkinson disease. Neurotherapeutics 2014, 11, 6–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonini, A.; Odin, P.; Pahwa, R.; Aldred, J.; Alobaidi, A.; Jalundhwala, Y.J.; Kukreja, P.; Bergmann, L.; Inguva, S.; Bao, Y.; et al. The long-term impact of levodopa/carbidopa intestinal gel on ‘off’-time in patients with advanced parkinson’s disease: A systematic review. Adv. Ther. 2021, 10. [Google Scholar] [CrossRef]
- Prasad, E.M.; Hung, S.Y. Behavioral tests in neurotoxin-induced animal models of parkinson’s disease. Antioxidants 2020, 9, 7. [Google Scholar] [CrossRef]
- Step 3: Clinical Research. Available online: https://www.fda.gov/patients/drug-development-process/step-3-clinical-research (accessed on 26 January 2021).
- Velentgas, P.; Dreyer, N.A.; Wu, A.W. Outcome definition and measurement. In Developing a Protocol for Observational Comparative Effectiveness Research: A User’s Guide; Agency for Healthcare Research and Quality (US): Rockville, MD, USA, 2013. [Google Scholar]
- Merchant, K.M.; Cedarbaum, J.M.; Brundin, P.; Dave, K.D.; Eberling, J.; Espay, A.J.; Hutten, S.J.; Javidnia, M.; Luthman, J.; Maetzler, W.; et al. A proposed roadmap for Parkinson’s disease proof of concept clinical trials investigating compounds targeting alpha-synuclein. J. Parkinson’s Dis. 2019, 9, 31–61. [Google Scholar] [CrossRef] [Green Version]
- Grimes, J.D.; Hassan, M.N.; Thakar, J. Antioxidant therapy in Parkinson’s disease. Can. J. Neurol. Sci. J. Can. Des. Sci. Neurol. 1987, 14, 483–487. [Google Scholar] [CrossRef] [Green Version]
- Annus, Á.; Vécsei, L. Spotlight on opicapone as an adjunct to levodopa in Parkinson’s disease: Design, development and potential place in therapy. Drug Des. Dev. Ther. 2017, 11, 143–151. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, E.V.; Gainetdinov, R.R.; Gurevich, V.V. G protein-coupled receptor kinases as regulators of dopamine receptor functions. Pharm. Res. 2016, 111, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaulieu, J.M.; Gainetdinov, R.R. The physiology, signaling and pharmacology of dopamine receptors. Pharm. Rev. 2011, 63, 182–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Undieh, A.S. Pharmacology of signaling induced by dopamine D(1)-like receptor activation. Pharm. Ther. 2010, 128, 37–60. [Google Scholar] [CrossRef] [Green Version]
- Ghiglieri, V.; Bagetta, V.; Pendolino, V.; Picconi, B.; Calabresi, P. Corticostriatal plastic changes in experimental L-DOPA-induced dyskinesia. Parkinson’s Dis. 2012, 2012, 358176. [Google Scholar] [CrossRef] [Green Version]
- Papapetropoulos, S.; Liu, W.; Duvvuri, S.; Thayer, K.; Gray, D.L. Evaluation of D1/D5 partial agonist PF-06412562 in Parkinson’s disease following oral administration. Neurodegener. Dis. 2018, 18, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Lewis, M.M.; Van Scoy, L.J.; De Jesus, S.; Eslinger, P.J.; Arnold, A.C.; Miller, A.J.; Fernandez-Mendoza, J.; Snyder, B.; Harrington, W.; et al. The D1/D5 dopamine partial agonist PF-06412562 in advanced-stage Parkinson’s disease: A feasibility study. J. Parkinson’s Dis. 2020, 10, 1515–1527. [Google Scholar] [CrossRef] [PubMed]
- Gurrell, R.; Duvvuri, S.; Sun, P.; DeMartinis, N. A phase I study of the safety, tolerability, pharmacokinetics and pharmacodynamics of the novel dopamine D1 receptor partial agonist, PF-06669571, in subjects with idiopathic Parkinson’s disease. Clin. Drug Investig. 2018, 38, 509–517. [Google Scholar] [CrossRef]
- Sohur, U.S.; Gray, D.L.; Duvvuri, S.; Zhang, Y.; Thayer, K.; Feng, G. Phase 1 Parkinson’s disease studies show the dopamine D1/D5 agonist PF-06649751 is safe and well tolerated. Neurol. Ther. 2018, 7, 307–319. [Google Scholar] [CrossRef] [Green Version]
- Riesenberg, R.; Werth, J.; Zhang, Y.; Duvvuri, S.; Gray, D. PF-06649751 efficacy and safety in early Parkinson’s disease: A randomized, placebo-controlled trial. Adv. Neurol. Disord. 2020, 13. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Horner, K.A. Dopamine agonists. In StatPearls; StatPearls Publishing, Copyright © 2020; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2020. [Google Scholar]
- Merims, D.; Giladi, N. Dopamine dysregulation syndrome, addiction and behavioral changes in Parkinson’s disease. Parkinsonism Relat. Disord. 2008, 14, 273–280. [Google Scholar] [CrossRef]
- Constantinescu, R. Update on the use of pramipexole in the treatment of Parkinson’s disease. Neuropsychiatr Dis. Treat. 2008, 4, 337–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silveira, C.R.A.; MacKinley, J.; Coleman, K.; Li, Z.; Finger, E.; Bartha, R.; Morrow, S.A.; Wells, J.; Borrie, M.; Tirona, R.G.; et al. Ambroxol as a novel disease-modifying treatment for Parkinson’s disease dementia: Protocol for a single-centre, randomized, double-blind, placebo-controlled trial. BMC Neurol. 2019, 19, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, T.; Sardi, S.P.; Shihabuddin, L.; Rudin, D.; Sharma, J.; Araujo, R.; Li, J.; Peterschmitt, M.J. Evaluation of glucosylceramide synthase (GCS) inhibition for GBA-associated Parkinson’s disease (P3.051). Neurology 2018, 90. [Google Scholar]
- Goetz, C.G.; Laska, E.; Hicking, C.; Damier, P.; Müller, T.; Nutt, J.; Warren Olanow, C.; Rascol, O.; Russ, H. Placebo influences on dyskinesia in Parkinson’s disease. Mov. Disord. 2008, 23, 700–707. [Google Scholar] [CrossRef] [Green Version]
- Parashos, S.A.; Luo, S.; Biglan, K.M.; Bodis-Wollner, I.; He, B.; Liang, G.S.; Ross, G.W.; Tilley, B.C.; Shulman, L.M. Measuring disease progression in early Parkinson disease: The national institutes of health exploratory trials in Parkinson disease (NET-PD) experience. JAMA Neurol. 2014, 71, 710–716. [Google Scholar] [CrossRef] [PubMed]
- Schwarzschild, M.A.; Schwid, S.R.; Marek, K.; Watts, A.; Lang, A.E.; Oakes, D.; Shoulson, I.; Ascherio, A.; Hyson, C.; Gorbold, E.; et al. Serum urate as a predictor of clinical and radiographic progression in Parkinson disease. Arch. Neurol. 2008, 65, 716–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Postuma, R.B.; Anang, J.; Pelletier, A.; Joseph, L.; Moscovich, M.; Grimes, D.; Furtado, S.; Munhoz, R.P.; Appel-Cresswell, S.; Moro, A.; et al. Caffeine as symptomatic treatment for Parkinson disease (Café-PD): A randomized trial. Neurology 2017, 89, 1795–1803. [Google Scholar] [CrossRef]
- Stefanis, L. α-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef] [Green Version]
- Iwatsubo, T. Aggregation of alpha-synuclein in the pathogenesis of Parkinson’s disease. J. Neurol. 2003, 250, iii11–iii14. [Google Scholar] [CrossRef]
- Bengoa-Vergniory, N.; Roberts, R.F.; Wade-Martins, R.; Alegre-Abarrategui, J. Alpha-synuclein oligomers: A new hope. Acta Neuropathol. 2017, 134, 819–838. [Google Scholar] [CrossRef] [Green Version]
- Fields, C.R.; Bengoa-Vergniory, N.; Wade-Martins, R. Targeting alpha-synuclein as a therapy for Parkinson’s disease. Front. Mol. Neurosci. 2019, 12, 299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobela, W.; Aebischer, P.; Schneider, B.L. Alpha-synuclein as a mediator in the interplay between aging and Parkinson’s disease. Biomolecules 2015, 5, 2675–2700. [Google Scholar] [CrossRef] [Green Version]
- Weihofen, A.; Liu, Y.; Arndt, J.W.; Huy, C.; Quan, C.; Smith, B.A.; Baeriswyl, J.L.; Cavegn, N.; Senn, L.; Su, L.; et al. Development of an aggregate-selective, human-derived α-synuclein antibody BIIB054 that ameliorates disease phenotypes in Parkinson’s disease models. Neurobiol. Dis. 2019, 124, 276–288. [Google Scholar] [CrossRef]
- Jankovic, J.; Goodman, I.; Safirstein, B.; Marmon, T.K.; Schenk, D.B.; Koller, M.; Zago, W.; Ness, D.K.; Griffith, S.G.; Grundman, M.; et al. Safety and tolerability of multiple ascending doses of PRX002/RG7935, an anti-α-synuclein monoclonal antibody, in patients with Parkinson disease: A randomized clinical trial. JAMA Neurol. 2018, 75, 1206–1214. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, J. Results Set Stage for Phase 2 Trial of Investigational Vaccine Affitope PD01A, Affiris Says. Parkinson’s News Today, 24 June 2020. [Google Scholar]
- McFarthing, K.; Simuni, T. Clinical trial highlights: Targeting alpha-synuclein. J. Parkinson’s Dis. 2019, 9, 5–16. [Google Scholar] [CrossRef] [Green Version]
- Analogue-based drug discovery. Chem. Int. 2010, 32, 12–15. [CrossRef]
- Magalhaes, J.; Gegg, M.E.; Migdalska-Richards, A.; Schapira, A.H. Effects of ambroxol on the autophagy-lysosome pathway and mitochondria in primary cortical neurons. Sci. Rep. 2018, 8, 1385. [Google Scholar] [CrossRef]
- Mullin, S.; Smith, L.; Lee, K.; D’Souza, G.; Woodgate, P.; Elflein, J.; Hällqvist, J.; Toffoli, M.; Streeter, A.; Hosking, J.; et al. Ambroxol for the treatment of patients with Parkinson disease with and without glucocerebrosidase gene mutations: A nonrandomized, noncontrolled trial. JAMA Neurol. 2020, 77, 427–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, L.W.; Villemagne, V.L.; Cheng, L.; Sherratt, N.A.; Ayton, S.; White, A.R.; Crouch, P.J.; Lim, S.; Leong, S.L.; Wilkins, S.; et al. The hypoxia imaging agent CuII(atsm) is neuroprotective and improves motor and cognitive functions in multiple animal models of Parkinson’s disease. J. Exp. Med. 2012, 209, 837–854. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.E.; Martinez, A.; Deutsch, G.K.; Prabhakar, V.; Lising, M.; Kapphahn, K.I.; Anidi, C.M.; Neuville, R.; Coburn, M.; Shah, N.; et al. Safety of plasma infusions in Parkinson’s disease. Mov. Disord. 2020. [Google Scholar] [CrossRef] [PubMed]
- Gross, R.E.; Watts, R.L.; Hauser, R.A.; Bakay, R.A.; Reichmann, H.; von Kummer, R.; Ondo, W.G.; Reissig, E.; Eisner, W.; Steiner-Schulze, H.; et al. Intrastriatal transplantation of microcarrier-bound human retinal pigment epithelial cells versus sham surgery in patients with advanced Parkinson’s disease: A double-blind, randomised, controlled trial. Lancet Neurol. 2011, 10, 509–519. [Google Scholar] [CrossRef]
- Ma, Y.; Tang, C.; Chaly, T.; Greene, P.; Breeze, R.; Fahn, S.; Freed, C.; Dhawan, V.; Eidelberg, D. Dopamine cell implantation in Parkinson’s disease: Long-term clinical and (18)F-FDOPA PET outcomes. J. Nucl. Med. 2010, 51, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Kaplitt, M.G.; Feigin, A.; Tang, C.; Fitzsimons, H.L.; Mattis, P.; Lawlor, P.A.; Bland, R.J.; Young, D.; Strybing, K.; Eidelberg, D.; et al. Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson’s disease: An open label, phase I trial. Lancet 2007, 369, 2097–2105. [Google Scholar] [CrossRef]
- Bartus, R.T.; Baumann, T.L.; Siffert, J.; Herzog, C.D.; Alterman, R.; Boulis, N.; Turner, D.A.; Stacy, M.; Lang, A.E.; Lozano, A.M.; et al. Safety/feasibility of targeting the substantia nigra with AAV2-neurturin in Parkinson patients. Neurology 2013, 80, 1698–1701. [Google Scholar] [CrossRef] [Green Version]
- Marks, W.J., Jr.; Bartus, R.T.; Siffert, J.; Davis, C.S.; Lozano, A.; Boulis, N.; Vitek, J.; Stacy, M.; Turner, D.; Verhagen, L.; et al. Gene delivery of AAV2-neurturin for Parkinson’s disease: A double-blind, randomised, controlled trial. Lancet Neurol. 2010, 9, 1164–1172. [Google Scholar] [CrossRef]
- Ghosh, S.; Won, S.J.; Wang, J.; Fong, R.; Butler, N.J.M.; Moss, A.; Wong, C.; Pan, J.; Sanchez, J.; Huynh, A.; et al. α-Synuclein aggregates induce c-Abl activation and dopaminergic neuronal loss by a feed-forward redox stress mechanism. Prog. Neurobiol. 2021, 202, 102070. [Google Scholar] [CrossRef]
- Outeiro, T.F. Alpha-Synuclein. In Reference Module in Neuroscience and Biobehavioral Psychology; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar] [CrossRef]
- Wang, P.; Li, X.; Li, X.; Yang, W.; Yu, S. Blood plasma of patients with Parkinson’s disease increases alpha-synuclein aggregation and neurotoxicity. Parkinson’s Dis. 2016, 2016, 7596482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hisamichi, M.; Kawarazaki, H.; Oroku, M.; Tsuruoka, K.; Sakurada, T.; Shirai, S.; Kido, R.; Kimura, K.; Shibagaki, Y. Risk factors for allergic reaction at initial therapeutic plasma exchange in a single-center study: Beware of high rates of severe allergic reaction. Ren. Replace. Ther. 2016, 2, 67. [Google Scholar] [CrossRef] [Green Version]
- Langston, J.W.; Wiley, J.C.; Tagliati, M. Optimizing Parkinson’s disease diagnosis: The role of a dual nuclear imaging algorithm. NPJ Parkinson’s Dis. 2018, 4, 5. [Google Scholar] [CrossRef]
- Snow, B.; Mulroy, E.; Bok, A.; Simpson, M.; Smith, A.; Taylor, K.; Lockhart, M.; Lam, B.B.J.; Frampton, C.; Schweder, P.; et al. A phase IIb, randomised, double-blind, placebo-controlled, dose-ranging investigation of the safety and efficacy of NTCELL® [immunoprotected (alginate-encapsulated) porcine choroid plexus cells for xenotransplantation] in patients with Parkinson’s disease. Parkinsonism Relat. Disord. 2019, 61, 88–93. [Google Scholar] [CrossRef] [PubMed]
- NTcell. Available online: https://lctglobal.com/research/ntcell (accessed on 26 May 2021).
- Freed, C.R.; Greene, P.E.; Breeze, R.E.; Tsai, W.Y.; DuMouchel, W.; Kao, R.; Dillon, S.; Winfield, H.; Culver, S.; Trojanowski, J.Q.; et al. Transplantation of embryonic dopamine neurons for severe Parkinson’s disease. N. Engl. J. Med. 2001, 344, 710–719. [Google Scholar] [CrossRef]
- Nakamura, T.; Dhawan, V.; Chaly, T.; Fukuda, M.; Ma, Y.; Breeze, R.; Greene, P.; Fahn, S.; Freed, C.; Eidelberg, D. Blinded positron emission tomography study of dopamine cell implantation for Parkinson’s disease. Ann. Neurol. 2001, 50, 181–187. [Google Scholar] [CrossRef]
- Ma, Y.; Feigin, A.; Dhawan, V.; Fukuda, M.; Shi, Q.; Greene, P.; Breeze, R.; Fahn, S.; Freed, C.; Eidelberg, D. Dyskinesia after fetal cell transplantation for parkinsonism: A PET study. Ann. Neurol. 2002, 52, 628–634. [Google Scholar] [CrossRef]
- Trott, C.T.; Fahn, S.; Greene, P.; Dillon, S.; Winfield, H.; Winfield, L.; Kao, R.; Eidelberg, D.; Freed, C.R.; Breeze, R.E.; et al. Cognition following bilateral implants of embryonic dopamine neurons in PD: A double blind study. Neurology 2003, 60, 1938–1943. [Google Scholar] [CrossRef]
- Björklund, A.; Dunnett, S.B.; Brundin, P.; Stoessl, A.J.; Freed, C.R.; Breeze, R.E.; Levivier, M.; Peschanski, M.; Studer, L.; Barker, R. Neural transplantation for the treatment of Parkinson’s disease. Lancet Neurol. 2003, 2, 437–445. [Google Scholar] [CrossRef]
- Freed, C.R.; Leehey, M.A.; Zawada, M.; Bjugstad, K.; Thompson, L.; Breeze, R.E. Do patients with Parkinson’s disease benefit from embryonic dopamine cell transplantation? J. Neurol. 2003, 250 (Suppl. 3), iii44–iii46. [Google Scholar] [CrossRef]
- Gordon, P.H.; Yu, Q.; Qualls, C.; Winfield, H.; Dillon, S.; Greene, P.E.; Fahn, S.; Breeze, R.E.; Freed, C.R.; Pullman, S.L. Reaction time and movement time after embryonic cell implantation in Parkinson disease. Arch. Neurol. 2004, 61, 858–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McRae, C.; Cherin, E.; Yamazaki, T.G.; Diem, G.; Vo, A.H.; Russell, D.; Ellgring, J.H.; Fahn, S.; Greene, P.; Dillon, S.; et al. Effects of perceived treatment on quality of life and medical outcomes in a double-blind placebo surgery trial. Arch. Gen. Psychiatry 2004, 61, 412–420. [Google Scholar] [CrossRef] [Green Version]
- Politis, M.; Lindvall, O. Clinical application of stem cell therapy in Parkinson’s disease. BMC Med. 2012, 10, 1. [Google Scholar] [CrossRef] [Green Version]
- Axelsen, T.M.; Woldbye, D.P.D. Gene therapy for Parkinson’s disease, an update. J. Parkinson’s Dis. 2018, 8, 195–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, A.E.; Gill, S.; Patel, N.K.; Lozano, A.; Nutt, J.G.; Penn, R.; Brooks, D.J.; Hotton, G.; Moro, E.; Heywood, P.; et al. Randomized controlled trial of intraputamenal glial cell line-derived neurotrophic factor infusion in Parkinson disease. Ann. Neurol. 2006, 59, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Isacson, O.; Kordower, J.H. Future of cell and gene therapies for Parkinson’s disease. Ann. Neurol. 2008, 64, S122–S138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, H.D.; Banasr, M.; Duman, R.S. Future antidepressant targets: Neurotrophic factors and related signaling cascades. Drug Discov. Today Strat. 2008, 5, 151–156. [Google Scholar] [CrossRef] [Green Version]
- Valdés, P.; Schneider, B.L. Gene therapy: A promising approach for neuroprotection in Parkinson’s disease? Front. Neuroanat. 2016, 10, 123. [Google Scholar] [CrossRef] [Green Version]
- Bryson, S. Gene Therapy Trial Patients, in Death, Helping Show What Did and Didn’t Work. Parkinson’s News Today, 26 March 2020. [Google Scholar]
- Time to Try Again: Gene-Based Therapy for Neurodegeneration: Society for Neuroscience Annual Meeting 2019. Available online: https://www.alzforum.org/news/conference-coverage/time-try-again-gene-based-therapy-neurodegeneration (accessed on 25 May 2020).
- Coune, P.G.; Schneider, B.L.; Aebischer, P. Parkinson’s disease: Gene therapies. Cold Spring Harb. Perspect. Med. 2012, 2, a009431. [Google Scholar] [CrossRef] [PubMed]
- Hudry, E.; Vandenberghe, L.H. Therapeutic AAV gene transfer to the nervous system: A clinical reality. Neuron 2019, 101, 839–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinderer, C.; Katz, N.; Buza, E.L.; Dyer, C.; Goode, T.; Bell, P.; Richman, L.K.; Wilson, J.M. Severe toxicity in nonhuman primates and piglets following high-dose intravenous administration of an adeno-associated virus vector expressing human SMN. Hum. Gene Ther. 2018, 29, 285–298. [Google Scholar] [CrossRef] [Green Version]
- Sangkuhl, K.; Klein, T.E.; Altman, R.B. Selective serotonin reuptake inhibitors pathway. Pharm. Genom. 2009, 19, 907–909. [Google Scholar] [CrossRef] [Green Version]
- Baumgarten, H.G.; Grozdanovic, Z. Psychopharmacology of central serotonergic systems. Pharmacopsychiatry 1995, 28, 73–79. [Google Scholar] [CrossRef]
- Tani, Y.; Ogata, A.; Koyama, M.; Inoue, T. Effects of piclozotan (SUN N4057), a partial serotonin 1A receptor agonist, on motor complications induced by repeated administration of levodopa in parkinsonian rats. Eur. J. Pharmacol. 2010, 649, 218–223. [Google Scholar] [CrossRef]
- Abdala, A.P.; Lioy, D.T.; Garg, S.K.; Knopp, S.J.; Paton, J.F.R.; Bissonnette, J.M. Effect of sarizotan, a 5-HT1a and D2-like receptor agonist, on respiration in three mouse models of rett syndrome. Am. J. Respir. Cell Mol. Biol. 2014, 50, 1031–1039. [Google Scholar] [CrossRef] [Green Version]
- SYN120 Fails to Show Efficacy Against Parkinson Dementia in Phase 2a SYNAPSE Trial. Available online: https://consultqd.clevelandclinic.org/syn120-fails-to-show-efficacy-against-parkinson-dementia-in-phase-2a-synapse-trial/ (accessed on 20 November 2020).
- Brea, J.; Castro-Palomino, J.; Yeste, S.; Cubero, E.; Párraga, A.; Domínguez, E.; Loza, M.I. Emerging opportunities and concerns for drug discovery at serotonin 5-HT2B receptors. Curr. Top. Med. Chem. 2010, 10, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in Parkinson’s disease. J. Parkinson’s Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C. Targeting the cholinergic system in Parkinson’s disease. Acta Pharmacol. Sin. 2020, 41, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Rascol, O.; Poewe, W.; Lees, A.; Aristin, M.; Salin, L.; Juhel, N.; Waldhauser, L.; Schindler, T.; Group, A.S. Tesofensine (NS 2330), a monoamine reuptake inhibitor, in patients with advanced Parkinson disease and motor fluctuations: The ADVANS study. Arch. Neurol. 2008, 65, 577–583. [Google Scholar] [CrossRef] [Green Version]
- Hauser, R.A.; Salin, L.; Juhel, N.; Konyago, V.L. Randomized trial of the triple monoamine reuptake inhibitor NS 2330 (tesofensine) in early Parkinson’s disease. Mov. Disord. 2007, 22, 359–365. [Google Scholar] [CrossRef]
- Bara-Jimenez, W.; Dimitrova, T.; Sherzai, A.; Favit, A.; Mouradian, M.M.; Chase, T.N. Effect of monoamine reuptake inhibitor NS 2330 in advanced Parkinson’s disease. Mov. Disord. 2004, 19, 1183–1186. [Google Scholar] [CrossRef] [Green Version]
- Carlson, A.B.; Kraus, G.P. Physiology, cholinergic receptors. In StatPearls; StatPearls Publishing, Copyright © 2020. StatPearls Publishing LLC.: Treasure Island, FL, USA, 2020. [Google Scholar]
- Villard, V.; Espallergues, J.; Keller, E.; Vamvakides, A.; Maurice, T. Anti-amnesic and neuroprotective potentials of the mixed muscarinic receptor/sigma1 (σ1) ligand ANAVEX2-73, a novel aminotetrahydrofuran derivative. J. Psychopharmacol. 2010, 25, 1101–1117. [Google Scholar] [CrossRef]
- Villafane, G.; Thiriez, C.; Audureau, E.; Straczek, C.; Kerschen, P.; Cormier-Dequaire, F.; Van Der Gucht, A.; Gurruchaga, J.M.; Quéré-Carne, M.; Evangelista, E.; et al. High-dose transdermal nicotine in Parkinson’s disease patients: A randomized, open-label, blinded-endpoint evaluation phase 2 study. Eur. J. Neurol. 2018, 25, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, M.M. Medical management of Parkinson’s disease. Pharm. Ther. 2008, 33, 590–606. [Google Scholar]
- Barth, A.L.; Schneider, J.S.; Johnston, T.H.; Hill, M.P.; Brotchie, J.M.; Moskal, J.R.; Cearley, C.N. NYX-458 improves cognitive performance in a primate Parkinson’s disease model. Mov. Disord. 2020, 35, 640–649. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Houck, D.R.; Gross, A.L.; Zhang, X.L.; Cearley, C.; Madsen, T.M.; Kroes, R.A.; Stanton, P.K.; Burgdorf, J.; Moskal, J.R. NYX-2925 Is a novel NMDA receptor-specific spirocyclic-β-lactam that modulates synaptic plasticity processes associated with learning and memory. Int J. Neuropsychopharmacol 2018, 21, 242–254. [Google Scholar] [CrossRef] [PubMed]
- Wood, P.L. NMDA antagonists for stroke and head trauma: Current status. Expert Opin. Investig. Drugs 1998, 7, 1505–1508. [Google Scholar] [CrossRef]
- Olivares, D.; Deshpande, V.K.; Shi, Y.; Lahiri, D.K.; Greig, N.H.; Rogers, J.T.; Huang, X. N-methyl D-aspartate (NMDA) receptor antagonists and memantine treatment for Alzheimer’s disease, vascular dementia and Parkinson’s disease. Curr. Alzheimer Res. 2012, 9, 746–758. [Google Scholar] [CrossRef] [PubMed]
- Andringa, G.; Eshuis, S.; Perentes, E.; Maguire, R.P.; Roth, D.; Ibrahim, M.; Leenders, K.L.; Cools, A.R. TCH346 prevents motor symptoms and loss of striatal FDOPA uptake in bilaterally MPTP-treated primates. Neurobiol. Dis. 2003, 14, 205–217. [Google Scholar] [CrossRef]
- Grotegut, P.; Perumal, N.; Kuehn, S.; Smit, A.; Dick, H.B.; Grus, F.H.; Joachim, S.C. Minocycline reduces inflammatory response and cell death in a S100B retina degeneration model. J. Neuroinflamm. 2020, 17, 375. [Google Scholar] [CrossRef] [PubMed]
- West, A.B. Achieving neuroprotection with LRRK2 kinase inhibitors in Parkinson disease. Exp. Neurol. 2017, 298, 236–245. [Google Scholar] [CrossRef]
- Maroney, A.C.; Finn, J.P.; Connors, T.J.; Durkin, J.T.; Angeles, T.; Gessner, G.; Xu, Z.; Meyer, S.L.; Savage, M.J.; Greene, L.A.; et al. Cep-1347 (KT7515), a semisynthetic inhibitor of the mixed lineage kinase family. J. Biol. Chem. 2001, 276, 25302–25308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mixed lineage kinase inhibitor CEP-1347 fails to delay disability in early Parkinson disease. Neurology 2007, 69, 1480–1490. [CrossRef]
- Mandhane, S.D.S.; Jani, K.; Sengupta, P.; Patel, A.; Bambal, R.; Ramanathan, V.; Zala, Y.; Dharmadhikari, N.; Rao, C.; Raghavan, A.; et al. K0706, a potent orally bioavailable brain-penetrating selective inhibitor of cABL protein tyrosine kinase, exhibits neuroprotective activity in preclinical models of Parkinson’s disease. In Proceedings of the MDS Abstracts, Agora 3 West, Level 3, Nice, France, 22–26 September 2019; p. 970. [Google Scholar]
- Lodish, M.B. Clinical review: Kinase inhibitors: Adverse effects related to the endocrine system. J. Clin. Endocrinol. Metab. 2013, 98, 1333–1342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, J.T.; Haap, M.; Kopp, H.G.; Lipp, H.P. Tyrosine kinase inhibitors-a review on pharmacology, metabolism and side effects. Curr. Drug Metab. 2009, 10, 470–481. [Google Scholar] [CrossRef]
- Jucaite, A.; Svenningsson, P.; Rinne, J.O.; Cselényi, Z.; Varnäs, K.; Johnström, P.; Amini, N.; Kirjavainen, A.; Helin, S.; Minkwitz, M.; et al. Effect of the myeloperoxidase inhibitor AZD3241 on microglia: A PET study in Parkinson’s disease. Brain 2015, 138, 2687–2700. [Google Scholar] [CrossRef] [Green Version]
- Gellhaar, S.; Sunnemark, D.; Eriksson, H.; Olson, L.; Galter, D. Myeloperoxidase-immunoreactive cells are significantly increased in brain areas affected by neurodegeneration in Parkinson’s and Alzheimer’s disease. Cell Tissue Res. 2017, 369, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Navarro, G. Adenosine A2A receptor antagonists in neurodegenerative diseases: Huge potential and huge challenges. Front. Psychiatry 2018, 9, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vernalis Licenses Proprietary Adenosine Receptor Antagonist Technology, Including Lead Compound V81444, For, Development and Commercialisation. Available online: https://www.vernalis.com/vernalis-licenses-proprietary-adenosine-receptor-antagonist-technology-including-lead-compound-v81444-for-development-and-commercialisation/ (accessed on 26 November 2020).
- Cheong, S.L.; Federico, S.; Spalluto, G.; Klotz, K.-N.; Pastorin, G. The current status of pharmacotherapy for the treatment of Parkinson’s disease: Transition from single-target to multitarget therapy. Drug Discov. Today 2019, 24, 1769–1783. [Google Scholar] [CrossRef]
- Chen, J.-F.; Xu, K.; Petzer, J.P.; Staal, R.; Xu, Y.-H.; Beilstein, M.; Sonsalla, P.K.; Castagnoli, K.; Castagnoli, N.; Schwarzschild, M.A. Neuroprotection by caffeine and A2A adenosine receptor inactivation in a model of Parkinson’s disease. J. Neurosci. 2001, 21, RC143. [Google Scholar] [CrossRef] [Green Version]
- Kumar, H.; Lim, H.-W.; More, S.V.; Kim, B.-W.; Koppula, S.; Kim, I.S.; Choi, D.-K. The role of free radicals in the aging brain and Parkinson’s disease: Convergence and parallelism. Int. J. Mol. Sci. 2012, 13, 10478–10504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.-W.; Lin, C.C.; Chen, Y.-H.; Yang, H.-B.; Hung, S.-Y. Celastrol inhibits dopaminergic neuronal death of Parkinson’s disease through activating mitophagy. Antioxidants 2020, 9, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sechi, G.; Deledda, M.G.; Bua, G.; Satta, W.M.; Deiana, G.A.; Pes, G.M.; Rosati, G. Reduced intravenous glutathione in the treatment of early Parkinson’s disease. Prog. Neuro Psychopharmacol. Biol. Psychiatry 1996, 20, 1159–1170. [Google Scholar] [CrossRef]
- Lobo, V.; Patil, A.; Phatak, A.; Chandra, N. Free radicals, antioxidants and functional foods: Impact on human health. Pharm. Rev. 2010, 4, 118–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Li, H.; Kim, H.; Yang, H.O. Protective effect of WIN-1001X, an herbal extract, on Parkinson’s disease experimental model via autophagy enhancement. Planta Med. 2014, 80, P1L98. [Google Scholar] [CrossRef]
- Ojo-Amaize, E.A.; Cottam, H.B. Short review of current research on the development of hypoestoxide as a therapeutic agent for Parkinson’s disease. J. Neurol Neurophysiol. 2016, 7, 2. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Ojo-Amaize, E.; Spencer, B.; Rockenstein, E.; Mante, M.; Desplats, P.; Wrasidlo, W.; Adame, A.; Nchekwube, E.; Oyemade, O.; et al. Hypoestoxide reduces neuroinflammation and α-synuclein accumulation in a mouse model of Parkinson’s disease. J. Neuroinflammation 2015, 12, 236. [Google Scholar] [CrossRef] [Green Version]
- Phase 2A Study of GM 608 in Mild to Moderate Parkinson Disease: GM 608 in A Phase IIA Pilot Double-blinded, Randomized, Placebo Controlled Trial in Mild to Moderate Parkinson Disease. Available online: https://ichgcp.net/clinical-trials-registry/NCT01850381 (accessed on 24 December 2020).
- McFarthing, K.; Larson, D.; Simuni, T. Clinical trial highlights-GLP-1 agonists. J. Parkinson’s Dis. 2020, 10, 355–368. [Google Scholar] [CrossRef] [Green Version]
- Dorsey, E.R.; Sherer, T.; Okun, M.S.; Bloem, B.R. The emerging evidence of the parkinson pandemic. J. Parkinson’s Dis. 2018, 8, S3–S8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, K.L. Diagnosis and Differential Diagnosis of Parkinson Disease; UpToDate: Waltham, MA, USA, 2017. [Google Scholar]
- Manfredsson, F.P.; Polinski, N.K.; Subramanian, T.; Boulis, N.; Wakeman, D.R.; Mandel, R.J. The future of GDNF in Parkinson’s disease. Front. Aging Neurosci. 2020, 12, 388. [Google Scholar] [CrossRef]
- Verschuur, C.V.M.; Suwijn, S.R.; Boel, J.A.; Post, B.; Bloem, B.R.; van Hilten, J.J.; van Laar, T.; Tissingh, G.; Munts, A.G.; Deuschl, G.; et al. Randomized delayed-start trial of levodopa in Parkinson’s disease. N. Engl. J. Med. 2019, 380, 315–324. [Google Scholar] [CrossRef]
- DeMaagd, G.; Philip, A. Parkinson’s disease and its management: Part 3: Nondopaminergic and nonpharmacological treatment options. Pharm. Ther. 2015, 40, 668–679. [Google Scholar]
- Münchau, A.; Bhatia, K.P. Pharmacological treatment of Parkinson’s disease. Postgrad. Med. J. 2000, 76, 602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, J.; Cui, Y.; Li, S.; Le, W. Current pharmaceutical treatments and alternative therapies of Parkinson’s disease. Curr. Neuropharmacol. 2016, 14, 339–355. [Google Scholar] [CrossRef] [PubMed]
- Dodel, R.C.; Eggert, K.M.; Singer, M.S.; Eichhorn, T.E.; Pogarell, O.; Oertel, W.H. Costs of drug treatment in Parkinson’s disease. Mov. Disord. 1998, 13, 249–254. [Google Scholar] [CrossRef]
- Charles, D.; Tolleson, C.; Davis, T.L.; Gill, C.E.; Molinari, A.L.; Bliton, M.J.; Tramontana, M.G.; Salomon, R.M.; Kao, C.; Wang, L.; et al. Pilot study assessing the feasibility of applying bilateral subthalamic nucleus deep brain stimulation in very early stage Parkinson’s disease: Study design and rationale. J. Parkinson’s Dis. 2012, 2, 215–223. [Google Scholar] [CrossRef] [Green Version]
- Hamani, C.; Pilitsis, J.; Rughani, A.I.; Rosenow, J.M.; Patil, P.G.; Slavin, K.S.; Abosch, A.; Eskandar, E.; Mitchell, L.S.; Kalkanis, S. Deep brain stimulation for obsessive-compulsive disorder: Systematic review and evidence-based guideline sponsored by the American society for stereotactic and functional neurosurgery and the congress of neurological surgeons (CNS) and endorsed by the CNS and American association of neurological surgeons. Neurosurgery 2014, 75, 327–333. [Google Scholar] [CrossRef]
- Follett, K.A.; Weaver, F.M.; Stern, M.; Hur, K.; Harris, C.L.; Luo, P.; Marks, W.J.; Rothlind, J.; Sagher, O.; Moy, C.; et al. Pallidal versus subthalamic deep-brain stimulation for Parkinson’s disease. N. Engl. J. Med. 2010, 362, 2077–2091. [Google Scholar] [CrossRef] [Green Version]
- Weaver, F.M.; Follett, K.A.; Stern, M.; Luo, P.; Harris, C.L.; Hur, K.; Marks, W.J., Jr.; Rothlind, J.; Sagher, O.; Moy, C.; et al. Randomized trial of deep brain stimulation for Parkinson disease: Thirty-six-month outcomes. Neurology 2012, 79, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Lozano, A.M.; Lipsman, N.; Bergman, H.; Brown, P.; Chabardes, S.; Chang, J.W.; Matthews, K.; McIntyre, C.C.; Schlaepfer, T.E.; Schulder, M.; et al. Deep brain stimulation: Current challenges and future directions. Nat. Rev. Neurol. 2019, 15, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Dang, T.T.H.; Rowell, D.; Connelly, L.B. Cost-effectiveness of deep brain stimulation with movement disorders: A systematic review. Mov. Disord. Clin. Pract. 2019, 6, 348–358. [Google Scholar] [CrossRef]
- Nirenberg, M.J.; Waters, C. Compulsive eating and weight gain related to dopamine agonist use. Mov. Disord. 2006, 21, 524–529. [Google Scholar] [CrossRef]
- Bonuccelli, U.; Del Dotto, P.; Rascol, O. Role of dopamine receptor agonists in the treatment of early Parkinson’s disease. Parkinsonism Relat. Disord. 2009, 15, S44–S53. [Google Scholar] [CrossRef]
- Kujawa, K.; Leurgans, S.; Raman, R.; Blasucci, L.; Goetz, C.G. Acute orthostatic hypotension when starting dopamine agonists in Parkinson’s disease. Arch. Neurol. 2000, 57, 1461–1463. [Google Scholar] [CrossRef] [Green Version]
- Hypersexuality due to dopaminergic drugs. Prescrire Int. 2005, 14, 224.
- Pinder, R.M. Pathological gambling and dopamine agonists: A phenotype? Neuropsychiatr. Dis. Treat. 2007, 3, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Borovac, J.A. Side effects of a dopamine agonist therapy for Parkinson’s disease: A mini-review of clinical pharmacology. Yale J. Biol. Med. 2016, 89, 37–47. [Google Scholar] [PubMed]
- Chia, R.; Sabir, M.S.; Bandres-Ciga, S.; Saez-Atienzar, S.; Reynolds, R.H.; Gustavsson, E.; Walton, R.L.; Ahmed, S.; Viollet, C.; Ding, J.; et al. Genome sequencing analysis identifies new loci associated with Lewy body dementia and provides insights into its genetic architecture. Nat. Genet. 2021, 53, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Goswami, R.; Subramanian, G.; Silayeva, L.; Newkirk, I.; Doctor, D.; Chawla, K.; Chattopadhyay, S.; Chandra, D.; Chilukuri, N.; Betapudi, V. Gene therapy leaves a vicious cycle. Front. Oncol. 2019, 9, 297. [Google Scholar] [CrossRef]
- Qian, H.-Z.; Vermund, S.H.; Kaslow, R.A.; Coffey, C.S.; Chamot, E.; Yang, Z.; Qiao, X.; Zhang, Y.; Shi, X.; Jiang, Y.; et al. Co-infection with HIV and hepatitis C virus in former plasma/blood donors: Challenge for patient care in rural China. AIDS 2006, 20, 1429–1435. [Google Scholar] [CrossRef] [Green Version]
- Kleinman, S.H.; Lelie, N.; Busch, M.P. Infectivity of human immunodeficiency virus-1, hepatitis C virus, and hepatitis B virus and risk of transmission by transfusion. Transfusion 2009, 49, 2454–2489. [Google Scholar] [CrossRef]
- Cicchetti, A.; Berrino, A.; Casini, M.; Codella, P.; Facco, G.; Fiore, A.; Marano, G.; Marchetti, M.; Midolo, E.; Minacori, R.; et al. Health technology assessment of pathogen reduction technologies applied to plasma for clinical use. Blood Transfus. 2016, 14, 287–386. [Google Scholar] [CrossRef]
- From the centers for disease control and prevention. Outbreak of hepatitis c associated with intravenous immunoglobulin administration--United States, October 1993–June 1994. JAMA 1994, 272, 424–425. [CrossRef]
- Hellstern, P.; Solheim, B.G. The Use of solvent/detergent treatment in pathogen reduction of plasma. Transfus. Med. Hemother. 2011, 38, 65–70. [Google Scholar] [CrossRef] [PubMed]
- GrÖNer, A. Pathogen safety of plasma-derived products–Haemate® P/Humate-P®. Haemophilia 2008, 14, 54–71. [Google Scholar] [CrossRef]
- Guo, Y.; Tian, X.; Wang, X.; Xiao, Z. Adverse effects of immunoglobulin therapy. Front. Immunol. 2018, 9, 1299. [Google Scholar] [CrossRef]
- Brecher, M.E.; Hay, S.N. Bacterial contamination of blood components. Clin. Microbiol. Rev. 2005, 18, 195–204. [Google Scholar] [CrossRef] [Green Version]
- Hingorani, A.D.; Kuan, V.; Finan, C.; Kruger, F.A.; Gaulton, A.; Chopade, S.; Sofat, R.; MacAllister, R.J.; Overington, J.P.; Hemingway, H.; et al. Improving the odds of drug development success through human genomics: Modelling study. Sci. Rep. 2019, 9, 18911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becerra, J.E.; Zorro, O.; Ruiz-Gaviria, R.; Castañeda-Cardona, C.; Otálora-Esteban, M.; Henao, S.; Navarrete, S.; Acevedo, J.C.; Rosselli, D. Economic analysis of deep brain stimulation in Parkinson disease: Systematic review of the literature. World Neurosurg. 2016, 93, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Marras, C.; Beck, J.C.; Bower, J.H.; Roberts, E.; Ritz, B.; Ross, G.W.; Abbott, R.D.; Savica, R.; Van Den Eeden, S.K.; Willis, A.W.; et al. Prevalence of Parkinson’s disease across North America. NPJ Parkinson’s Dis. 2018, 4, 21. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapeutic Strategy | Classification | Name | PD Subjects | Trial Status | Reasons for Discontinuation | Sponsor | ClinicalTrials.gov Identifier and Reference |
|---|---|---|---|---|---|---|---|
| Dopamine receptor agonists | Small molecular Dopamine D1/D5 partial agonist | PF-06412562 | Advanced-stage PD | Phase I | Not applicable | Milton S. Hershey Medical Center | NCT03665454 [30] |
| Dopamine receptor agonists | Small molecular Dopamine D1 partial agonist | PF-06669571 | Idiopathic PD | Phase I | Not applicable | Pfizer | NCT02565628 [31] |
| Dopamine receptor agonists | Small molecular Dopamine D1/D5 partial agonist | PF-06649751 | Idiopathic PD | Phase I | Not applicable | Pfizer | NCT02224664 [32] |
| Dopamine receptor agonists | Small molecular Dopamine D1/D5 partial agonist | PF-06649751 | Early stage PD | Phase II discontinued | Terminated due to lack of efficacy in moderate/advanced PD. | Pfizer | NCT02847650 [33] |
| Dopamine receptor agonists | Small molecular Dopamine D1/D5 partial agonist | PF-06649751 | PD with motor fluctuations | Phase II discontinued | Terminated due to insufficient efficacy. | Pfizer | NCT02687542 |
| Dopamine receptor agonists | Small molecular Dopamine D2 agonist | CJH1 (CLR4001) | PD patients | Phase I/II | Unknown | Alexandra Marine and General Hospital | NCT01684475 |
| Anti-α-synuclein aggregation therapy | Small molecular Dock with β-glucocerebrosidase to increase its levels and decrease the cerebrospinal fluid α-synuclein level | Ambroxol | PD with dementia | Phase II | Not applicable | Lawson Health Research Institute | NCT02914366 [49] |
| Anti-α-synuclein aggregation therapy | Small molecular Peroxynitrite scavenger | Cu(II)ATSM | Early idiopathic PD | Phase I | Not applicable | Collaborative Medicinal Development Pty Limited | NCT03204929 |
| Gene therapy | Small molecular glucocerebrosidase (GBA) gene mutating therapy | GZ/SAR402671 | Early stage PD | Phase II discontinued | Terminated due to not meeting the primary and secondary endpoints. | Genzyme | NCT02906020 [53] |
| Serotonin receptor agonists or antagonists | Small molecular Dual 5-HT6/5-HT2 antagonist | SYN120 | PD with dementia | Phase II | Not applicable | Biotie Therapies Inc. | NCT02258152 |
| Serotonin receptor agonists or antagonists | Small molecular Selective 5-HT1A partial agonist | Piclozotan (SUN N4057) | Idiopathic PD | Phase II | Not applicable | Daiichi Sankyo, Inc. | NCT00623363 |
| Serotonin receptor agonists or antagonists | Small molecular Selective 5-HT1A agonist and D2 antagonist | Sarizotan | Idiopathic PD | Phase III | Not applicable | EMD Serono | NCT00105508 [54] |
| Monoamine reuptake inhibitors | Small molecular Triple monoamine reuptake inhibitor (serotonin, noradrenaline, and dopamine reuptake inhibitor) | NS 2330 (tesofensine) | Early stage PD | Phase II | Not applicable | Boehringer Ingelheim | NCT00148486 |
| Muscarinic and nicotinic acetylcholine receptor agonists | Small molecular Muscarinic agonist and sigma1 agonist | ANAVEX2-73 | PD with dementia | Phase II | Not applicable | Anavex Life Sciences Corp. | NCT04575259 |
| Muscarinic and nicotinic acetylcholine receptor agonists | Small molecular Nicotinic agonist | Nicotine transdermal patch | Early stage PD | Phase II | Unknown | James BOYD MD | NCT01560754 |
| Muscarinic and nicotinic acetylcholine receptor agonists | Small molecular Nicotinic agonist | Nicotine nasal spray | PD (Hoehn and Yahr stage 2–3) | Phase II | Not applicable | El Instituto Nacional de Neurologia y Neurocirugia Manuel Velasco Suarez | NCT03865121 |
| N-methyl-D-aspartate receptor (NMDAR) modulators | Small molecular NMDAR modulator | NYX-458 | Mild cognitive impairment associated with PD | Phase II | Not applicable | Aptinyx | NCT04148391 |
| NMDAR modulator | Small molecular D-amino acid oxidase inhibitor | DAAOI-P | PD with dementia | Phase II | Not applicable | China Medical University Hospital | NCT04470037 |
| Anti-apoptotic drugs | Small molecular Dibenz[b,f]oxepin-10-ylmethyl-prop-2-ynyl-amine, hydrogen maleate salt | TCH346 | Early stage PD | Phase I/II | Not applicable | Novartis | NCT00407212 |
| Anti-apoptotic drugs | Small molecular Synthetic tetracycline derivative | Minocycline | Early stage untreated PD | Phase II | Not applicable | University of Rochester | NCT00063193 [55] |
| Kinase inhibitors | Small molecular Semisynthetic inhibitor of the mixed lineage kinase family | CEP-1347 (KT7515) | Early stage PD | Phase II/III discontinued | Terminated due to insufficient efficacy. | Cephalon | NCT00040404 [56] |
| Kinase inhibitors | Small molecular Orally selective inhibitor of cABL protein tyrosine kinase | K0706 | Early stage PD | Phase II | Not applicable | Sun Pharma Advanced Research Company Limited | NCT03655236 |
| Myeloperoxidase inhibitors | Small molecular Irreversible myeloperoxidase inhibitor | AZD3241 | Idiopathic PD | Phase II | Not applicable | AstraZeneca | NCT01603069 |
| Adenosine A2A receptor antagonists | Small molecular Adenosine A2A antagonist | V81444 | PD patients | Phase I | Not applicable | Vernalis (R&D) Ltd. | NCT02764892 |
| Adenosine A2A receptor antagonists | Small molecular Selective Adenosine A2A antagonist | Caffeine | PD (Hoehn and Yahr stage 1–3) | Phase III | Not applicable | McGill University Health Centre/Research Institute of the McGill University Health Centre | NCT01738178 [57] |
| Antioxidants | Small molecular Intranasal glutathione therapy | Reduced glutathione | PD (modified Hoehn and Yahr stage < 3) | Phase I | Not applicable | Bastyr University | NCT01398748 |
| Antioxidants | Small molecular Intranasal reduced glutathione | Reduced glutathione | PD (Hoehn and Yahr stage 2–3) | Phase I | Not applicable | University of Washington | NCT02324426 |
| Others | Small molecular Synthetic oligopeptide | GM 608 | Mild to moderate-stage PD | Phase II | Not applicable | Genervon Biopharmaceuticals, LLC | NCT01850381 |
| Others | Small molecular Glucagon-like peptide 1 receptor agonist | NLY01 | Early stage PD | Phase II | Not applicable | Neuraly, Inc. | NCT04154072 |
| Therapeutic Strategy | Classification | Name | PD Subjects | Trial Status | Reasons for Discontinuation | Sponsor | ClinicalTrials.gov Identifier and Reference |
|---|---|---|---|---|---|---|---|
| Anti-α-synuclein aggregation therapy | Monoclonal antibody | ABBV-0805 | Idiopathic PD | Phase I discontinued | Withdrawn due to strategic considerations. | AbbVie | NCT04127695 |
| Anti-α-synuclein aggregation therapy | Vaccine Short synthetic peptides | AFFITOPE® PD01A | Early stage PD | Phase I | Not applicable | Affiris AG | NCT01568099 [45] |
| Anti-α-synuclein aggregation therapy | Monoclonal antibody IgG1 protein produced from memory B cells | BIIB054 | PD patients | Phase II discontinued | Terminated due to lack of efficacy. | Biogen | NCT03318523 |
| Anti-α-synuclein aggregation therapy | Monoclonal antibody | PRX002 (Prasinezumab/ RO7046015) | Idiopathic PD | Phase I | Not applicable | Prothena Biosciences Limited | NCT02157714 [43] |
| Anti-α-synuclein aggregation therapy | Monoclonal antibody | PRX002 (Prasinezumab/ RO7046015) | Early stage PD | Phase II | Not applicable | Hoffmann-La Roche | NCT03100149 [43] |
| Convalescent plasma therapy | Young plasma infusions | Infusions of young plasma | Moderate-stage PD | Phase I | Not applicable | Stanford University | NCT02968433 [58] |
| Cell-based therapy | Injection cultured human retinal pigment epithelial cells into both hemispheres | Spheramine/ BAY86-5280 | Advanced-stage PD | Phase II discontinued | Terminated. The trial was completed, and only the lifelong extended follow-up phase was discontinued after 12 years. | Bayer | NCT00206687 [62] |
| Cell-based therapy | Xenotransplantation of immunoprotected (alginate-encapsulated) choroid plexus cells in the brain | NTCELL | Idiopathic PD | Phase I/II | Not applicable | Living Cell Technologies | NCT01734733 |
| Cell-based therapy | Xenotransplantation of immunoprotected (alginate-encapsulated) choroid plexus cells in the brain | NTCELL | Idiopathic PD | Phase II | Not applicable | Living Cell Technologies | NCT02683629 |
| Cell-based therapy | Neuronal progenitor stem cells | Adult neuronal progenitor stem cell | PD | Phase II discontinued | The study was withdrawn before participants were enrolled. | Rajavithi Hospital | NCT00927108 |
| Cell-based therapy | Embryonic dopamine cell implant | Embryonic dopamine cell implant surgery | Idiopathic PD | Phase III | Not applicable | University of Colorado, Denver | NCT00038116 [73] |
| Gene therapy | AAV2-GDNF delivered to the putamen | AAV2-GDNF | Mild to moderate and moderate to severe PD | Phase I | Not applicable | Brain Neurotherapy Bio, Inc. | NCT04167540 |
| Gene therapy | Surgical infusion of AAV-GAD into the subthalamic nucleus | Glutamic acid decarboxylase (GAD) gene therapy | Advanced-stage PD | Phase I | Not applicable | Neurologix, Inc. | NCT00195143 [80] |
| Gene therapy | Adeno-associated virus delivery of neurturin gene in the substantia nigra and putamen | CERE-120 | Idiopathic PD | Phase I/II | Not applicable | Sangamo Therapeutics | NCT00985517 [81] |
| Gene therapy | Glucocerebrosidase gene therapy by intra cisterna magna administration | PR001A | Moderate to severe PD | Phase I/IIa | Not applicable | Prevail Therapeutics | NCT04127578 |
| Gene therapy | AAV2-neurturin gene therapy | CERE-120 | Idiopathic PD | Phase II | Not applicable | Sangamo Therapeutics (Ceregene) | NCT00400634 [83] |
| Antioxidants and botanical-based medication | Plant-based herbal dry powder | Hypoestoxide | PD | Phase I/II | Not applicable | Adesola Ogunniyi, University of Ibadan | NCT04858074 |
| Antioxidants and botanical-based medication | Plant-based herbal extract | WIN-1001X | Early stage PD | Phase II | Not applicable | Medi Help Line | NCT04220762 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prasad, E.M.; Hung, S.-Y. Current Therapies in Clinical Trials of Parkinson’s Disease: A 2021 Update. Pharmaceuticals 2021, 14, 717. https://doi.org/10.3390/ph14080717
Prasad EM, Hung S-Y. Current Therapies in Clinical Trials of Parkinson’s Disease: A 2021 Update. Pharmaceuticals. 2021; 14(8):717. https://doi.org/10.3390/ph14080717
Chicago/Turabian StylePrasad, E. Maruthi, and Shih-Ya Hung. 2021. "Current Therapies in Clinical Trials of Parkinson’s Disease: A 2021 Update" Pharmaceuticals 14, no. 8: 717. https://doi.org/10.3390/ph14080717
APA StylePrasad, E. M., & Hung, S.-Y. (2021). Current Therapies in Clinical Trials of Parkinson’s Disease: A 2021 Update. Pharmaceuticals, 14(8), 717. https://doi.org/10.3390/ph14080717