
Fluorescence Cross-Correlation Spectroscopy Yields True Affinity and Binding Kinetics of Plasmodium Lactate Transport Inhibitors

 and
and
Abstract
:1. Introduction
2. Results
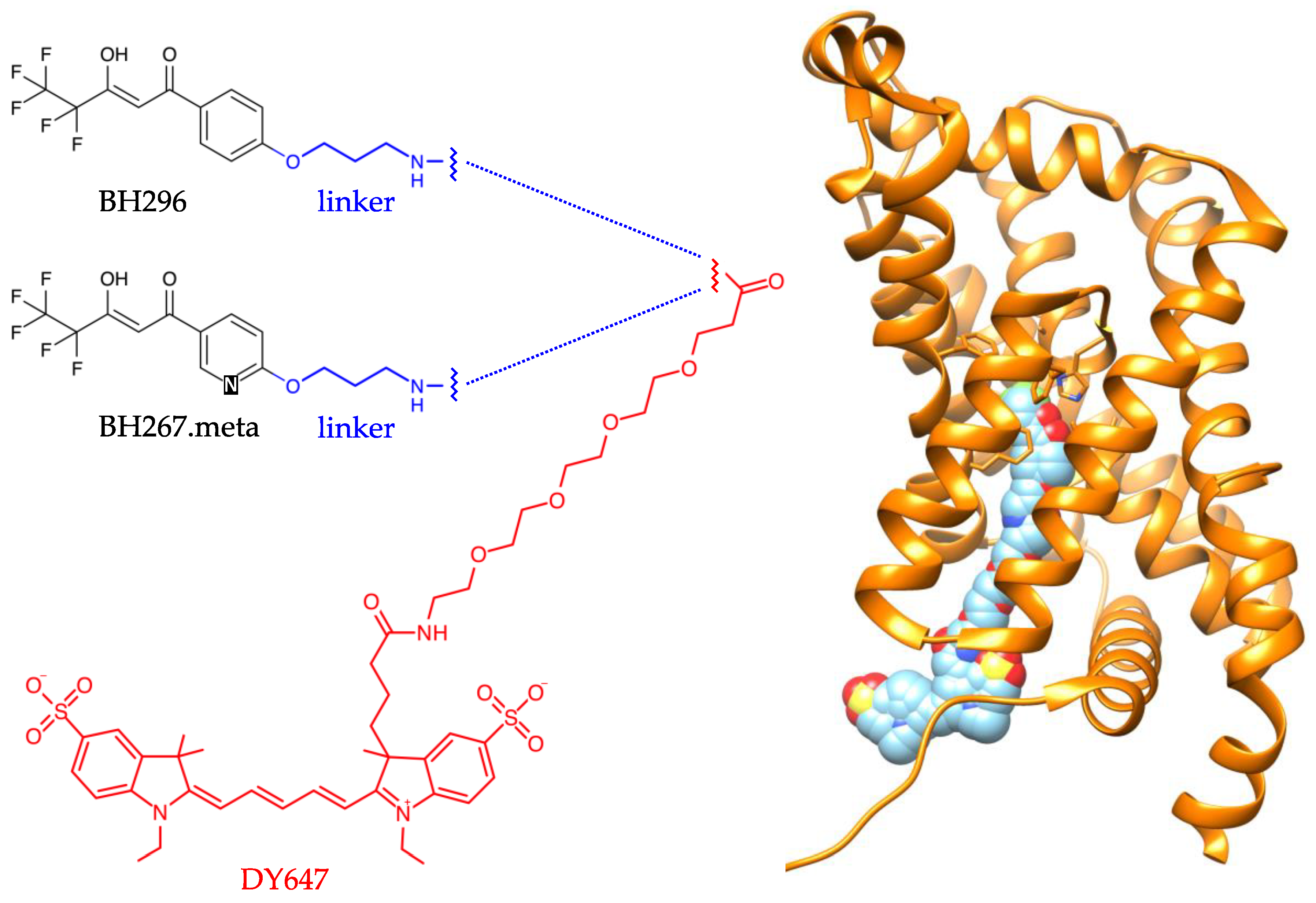
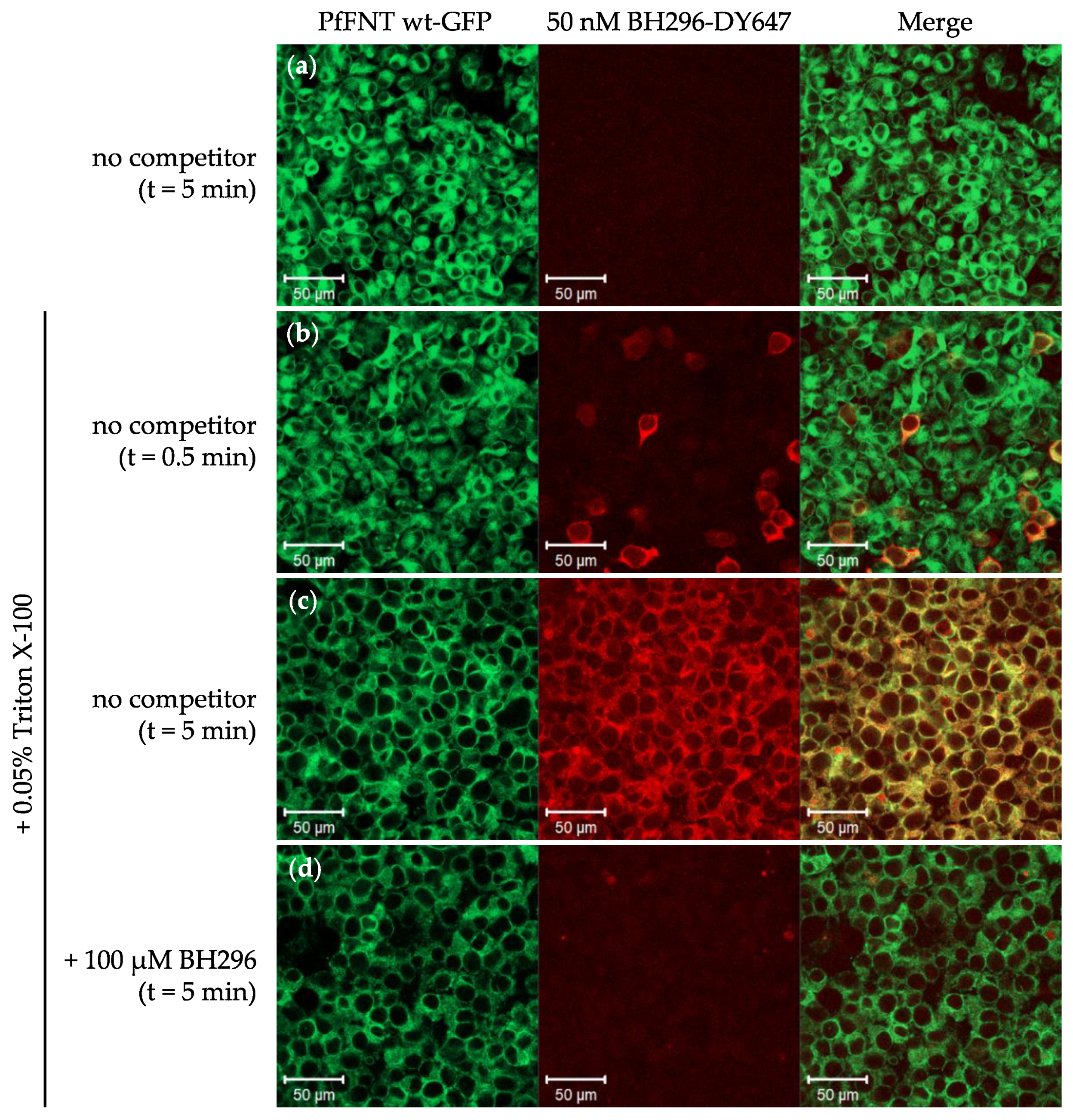
2.1. The Small Molecule Inhibitor BH296 Binds to the Cytoplasmic Side of PfFNT
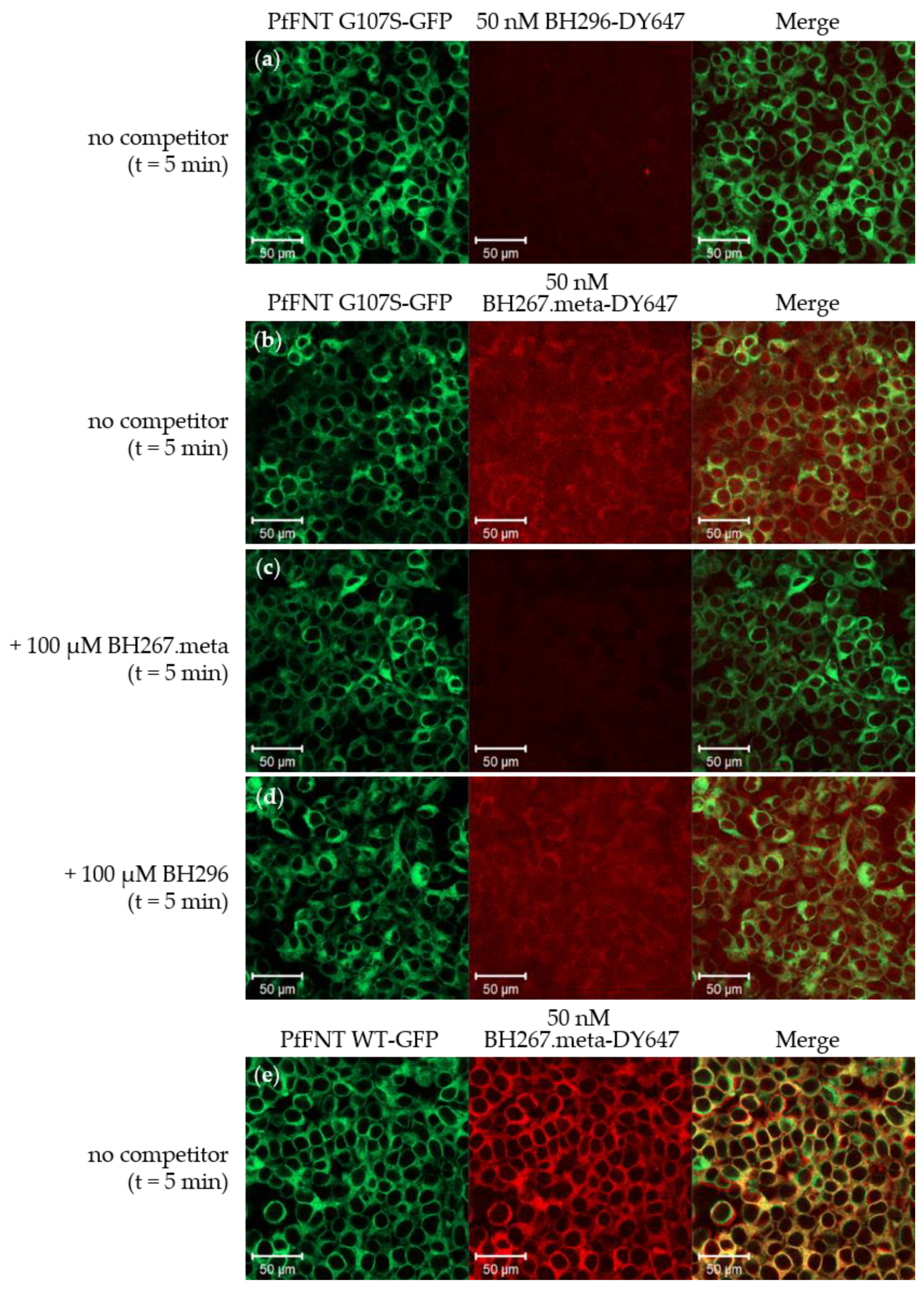
2.2. BH267.meta, but Not BH296, Bind to the PfFNT Mutant G107S
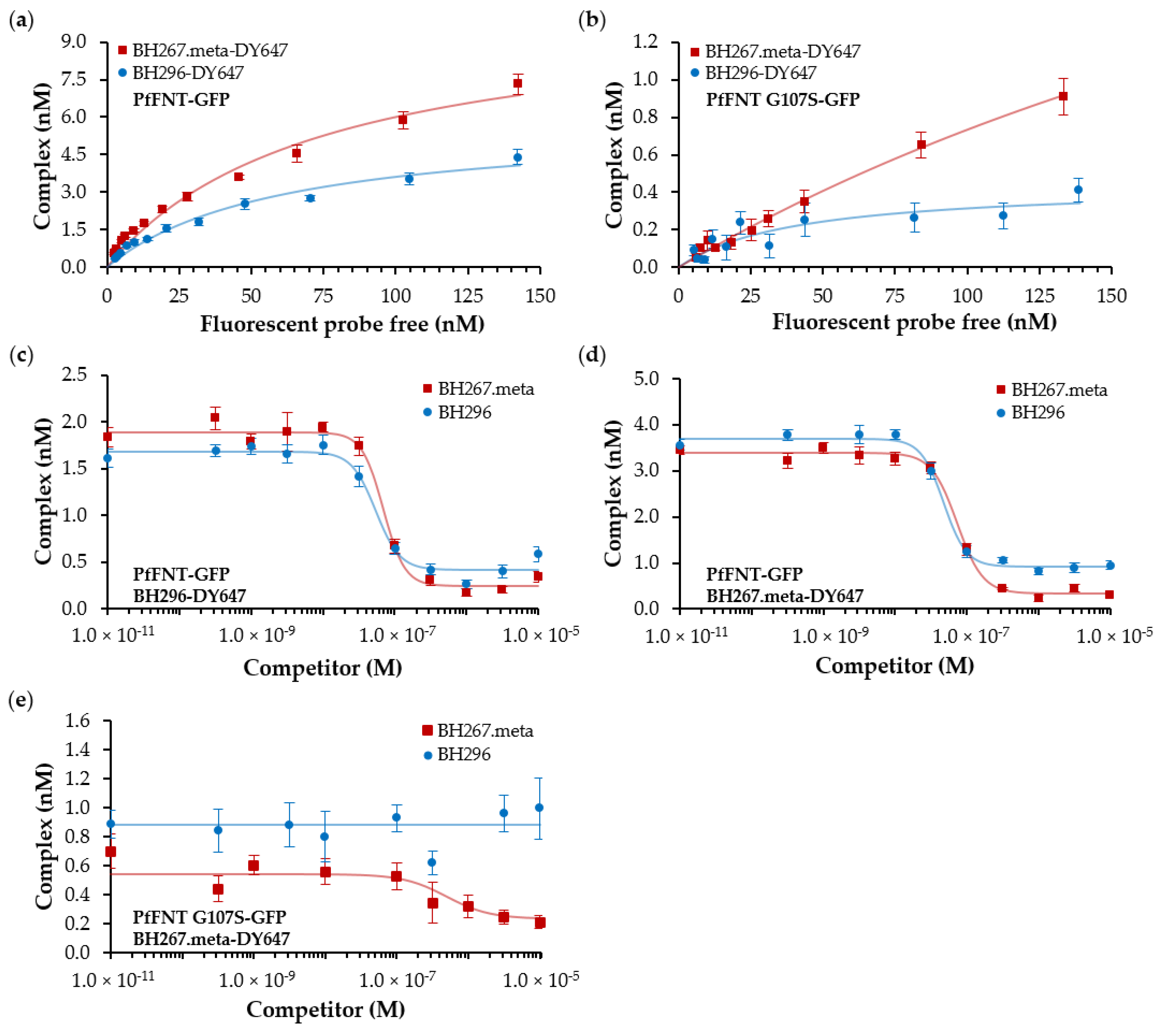
2.3. Fluorescence Cross-Correlation Spectroscopy (FCCS) Allows for Affinity Determinations of Drug Target Interactions
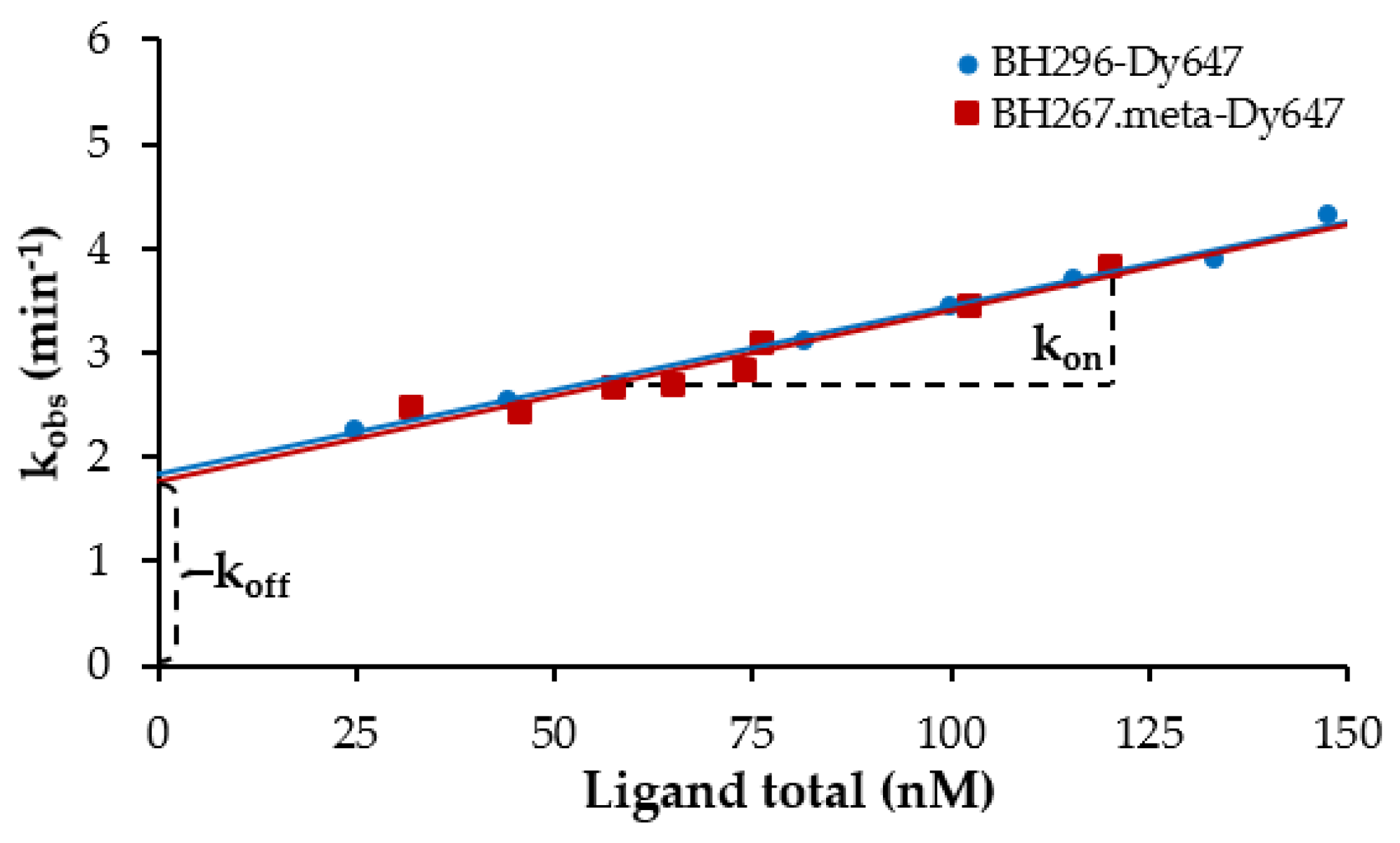
2.4. Binding Kinetics of Labeled BH296 and 267.meta to PfFNT by Time-Resolved FCCS Measurements
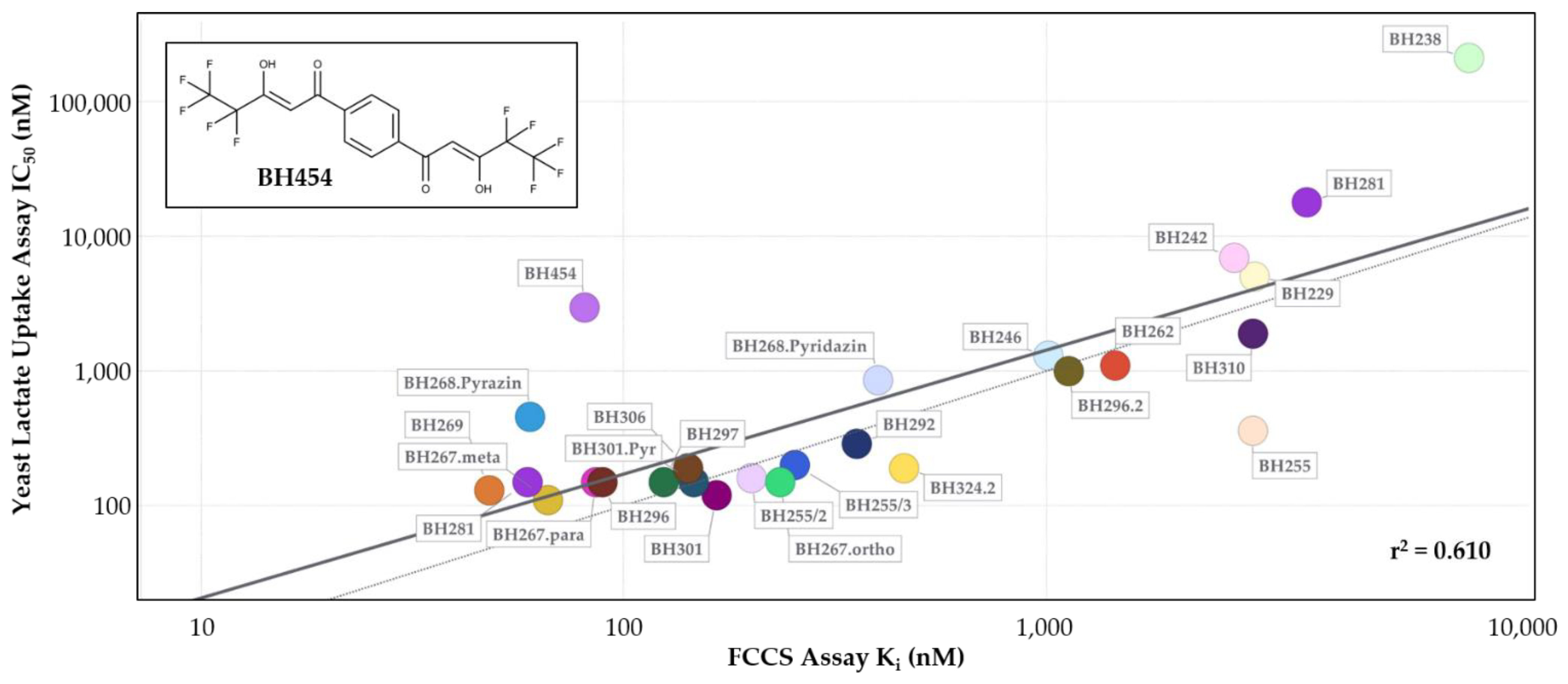
2.5. A comparison of Biophysical Affinity Data and IC50 Values from Functional Assays
3. Discussion
4. Materials and Methods
4.1. Synthesis of BH296 and BH267.meta with a 3-Aminopropoxy Linker for Fluorescence Labeling
4.2. Cell Culture Conditions
4.3. Expression of PfFNT-GFP
4.4. Generation of Cell Lysate
4.5. Fluorescent Labeling of the Tracer Molecule
4.6. Live Cell Imaging
4.7. FCCS Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McKee, R.W.; Ormsbee, R.A.; Anfinsen, C.B.; Geiman, Q.M.; Ball, E.G. Studies on malarial parasites: VI. The chemistry and metabolism of normal and parasitized (P. knowlesi) monkey blood. J. Exp. Med. 1946, 84, 569–582. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Rambow, J.; Bock, S.; Holm-Bertelsen, J.; Wiechert, M.; Blancke Soares, A.; Spielmann, T.; Beitz, E. Identity of a Plasmodium lactate/H+ symporter structurally unrelated to human transporters. Nat. Commun. 2015, 6, 6284. [Google Scholar] [CrossRef] [PubMed]
- Golldack, A.; Henke, B.; Bergmann, B.; Wiechert, M.; Erler, H.; Blancke Soares, A.; Spielmann, T.; Beitz, E. Substrate-analogous inhibitors exert antimalarial action by targeting the Plasmodium lactate transporter PfFNT at nanomolar scale. PLoS Pathog. 2017, 13, e1006172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spangenberg, T.; Burrows, J.N.; Kowalczyk, P.; McDonald, S.; Wells, T.N.; Willis, P. The open access malaria box: A drug discovery catalyst for neglected diseases. PLoS ONE 2013, 8, e62906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hapuarachchi, S.V.; Cobbold, S.A.; Shafik, S.H.; Dennis, A.S.; McConville, M.J.; Martin, R.E.; Kirk, K.; Lehane, A.M. The malaria parasite’s lactate transporter PfFNT is the target of antiplasmodial compounds identified in whole cell phenotypic screens. PLoS Pathog. 2017, 13, e1006180. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Huang, Y.; Wang, J.; Cheng, C.; Huang, W.; Lu, P.; Xu, Y.-N.; Wang, P.; Yan, N.; Shi, Y. Structure of the formate transporter FocA reveals a pentameric aquaporin-like channel. Nature 2009, 462, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Wiechert, M.; Beitz, E. Mechanism of formate-nitrite transporters by dielectric shift of substrate acidity. EMBO J. 2017, 36, 949–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helmstetter, F.; Arnold, P.; Höger, B.; Petersen, L.M.; Beitz, E. Formate-nitrite transporters carrying nonprotonatable amide amino acids instead of a central histidine maintain pH-dependent transport. J. Biol. Chem. 2019, 294, 623–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiechert, M.; Beitz, E. Formate-nitrite transporters: Monoacids ride the dielectric slide. Channels 2017, 11, 365–367. [Google Scholar] [CrossRef] [PubMed]
- Bader, A.; Beitz, E. Transmembrane facilitation of lactate/H+ instead of lactic acid is not a question of semantics but of cell viability. Membranes 2020, 10, 236. [Google Scholar] [CrossRef] [PubMed]
- Lyu, M.; Su, C.C.; Kazura, J.W.; Yu, E.W. Structural basis of transport and inhibition of the Plasmodium falciparum transporter PfFNT. EMBO Rep. 2021, 22, e51628. [Google Scholar] [CrossRef] [PubMed]
- Walloch, P.; Henke, B.; Häuer, S.; Bergmann, B.; Spielmann, T.; Beitz, E. Introduction of scaffold nitrogen atoms renders inhibitors of the malarial l-lactate transporter, PfFNT, effective against the Gly107Ser resistance mutation. J. Med. Chem. 2020, 63, 9731–9741. [Google Scholar] [CrossRef] [PubMed]
- Walloch, P.; Hansen, C.; Priegann, T.; Schade, D.; Beitz, E. Pentafluoro-3-hydroxy-pent-2-en-1-ones potently inhibit FNT-type lactate transporters from all five human-pathogenic Plasmodium species. ChemMedChem 2021, 16, 1283–1289. [Google Scholar] [CrossRef] [PubMed]
- Schwille, P.; Meyer-Almes, F.J.; Rigler, R. Dual-color fluorescence cross-correlation spectroscopy for multicomponent diffusional analysis in solution. Biophys. J. 1997, 72, 1878–1886. [Google Scholar] [CrossRef] [Green Version]
- Antoine, T.; Ott, D.; Ebell, K.; Hansen, K.; Henry, L.; Becker, F.; Hannus, S. Homogeneous time-resolved G protein-coupled receptor-ligand binding assay based on fluorescence cross-correlation spectroscopy. Anal. Biochem. 2016, 502, 24–35. [Google Scholar] [CrossRef] [Green Version]
- Koley, D.; Allen, J.; Bard, A.J. Triton X-100 concentration effects on membrane permeability of a single HeLa cell by scanning electrochemical microscopy (SECM). Proc. Natl. Acad. Sci. USA 2010, 107, 16783–16787; [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Prusoff, W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Fluorescent Probe | KD (nM) | Active Receptor (%) |
|---|---|---|---|
| PfFNT wt-GFP | BH296-DY647 BH267.meta-DY647 | 67 ± 6 72 ± 1 | 68 ± 3 100 |
| PfFNT G107S-GFP | BH296-DY647 BH267.meta-DY647 | >2000 405 ± 1 | n.d. 100 |
| Target | Fluorescent Probe | Ki Competitor (nM) | |
|---|---|---|---|
| BH296 | BH267.meta | ||
| PfFNT wt-GFP | BH296-DY647 BH267.meta-DY647 | 48 ± 2 47 ± 1 | 66 ± 6 56 ± 3 |
| PfFNT G107S-GFP | BH267.meta-DY647 | >10,000 | 417 ± 16 |
| Target | Fluorescent Probe | kon (nM−1 min−1) | koff (min−1) | KD (Kinetics, nM) | KD (Equilibrium, nM) |
|---|---|---|---|---|---|
| PfFNT wt-GFP | BH296-DY647 | 0.0167 ± 0.0006 | 1.8659 ± 0.0339 | 113 ± 2 | 67 ± 6 |
| BH267.meta-DY647 | 0.0161 ± 0.0005 | 1.7839 ± 0.0250 | 112 ± 5 | 72 ± 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jakobowska, I.; Becker, F.; Minguzzi, S.; Hansen, K.; Henke, B.; Epalle, N.H.; Beitz, E.; Hannus, S. Fluorescence Cross-Correlation Spectroscopy Yields True Affinity and Binding Kinetics of Plasmodium Lactate Transport Inhibitors. Pharmaceuticals 2021, 14, 757. https://doi.org/10.3390/ph14080757
Jakobowska I, Becker F, Minguzzi S, Hansen K, Henke B, Epalle NH, Beitz E, Hannus S. Fluorescence Cross-Correlation Spectroscopy Yields True Affinity and Binding Kinetics of Plasmodium Lactate Transport Inhibitors. Pharmaceuticals. 2021; 14(8):757. https://doi.org/10.3390/ph14080757
Chicago/Turabian StyleJakobowska, Iga, Frank Becker, Stefano Minguzzi, Kerrin Hansen, Björn Henke, Nathan Hugo Epalle, Eric Beitz, and Stefan Hannus. 2021. "Fluorescence Cross-Correlation Spectroscopy Yields True Affinity and Binding Kinetics of Plasmodium Lactate Transport Inhibitors" Pharmaceuticals 14, no. 8: 757. https://doi.org/10.3390/ph14080757
APA StyleJakobowska, I., Becker, F., Minguzzi, S., Hansen, K., Henke, B., Epalle, N. H., Beitz, E., & Hannus, S. (2021). Fluorescence Cross-Correlation Spectroscopy Yields True Affinity and Binding Kinetics of Plasmodium Lactate Transport Inhibitors. Pharmaceuticals, 14(8), 757. https://doi.org/10.3390/ph14080757