
Synthesis and Biological Activities of Pyrazino[1,2-a]indole and Pyrazino[1,2-a]indol-1-one Derivatives



Abstract
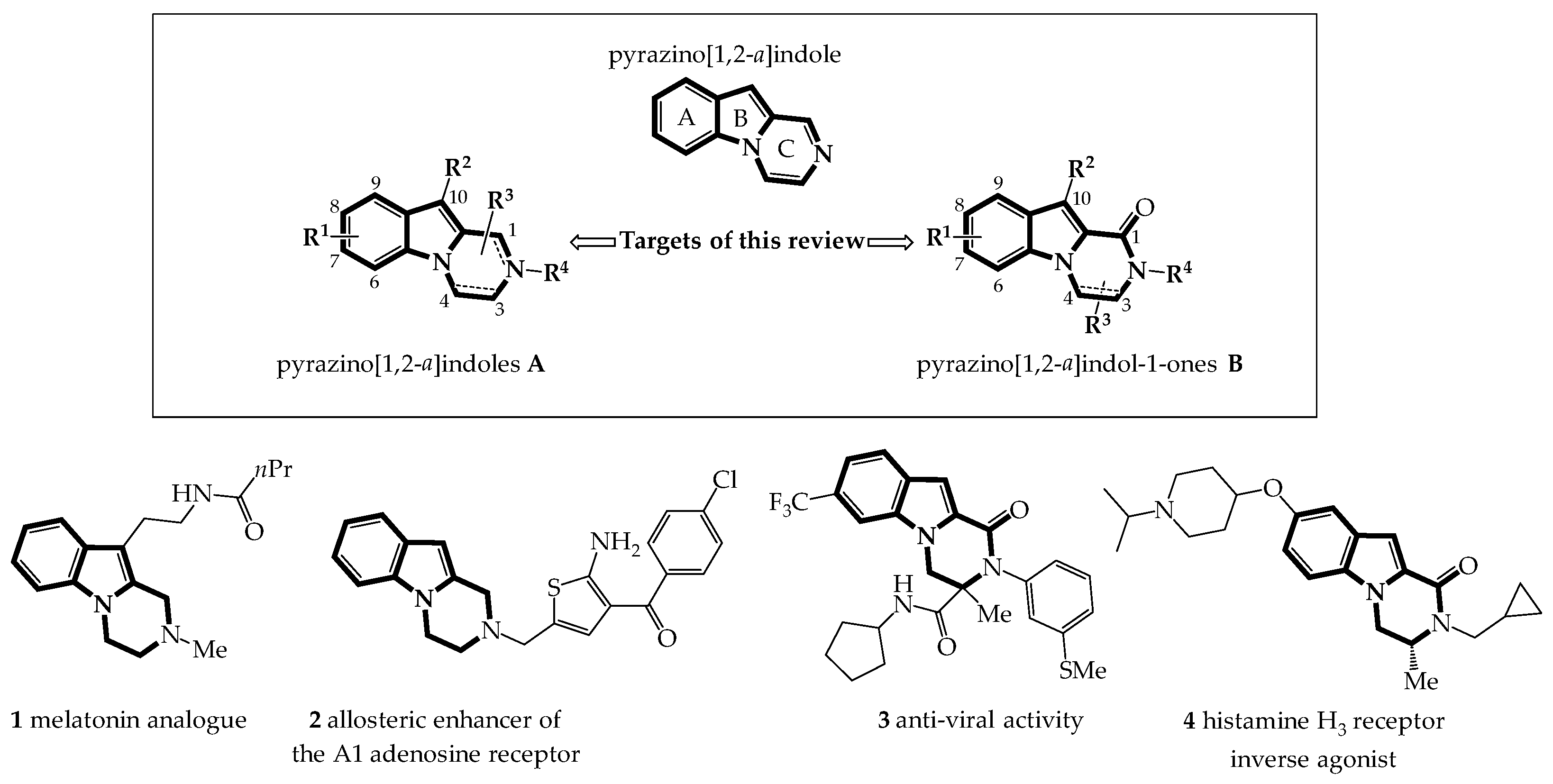
:1. Introduction
2. Pyrazinoindoles A: Synthesis and Biological Properties
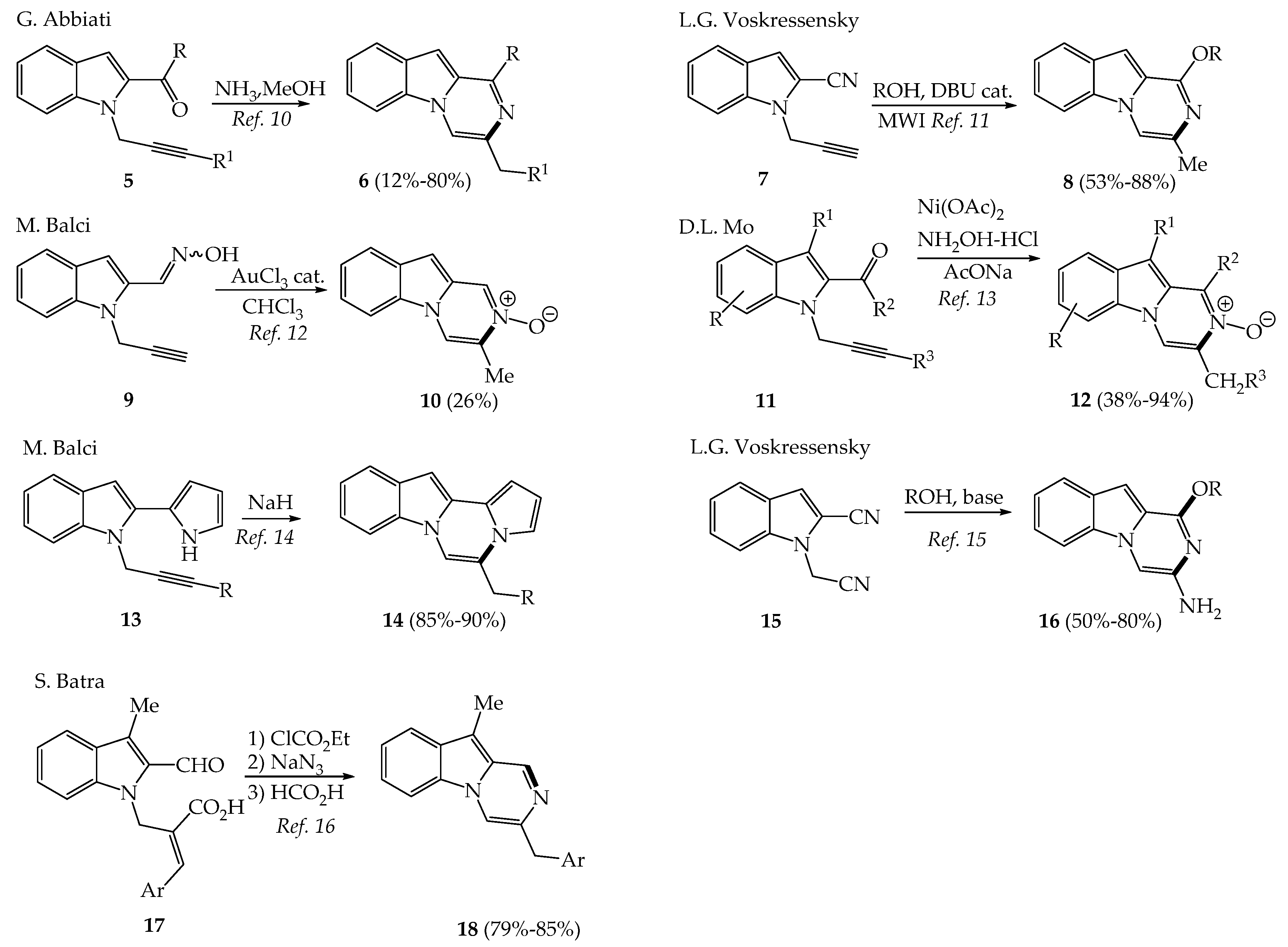
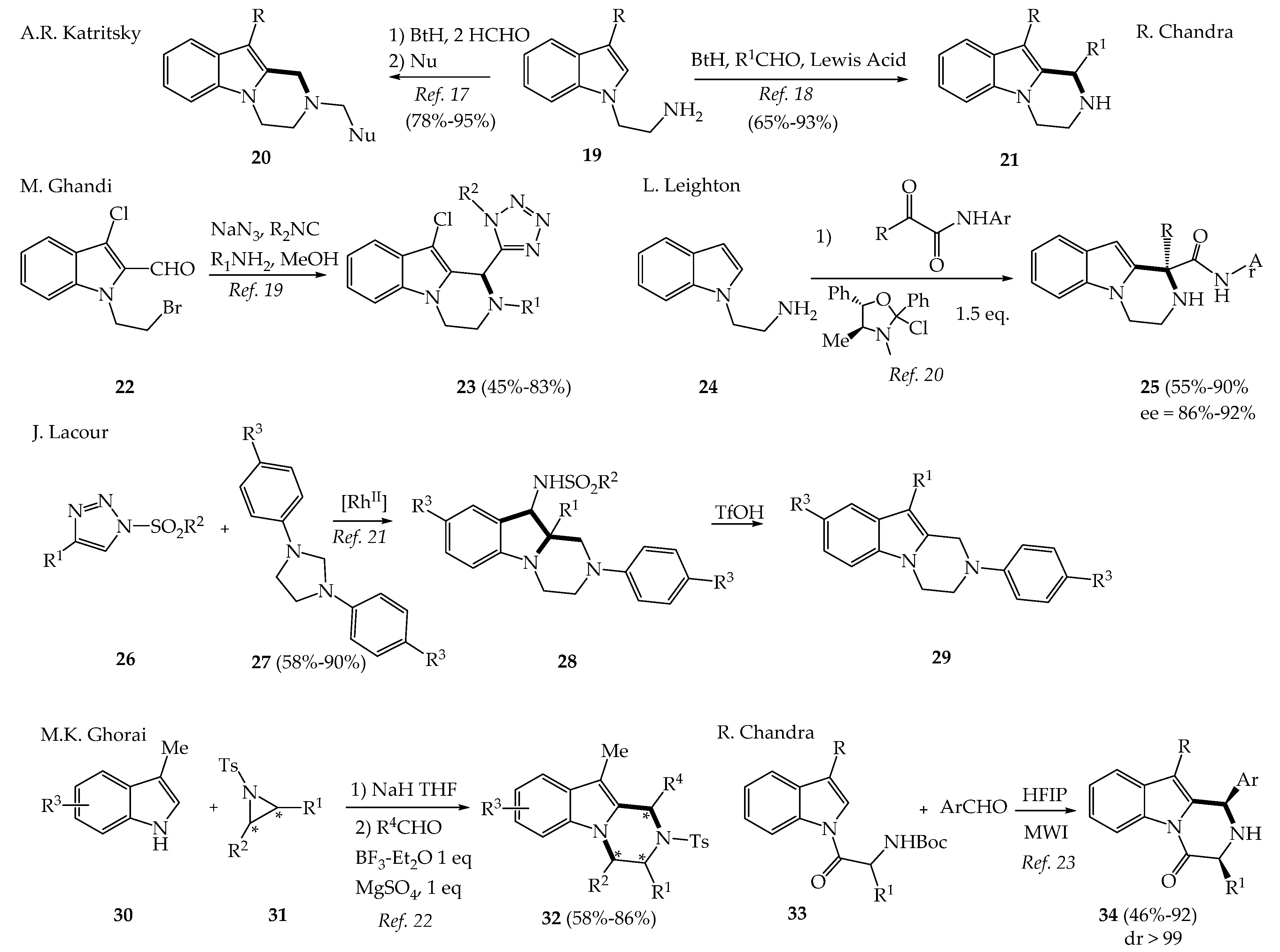
2.1. Recent Synthetic Approaches to Variously Substituted Pyrazinoindoles and 3,4-Dihydropyrazinoindoles
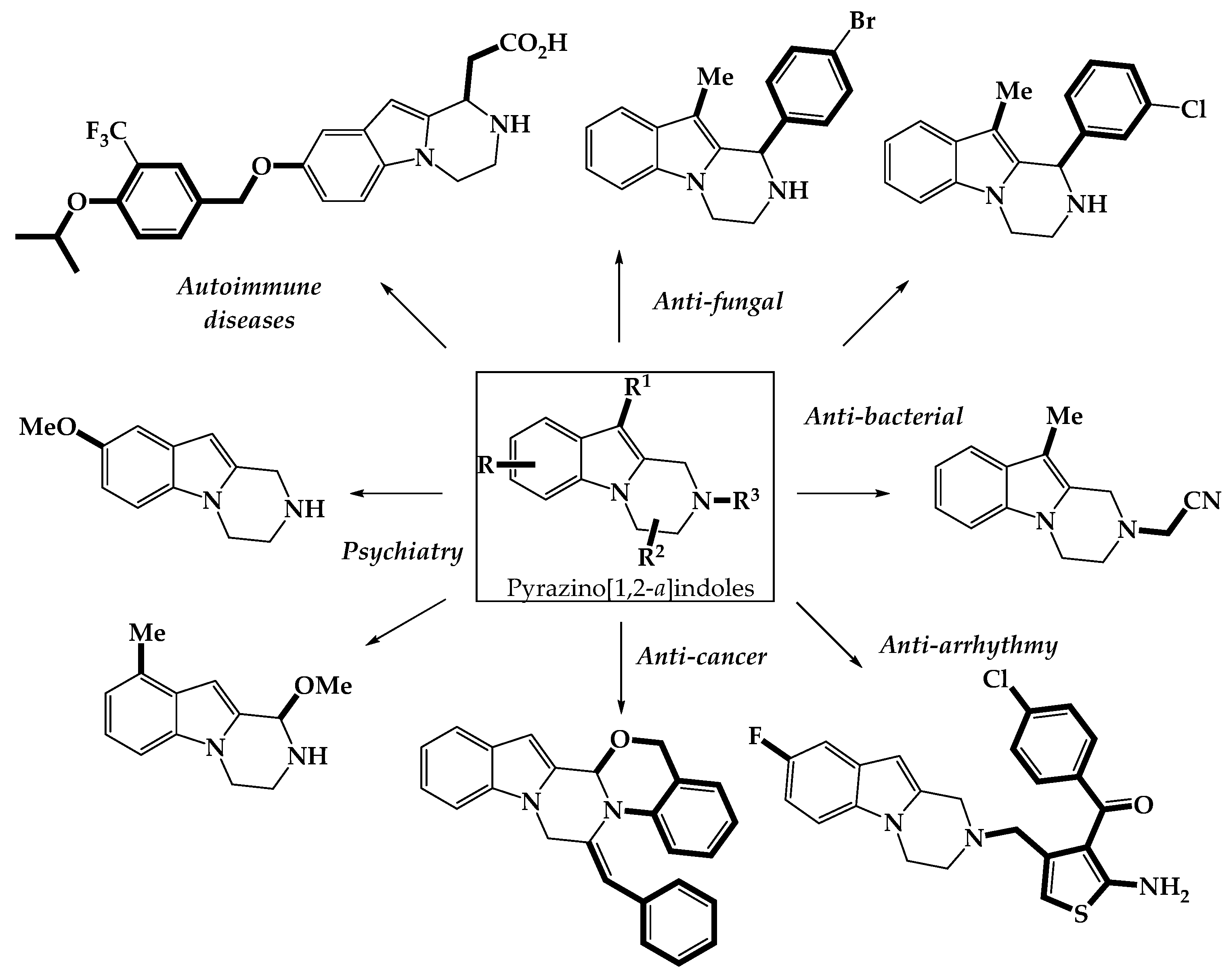
2.2. Biologically Active Pyrazino[1,2-a]indoles
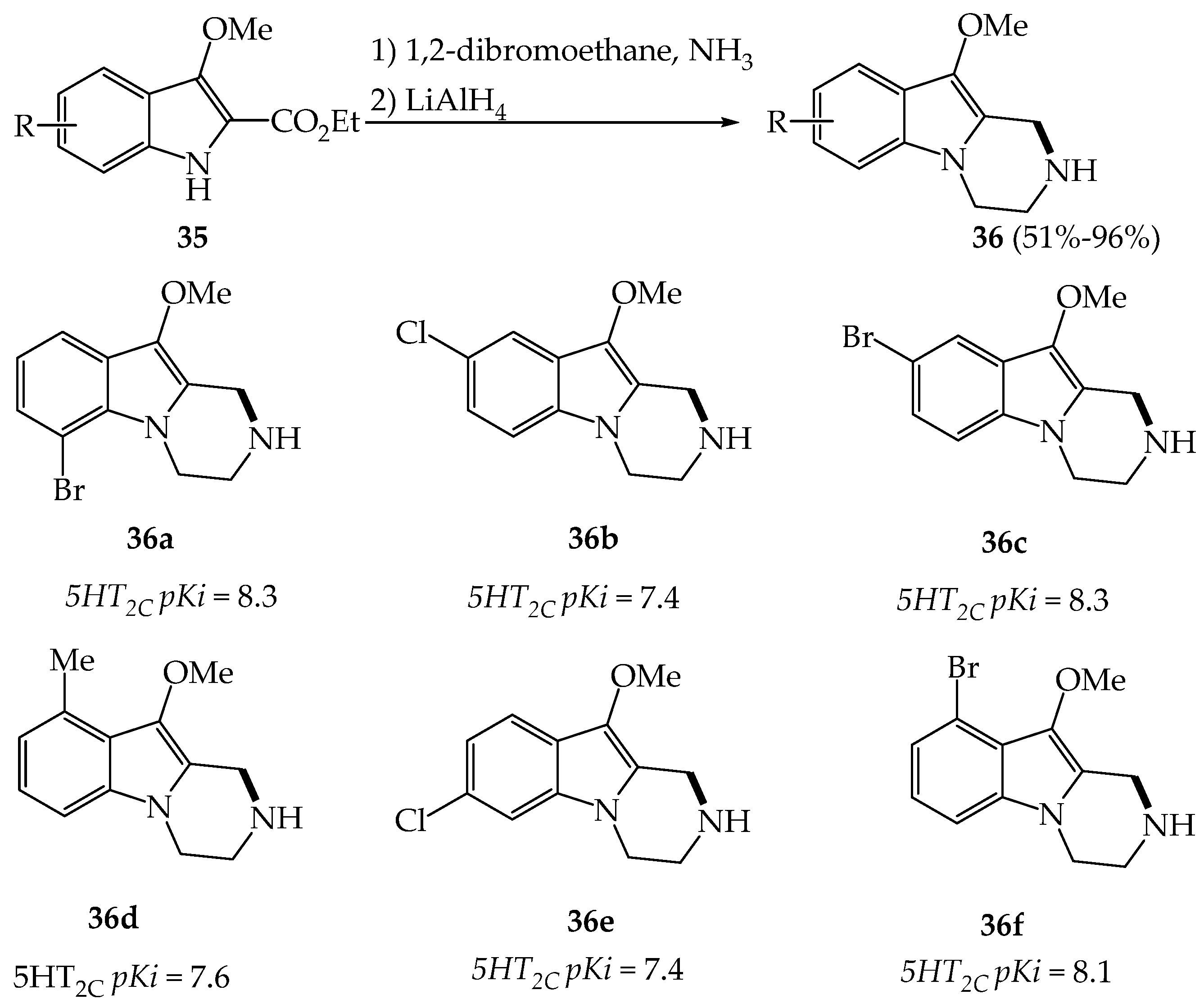
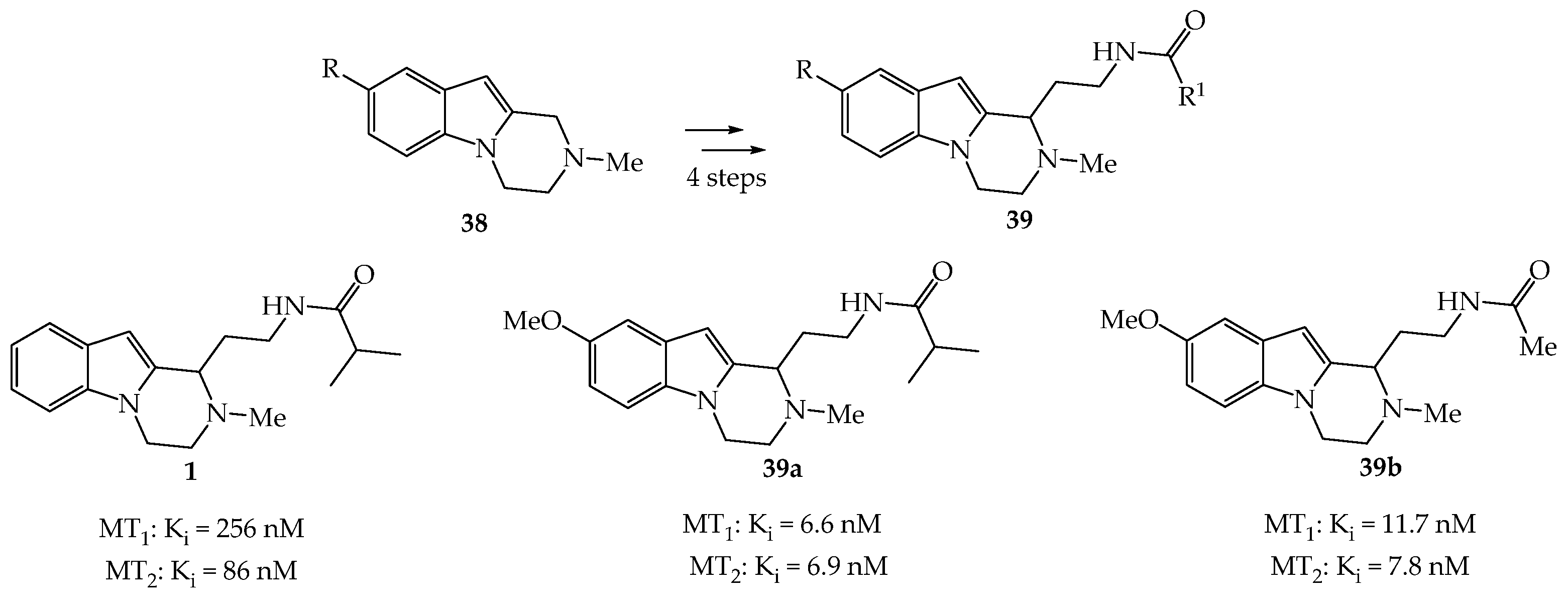
2.2.1. Neuropsychiatric Properties
2.2.2. Auto-Immune Properties
2.2.3. Anti-Bacterial and Anti-Fungal Properties
2.2.4. Anti-Arrhythmic, Anti-Lipolytic, Neuro- and Cardio-Protective Properties
2.2.5. Anti-Cancer Properties
3. Pyrazinoindol-1-Ones B: Synthesis and Biological Properties
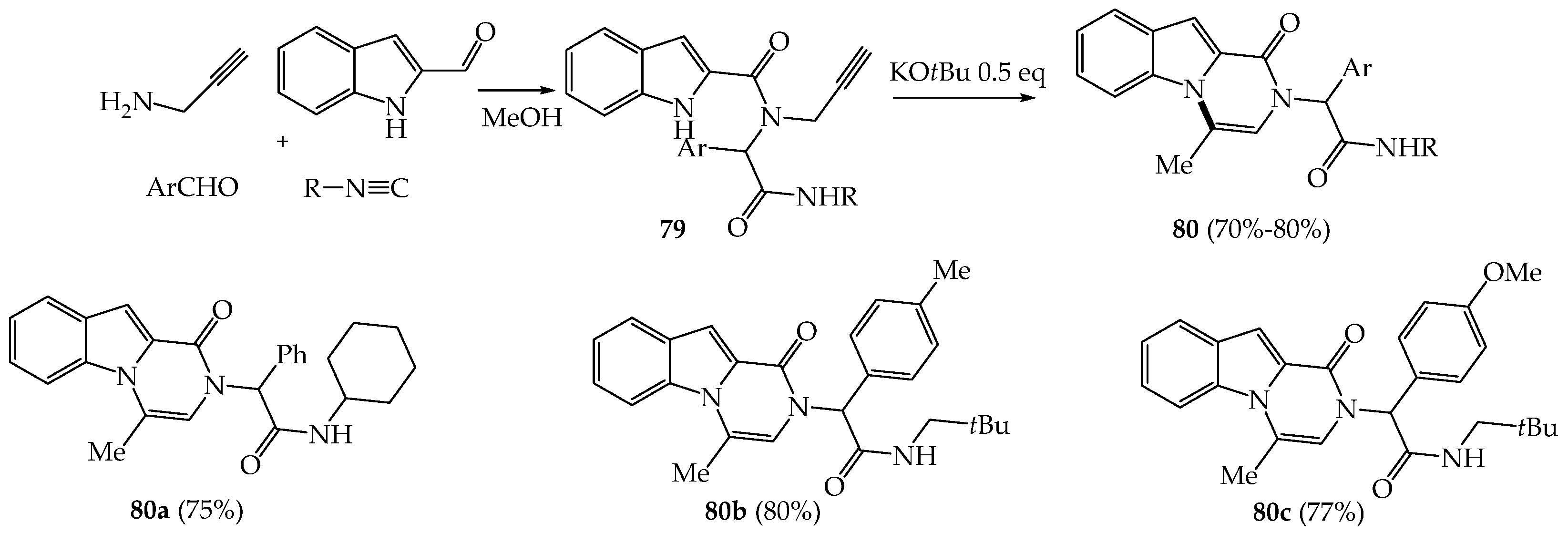
3.1. Recent Synthetic Approaches to Variously Substituted Pyrazinoindol-1-Ones of Type B
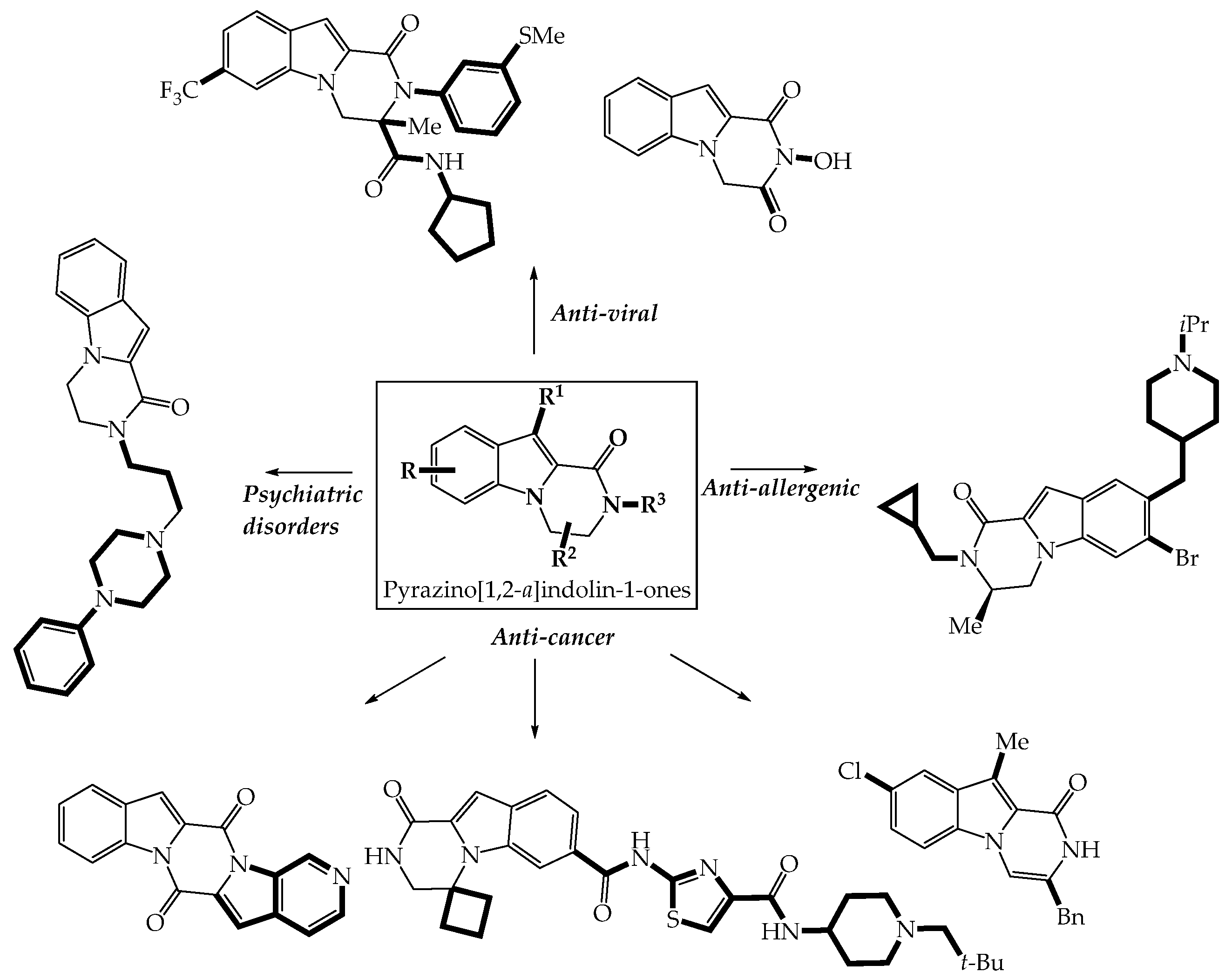
3.2. Biologically Active Pyrazino[1,2-a]indol-1-ones
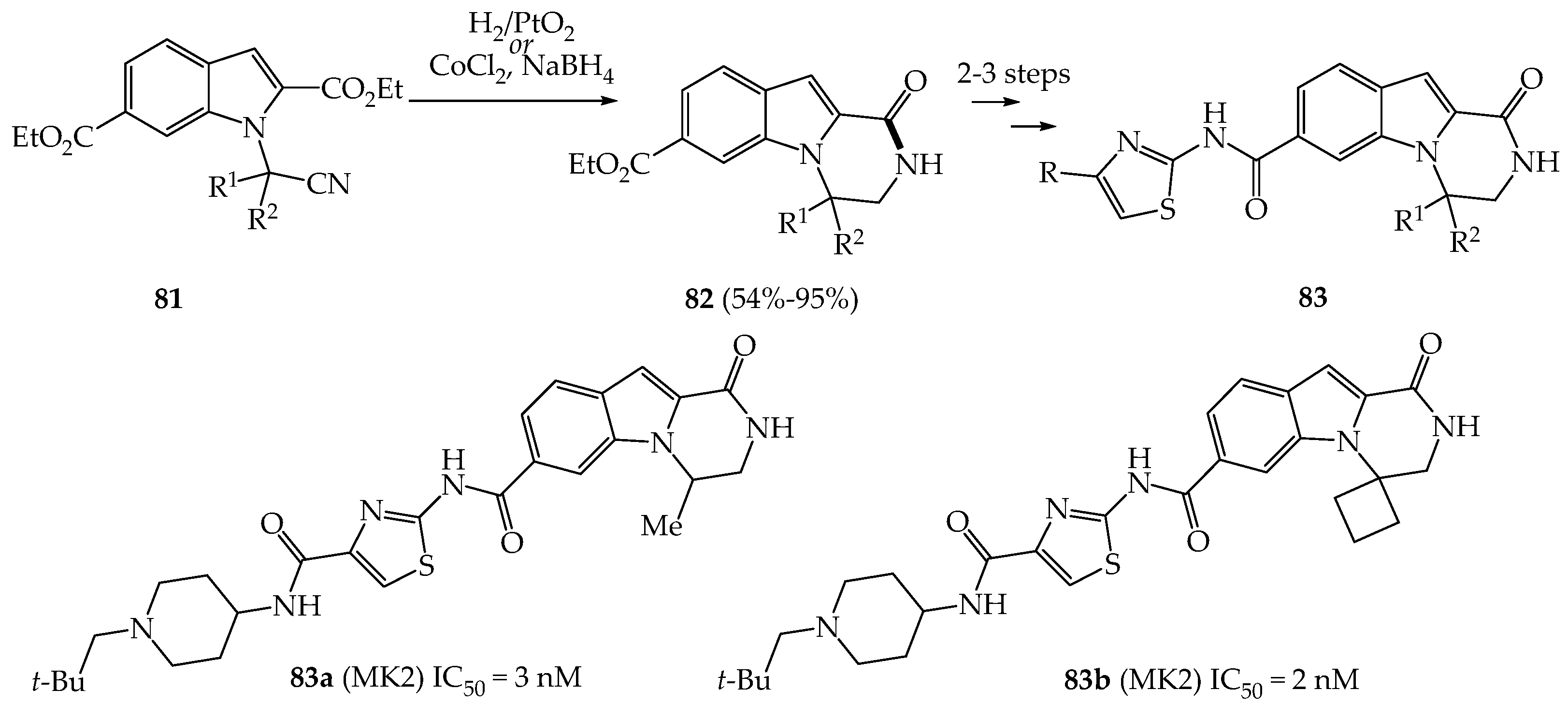
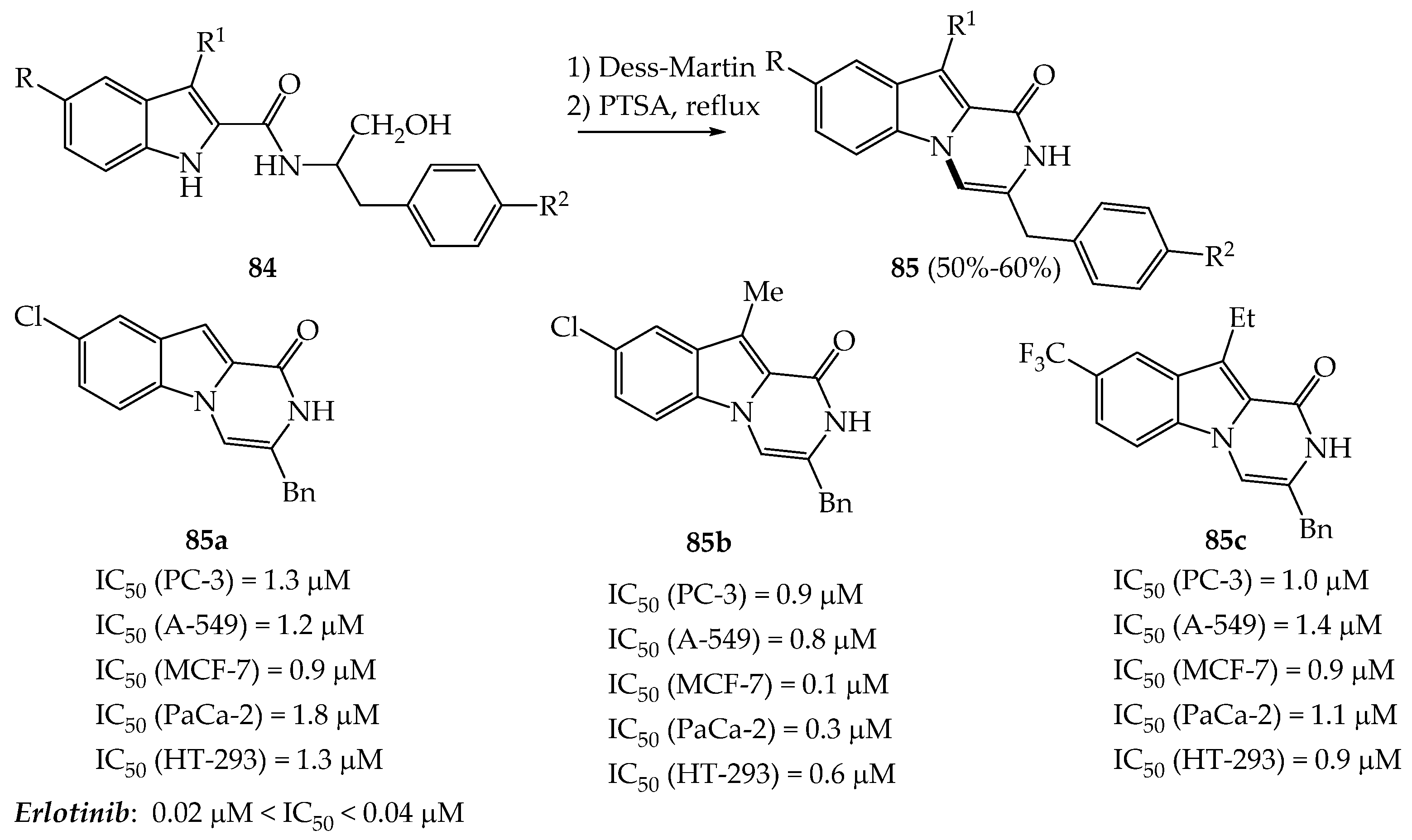
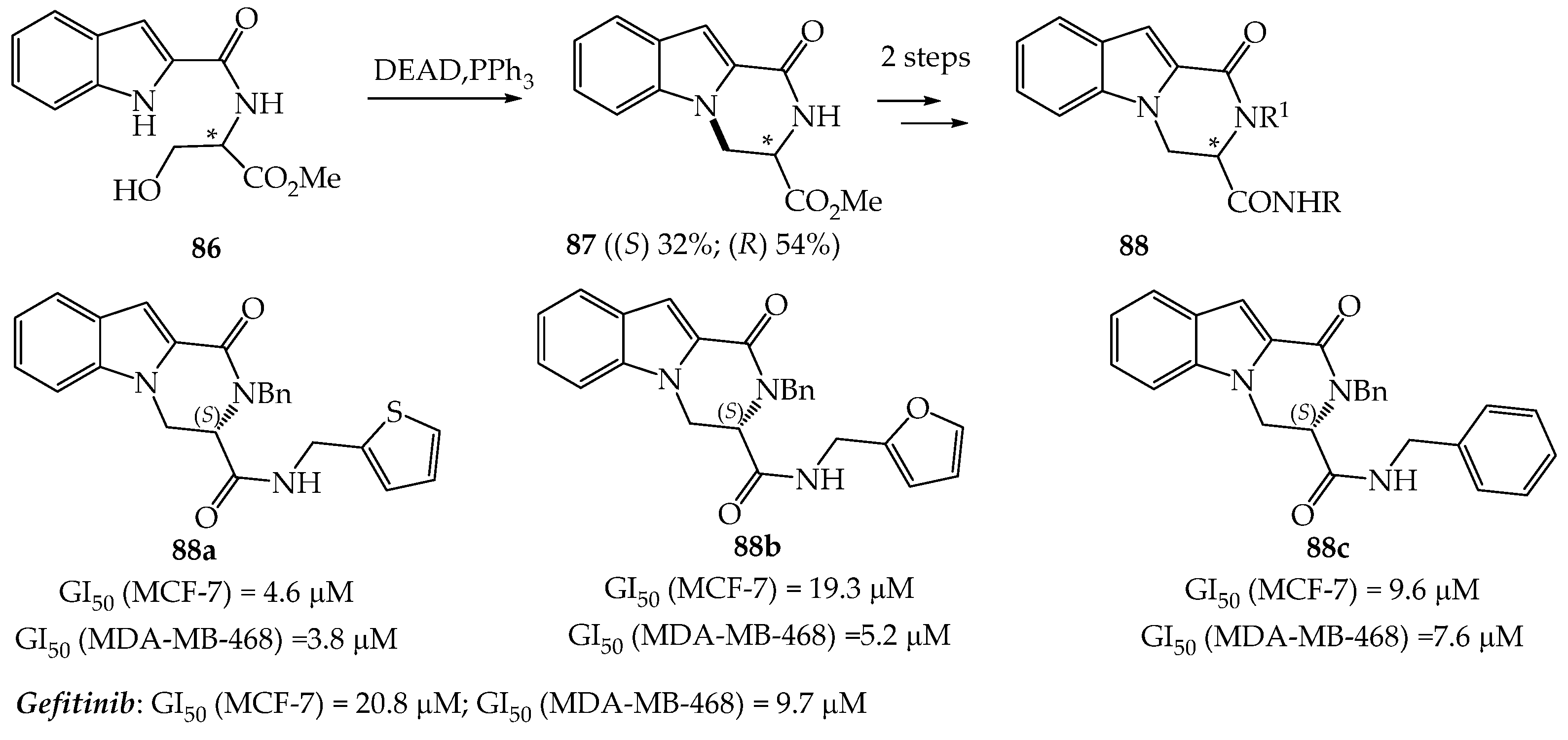
3.2.1. Anti-Cancer Properties
3.2.2. Anti-Infectious Properties
3.2.3. Neuropsychiatric Properties
3.2.4. Anti-Allergenic Properties
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singh, A.; Mahapatra, S.; Sewariya, S.; Singh, N.; Singh, S.; Kumar, Y.; Bandichhor, R.; Chandra, R. A mini-review on the synthesis of pyrazinoindole: Recent progress and perspectives. Mini Rev. Org. Chem. 2021, 18, 504–514. [Google Scholar] [CrossRef]
- Sokolova, E.A.; Festa, A.A. Synthesis of pyrazino[1,2-a] indoles and indolo [1,2-a] quinoxalines (microreview). Chem. Heterocycl. Comp. 2016, 52, 219–221. [Google Scholar] [CrossRef]
- Markl, C.; Attia, M.I.; Julius, J.; Sehti, S.; Witt-Enderby, P.A.; Zlotos, D.P. Synthesis and pharmacological evaluation of 1,2,3,4-tetrahydropyrazino[1,2-a] indole and 2-[(phenylmethylamino)methyl]-1H-indole analogues as novel melatoninergic ligands. Bioorg. Med. Chem. 2009, 17, 4583–4594. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, R.; Baraldi, P.G.; Carrion, M.D.; Cara, C.L.; Salvador, M.K.; Preti, D.; Tabrizi, M.A.; Moorman, A.R.; Vincenzi, F.; Borea, P.A.; et al. Synthesis and biological effects of novel 2-amino-3-(4-chlorobenzoyl)-4-substituted thiophenes as allosteric enhancers of the A1 adenosine receptor. Eur. J. Med. Chem. 2013, 67, 409–427. [Google Scholar] [CrossRef] [PubMed]
- Kounde, C.S.; Yeo, H.Q.; Wang, Q.Y.; Wan, K.F.; Dong, H.; Karuna, R.; Dix, I.; Wagner, T.; Zou, B.; Simon, O. Discovery of 2-oxopiperazine dengue inhibitors by scaffold morphing of a phenotypic high-throughput screening hit. Bioorg. Med. Chem. Lett. 2017, 27, 1385–1389. [Google Scholar] [CrossRef] [PubMed]
- Richter, H.G.F.; Freichel, C.; Huwyler, J.; Nakagawa, T.; Nettekoven, M.; Plancher, J.-M.; Raab, S.; Roche, O.; Schuler, F.; Taylor, S.; et al. Discovery of potent and selective histamine H3 receptor inverse agonists based on the 3,4-dihydro-2H-pyrazino[1,2-a] indol-1-one scaffold. Bioorg. Med. Chem. Lett. 2010, 20, 5713–5717. [Google Scholar] [CrossRef]
- Yao, Y.; Alami, M.; Hamze, A.; Provot, O. Recent advances in the synthesis of pyrido[1,2-a] indoles. Org. Biomol. Chem. 2021, 19, 3509–3526. [Google Scholar] [CrossRef]
- Pecnard, S.; Hamze, A.; Pozzo, J.-L.; Alami, M.; Provot, O. Synthesis of oxazino[4,3-a] indoles and their applications. Eur. J. Med. Chem. 2021, 224, 113728. [Google Scholar] [CrossRef]
- Abbiati, G.; Arcadi, A.; Beccalli, E.; Rossi, E. Novel intramolecular cyclization of N-alkynyl heterocycles containing proximate nucleophiles. Tetrahedron Lett. 2003, 44, 5331–5334. [Google Scholar] [CrossRef]
- Abbiati, G.; Arcadi, A.; Bellinazzi, A.; Beccalli, E.; Rossi, E.; Zanzola, S. Intramolecular cyclization of δ-iminoacetylenes: A new entry to pyrazino[1,2-a] indoles. J. Org. Chem. 2005, 70, 4088–4095. [Google Scholar] [CrossRef]
- Festa, A.A.; Zalte, R.R.; Golantsov, N.E.; Varlamov, A.V.; Van der Eycken, E.V.; Voskressensky, L.G. DBU-catalyzed alkyne—Imidate cyclization toward 1-alkoxypyrazino[1,2-a] indole synthesis. J. Org. Chem. 2018, 83, 9305–9311. [Google Scholar] [CrossRef]
- Guven, S.; Ozer, M.S.; Kaya, S.; Menges, N.; Balci, M. Gold-catalyzed oxime–oxime rearrangement. Org. Lett. 2015, 17, 2660–2663. [Google Scholar] [CrossRef] [PubMed]
- Bi, H.Y.; Du, M.; Pan, C.X.; Xiao, Y.; Su, G.F.; Mo, D.L. Nickel(II)-catalyzed [5 + 1] annulation of 2-carbonyl-1-propargylindoles with hydroxylamine to synthesize pyrazino[1,2-a] indole-2-oxides in water. J. Org. Chem. 2019, 84, 9859–9868. [Google Scholar] [CrossRef]
- Basceken, S. Kaya, S.; Balci, M. Intramolecular gold-catalyzed and NaH-supported cyclization reactions of N-propargyl indole derivatives with pyrazole and pyrrole rings: Synthesis of pyrazolodiazepinoindole, pyrazolopyrazinoindole, and pyrrolopyrazinoindole. J. Org. Chem. 2015, 80, 12552–12561. [Google Scholar] [CrossRef] [PubMed]
- Festa, A.A.; Golantsov, N.E.; Storozhenko, O.A.; Shumsky, A.N.; Varlamov, A.V.; Voskressensky, L.G. Alcohol-initiated dinitrile cyclization in basic media: A route toward pyrazino [1,2-a] indole-3-amines. Synlett 2018, 29, 898–903. [Google Scholar]
- Nayak, M.; Pandey, G.; Batra, S. Synthesis of pyrrolo[1,2-a] pyrazines and pyrazino[1,2-a] indoles by curtius reaction in morita–baylis–hillman derivatives. Tetrahedron 2011, 67, 7563–7569. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Verma, A.K.; He, H.Y.; Chandra, R. Novel synthesis of 1,2,3,4-tetrahydropyrazino[1,2-a] indoles. J. Org. Chem. 2003, 68, 4938–4940. [Google Scholar] [CrossRef]
- Tiwari, R.K.; Singh, J.; Singh, D.; Verma, A.K.; Chandra, R. Highly efficient one-pot synthesis of 1-substituted-1,2,3,4-tetrahydropyrazino [1,2-a] indoles. Tetrahedron 2005, 61, 9513–9518. [Google Scholar] [CrossRef]
- Salahi, S.; Ghandi, M.; Abbasi, A. An efficient ugi-azide four-component approach for the preparation of novel 1-(1H-tetrazol-5-yl)-10-chloro-1,2,3,4-tetrahydropyrazino[1,2-a] indoles. J. Heterocyclic Chem. 2019, 56, 1296–1305. [Google Scholar] [CrossRef]
- Schönherr, H.; Leighton, J.L. Direct and highly enantioselective iso-pictet-spengler reactions with α-ketoamides: Access to underexplored indole core structures. Org. Lett. 2012, 14, 2610–2613. [Google Scholar] [CrossRef]
- Milcendeau, P.; Zhang, Z.; Glinsky-Olivier, N.; van Elslande, E.; Guinchard, X. Au(I)-catalyzed pictet-spengler reactions all around the indole ring. J. Org. Chem. 2021, 86, 6406–6422. [Google Scholar] [CrossRef]
- Guarnieri-Ibáňez, A.; de Aguirre, A.; Besnard, C.; Poblador-Bahamonde, A.I.; Lacour, J. Regiodivergent synthesis of pyrazino-indolines vs. triazocines via α-imino carbenes addition to imidazolidines. Chem. Sci. 2021, 12, 1479–1485. [Google Scholar] [CrossRef]
- Wani, I.A.; Das, S.; Mondal, S.; Ghorai, M.K. Stereoselective construction of pyrazinoindoles and oxazinoindoles via ring-opening/pictet-spengler reaction of aziridines and epoxides with 3-methylindoles and carbonyls. J. Org. Chem. 2018, 83, 14553–14567. [Google Scholar] [CrossRef]
- Singh, A.; Singh, S.; Sewariya, S.; Singh, N.; Singh, P.; Kumar, A.; Bandichhor, R.; Chandra, R. Stereospecific N-acylation of indoles and corresponding microwave mediated synthesis of pyrazinoindoles using hexafluoroisopropanol. Tetrahedron 2021, 84, 132017. [Google Scholar] [CrossRef]
- Bos, M.; Jenck, F.; Martin, J.R.; Moreau, J.L.; Mutel, V.; Sleight, A.J.; Widmer, U. Synthesis, pharmacology and therapeutic potential of 10-methoxypyrazino[1,2-a] indoles, partial agonists at the 5HT2c receptor. Eur. J. Med. Chem. 1997, 32, 253–261. [Google Scholar] [CrossRef]
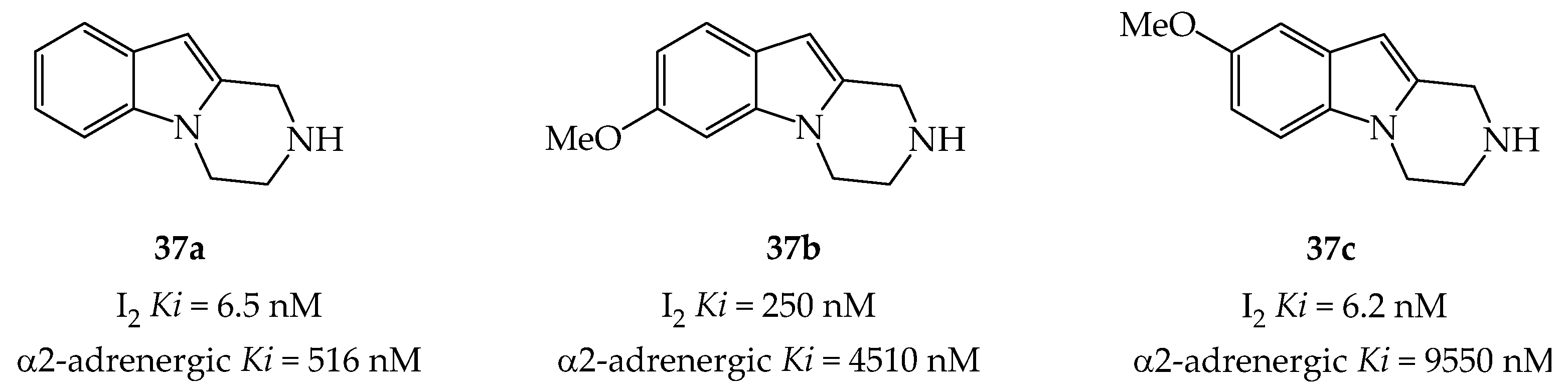
- Chang-Fong, J.; Tyacke, R.J.; Lau, A.; Westaway, J.; Hudson, A.L.; Glennon, R.A. Pyrazino [1,2-a] indoles as novel high-affinity and selective imidazoline I2 receptor ligands. Bioorg. Med. Chem. Lett. 2004, 14, 1003–1005. [Google Scholar] [CrossRef]
- Husbands, S.M.; Glennon, R.A.; Gorgerat, S.; Gough, R.; Tyacke, R.; Crosby, J.; Nutt, D.J.; Lewis, J.W.; Hudson, A.L. β-carboline binding to imidazoline receptors. Drug Alcohol Depend. 2001, 64, 203–208. [Google Scholar] [CrossRef]
- Glennon, R.A.; Grella, B.; Tyacke, R.J.; Lau, A.; Westaway, J.; Hudson, A.L. Binding of β-carbolines at imidazoline I2 receptors: A structure–affinity investigation. Bioorg. Med. Chem. Lett. 2004, 14, 999–1002. [Google Scholar] [CrossRef]
- Guandalini, L.; Martini, E.; Gualtieri, F.; Romanelli, M.N.; Varani, K. Design, synthesis and preliminary pharmacological evaluation of rigid analogues of the nicotinic agonist 1,1-dimethyl-4-phenylpiperazinium iodide (DMPP). Arkivoc 2004, 2004, 286–300. [Google Scholar] [CrossRef] [Green Version]
- Buzard, D.J.; Schrader, T.O.; Zhu, X.; Lehmann, J.; Johnson, B.; Kasem, M.; Kim, S.H.; Kawasaki, A.; Lopez, L.; Moody, J.; et al. Design and synthesis of new tricyclic indoles as potent modulators of the S1P1 receptor. Bioorg. Med. Chem. Lett. 2015, 25, 659–663. [Google Scholar] [CrossRef] [PubMed]
- Buzard, D.J.; Kim, S.H.; Lopez, L.; Kawasaki, A.; Zhu, X.; Moody, J.; Thoresen, T.; Calderon, I.; Ullman, B.; Han, S.; et al. Discovery of APD334: Design of a clinical stage functional antagonist of the sphingosine-1-phosphate-1 receptor. ACS Med. Chem. Lett. 2014, 5, 1313–1317. [Google Scholar] [CrossRef] [Green Version]
- Bit, R.A.; Davis, P.D.; Elliott, L.H.; Harris, W.; Hill, C.H.; Keech, E.; Kumar, H.; Lawton, G.; Maw, A.; Nixon, J.S.; et al. Inhibitors of protein kinase C. 3. Potent and highly selective bisindolylmaleimides by conformational restriction. J. Med. Chem. 1993, 36, 21–29. [Google Scholar] [CrossRef]
- Tiwari, R.K.; Singh, D.; Singh, J.; Yadav, V.; Pathak, A.K.; Dabur, R.; Chhillar, A.K.; Singh, R.; Sharma, G.L.; Chambra, R.; et al. Synthesis and antibacterial activity of substituted 1,2,3,4-tetrahydropyrazino [1,2-a] indoles. Bioorg. Med. Chem. Lett. 2006, 16, 413–416. [Google Scholar] [CrossRef]
- Tiwari, R.K.; Verma, A.K.; Chhillar, A.K.; Singh, D.; Singh, J.; Sankar, V.K.; Yadav, V.; Sharma, G.L.; Chandra, R. Synthesis and antibacterial activity of substituted 1,2,3,4-tetrahydropyrazino[1,2-a] indoles. Bioorg. Med. Chem. 2006, 14, 2747–2752. [Google Scholar] [CrossRef]
- Romagnoli, R.; Baraldi, P.G.; Carrion, M.D.; Cruz-Lopez, O.; Lopez Cara, C.; Preti, D.; Tabrizi, M.A. Balzarini, J.; Hamel, E.; Fabbri, E.; et al. Discovery of 8-methoxypyrazino [1,2-a] indole as a new potent antiproliferative agent against human leukemia K562 cells. A structure-activity relationship study. Lett. Drug Des. Discov. 2009, 6, 298–303. [Google Scholar] [CrossRef] [Green Version]
- Kumar, K.S.; Kumar, N.P.; Rajesham, B.; Kishan, G.; Akula, S.; Kancha, R.K. Silver-catalyzed synthesis of pyrrolopiperazine fused with oxazine/imidazole via a domino approach: Evaluation of anti-cancer activity. N. J. Chem. 2018, 42, 34–38. [Google Scholar] [CrossRef]
- An, J.; Chang, N.J.; Song, L.D.; Jin, Y. Ma, Y.Q.; Chen, J.R.; Xiao, W.J. Efficient and general synthesis of oxazino [4, 3-a] indoles by cascade addition-cyclization reactions of (1 H-indol-2-yl) methanols and vinyl sulfonium salts. Chem. Commun. 2011, 47, 1869–1871. [Google Scholar] [CrossRef] [PubMed]
- Pecnard, S.; Hamze, A.; Bignon, J.; Prost, B.; Deroussent, A.; Gallego-Yerga, L.; Peláez, R.; Paik, J.Y.; Diederich, M.; Alami, M.; et al. Anticancer properties of indole derivatives as IsoCombretastatin A-4 analogues. Eur. J. Med. Chem. 2021, 223, 113656. [Google Scholar] [CrossRef]
- Palomba, M.; Sancineto, L.; Marini, F.; Santi, C.; Bagnoli, L. A domino approach to pyrazino-indoles and pyrroles using vinyl selenones. Tetrahedron 2018, 74, 7156–7163. [Google Scholar] [CrossRef]
- Bandini, M.; Eichholzer, A.; Monari, M.; Piccinelli, F.; Umani-Ronchi, A. Versatile base-catalyzed route to polycyclic heteroaromatic compounds by intramolecular Aza-Michael addition. Eur. J. Org. Chem. 2007, 2007, 2917–2920. [Google Scholar] [CrossRef]
- Broggini, G.; Barbera, V.; Beccalli, E.M.; Borsini, E.; Galli, S.; Lanza, G.; Zecchi, G. Palladium(II)/copper halide/solvent combination for selective intramolecular domino reactions of indolecarboxylic acid allylamides: An unprecedented arylation/esterification sequence. Adv. Synth. Catal. 2012, 354, 159–170. [Google Scholar] [CrossRef]
- Abbiati, G.; Beccalli, E.M.; Broggini, G.; Zoni, C. Regioselectivity on the palladium-catalyzed intramolecular cyclization of indole derivatives. J. Org. Chem. 2003, 68, 7625–7628. [Google Scholar] [CrossRef]
- Abbiati, G.; Beccalli, E.; Broggini, G.; Martinelli, M.; Paladino, G. Pd-catalyzed cyclization of 1-allyl-2-indolecarboxamides by intramolecular amidation of unactivated ethylenic bond. Synlett 2006, 1, 0073–0076. [Google Scholar] [CrossRef]
- Boothe, J.B.; Shen, Y.; Wolfe, J.P. Synthesis of substituted γ-and δ-lactams via pd-catalyzed alkene carboamination reactions. J. Org. Chem. 2017, 82, 2777–2786. [Google Scholar] [CrossRef] [PubMed]
- Laliberté, S.; Dornan, P.K.; Chen, A. Palladium-catalyzed double allylic alkylation of indole-2-hydroxamates: Easy access to pyrazino [1,2-a] indole derivatives. Tetrahedron Lett. 2010, 51, 363–366. [Google Scholar] [CrossRef]
- Trost, B.M.; Osipov, M.; Dong, G. Palladium-catalyzed dynamic kinetic asymmetric transformations of vinyl aziridines with nitrogen heterocycles: Rapid access to biologically active pyrroles and indoles. J. Am. Chem. Soc. 2010, 132, 15800–15807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verniest, G.; Padwa, A. Gold- and silver-mediated cycloisomerizations of N-propargylamides. Org. Lett. 2008, 10, 4379–4382. [Google Scholar] [CrossRef]
- Sari, O.; Seybek, A.F.; Kaya, S.; Menges, N.; Erdem, S.; Balci, M. Mechanistic insights into the reaction of N-propargylated pyrrole- and indole-carbaldehyde with ammonia, alkyl amines, and branched amines: A synthetic and theoretical investigation. Eur. J. Org. Chem. 2019, 2019, 5261–5274. [Google Scholar] [CrossRef]
- Mahdavi, M.; Hassanzadeh-Soureshjan, R.; Saeedi, M.; Ariafard, A.; BabaAhmadi, R.; Ranjbar, P.R.; Shafiee, A. Experimental and computational evidence for KOt-Bu-promoted synthesis of oxopyrazino[1,2-a] indoles. RSC Adv. 2015, 5, 101353. [Google Scholar] [CrossRef]
- Goldberg, D.R.; Choi, Y.; Cogan, D.; Corson, M.; DeLeon, R.; Gao, A.; Gruenbaum, L.; Hao, M.H.; Joseph, D.; Kashem, M.A.; et al. Pyrazinoindolone inhibitors of MAPKAP-K2. Bioorg. Med. Chem. Lett. 2008, 18, 938–941. [Google Scholar] [CrossRef]
- Youssif, B.G.M.; Abdelrahman, M.H.; Abdelgawad, M.A.; Ibrahim, H.M.; Salem, O.I.A.; Mohamed, L. Treambleau, M.F.A.; Bukhari, S.N.A. Design, synthesis, mechanistic and histopathological studies of small-molecules of novel indole-2-carboxamides and pyrazino[1,2-a] indol-1(2H)-ones as potential anticancer agents effecting the reactive oxygen species production. Eur. J. Med. Chem. 2018, 146, 260–273. [Google Scholar] [PubMed]
- Kim, Y.J.; Pyo, J.S.; Jung, Y.S.; Kwak, J.H. Design, synthesis, and biological evaluation of novel 1-oxo-1,2,3,4-tetrahydropyrazino[1,2-a] indole-3-carboxamide analogs in MCF-7 and MDA-MB-468 breast cancer cell lines. Bioorg. Med. Chem. Lett. 2017, 27, 607–611. [Google Scholar] [CrossRef]
- Shiokawa, Z.; Kashiwabara, E.; Yoshidome, D.; Fukase, K.; Inuki, S.; Fujimoto, Y. Discovery of a novel scaffold as an indoleamine 2,3-dioxygenase 1 (IDO1) inhibitor based on the pyrrolopiperazinone alkaloid, longamide, b. ChemMedChem 2016, 11, 2682–2689. [Google Scholar] [CrossRef] [PubMed]
- Boger, D.L.; Fink, B.E.; Hendrick, M.P. A new class of highly cytotoxic diketopiperazines. Bioorg. Med. Chem. Lett. 2000, 10, 1019–1020. [Google Scholar] [CrossRef]
- Parrino, B.; Spano, V.; Carbone, A.; Barraja, P.; Diana, P.; Cirrincione, G.; Montalbano, A. Synthesis of the new ring system bispyrido [4′,3′:4, 5] pyrrolo [1,2-a: 1′,2′-d] pyrazine and its deaza analogue. Molecules 2014, 19, 13342–13357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akeng’a, T.O.; Read, R.W. Synthesis of Indoles: Tetrahydropyrazino[1,2-a]indole-1,4-dione and Pyrazino[1,2-a]indole-6,13-diones from Piperazine-2,5-diones. S. Afr. J. Chem. 2005, 58, 93–97. [Google Scholar]
- Vigushin, D.M.; Brooke, G.; Coombes, R.C.; Moody, C.J. Pyrazino [1,2-a] indole-1, 4-diones, simple analogues of gliotoxin, as selective inhibitors of geranylgeranyltransferase I. Bioorg. Med. Chem. Lett. 2003, 13, 3661–3663. [Google Scholar] [CrossRef]
- Liang, W.L.; Le, X.; Li, H.J.; Yang, H.L.; Chen, J.X.; Xu, J.; Liu, H.L.; Wang, L.Y.; Wang, K.T.; Hu, K.C.; et al. Exploring the chemodiversity and biological activities of the secondary metabolites from the marine fungus Neosartorya pseudofischeri. Mar. Drugs 2014, 12, 5657–5676. [Google Scholar] [CrossRef] [Green Version]
- Zoidis, G.; Giannakopoulou, E.; Stevaert, A.; Frakolaki, E.; Myrianthopoulos, V.; Fytas, G.; Mavromara, P.; Mikros, E.; Bartenschlager, R.; Vassilaki, N.; et al. Novel indole–flutimide heterocycles with activity against influenza PA endonuclease and hepatitis C. virus. MedChemComm 2016, 7, 447–456. [Google Scholar] [CrossRef]
- Edwards, T.C.; Lomonosova, E.; Patel, J.A.; Li, Q.; Villa, J.A.; Gupta, A.K.; Morrison, L.A.; Bailly, F.; Cotelle, P.; Giannakopoulou, E.; et al. Inhibition of hepatitis B virus replication by N-hydroxyisoquinolinediones and related polyoxygenated heterocycles. Antiviral Res. 2017, 143, 205–217. [Google Scholar] [CrossRef]
- Ilyn, A.P.; Kuzovkova, J.A.; Potapov, V.V.; Shkirando, A.M.; Kovrigin, D.I.; Tkachenko, S.E.; Ivachtchenko, A. An efficient synthesis of novel heterocycle-fused derivatives of 1-oxo-1,2,3,4-tetrahydropyrazine using Ugi condensation. Tetrahedron Lett. 2005, 46, 881–884. [Google Scholar] [CrossRef]
- Mokrosz, J.L.; Duszynska, B.; Paluchowska, M. Structure-activity relationship studies of CNS agents, XV: N-[ω-(4-aryl-1-piperazinyl)alkyl]-2-oxo-1,2,3,4-tetrahydroquinolines and -4-oxo-1,2,3,4-tetrahydropyrazino[1,2-a] indoles: New, highly potent 5-HT1A ligands ωN-[ω-(4-aryl-1-piperazinyl)alkyl]-2-oxo-1,2,3,4-tetrahydrochinoline und -4-oxo-1,2,3,4-tetrahydropyrazino-[1,2-a] indole: Neue starke 5-HT1A Liganden. Arch. Pharm. 1994, 327, 529–531. [Google Scholar]
- Campiani, G.; Butini, S.; Trotta, F.; Fattorusso, C.; Catalanotti, B.; Aiello, F.; Gemma, S.; Nacci, V.; Novellino, E.; Stark, J.A.; et al. Synthesis and pharmacological evaluation of potent and highly selective D3 receptor ligands: Inhibition of cocaine-seeking behavior and the role of dopamine D3/D2 receptors. J. Med. Chem. 2003, 46, 3822–3839. [Google Scholar] [CrossRef] [PubMed]

































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
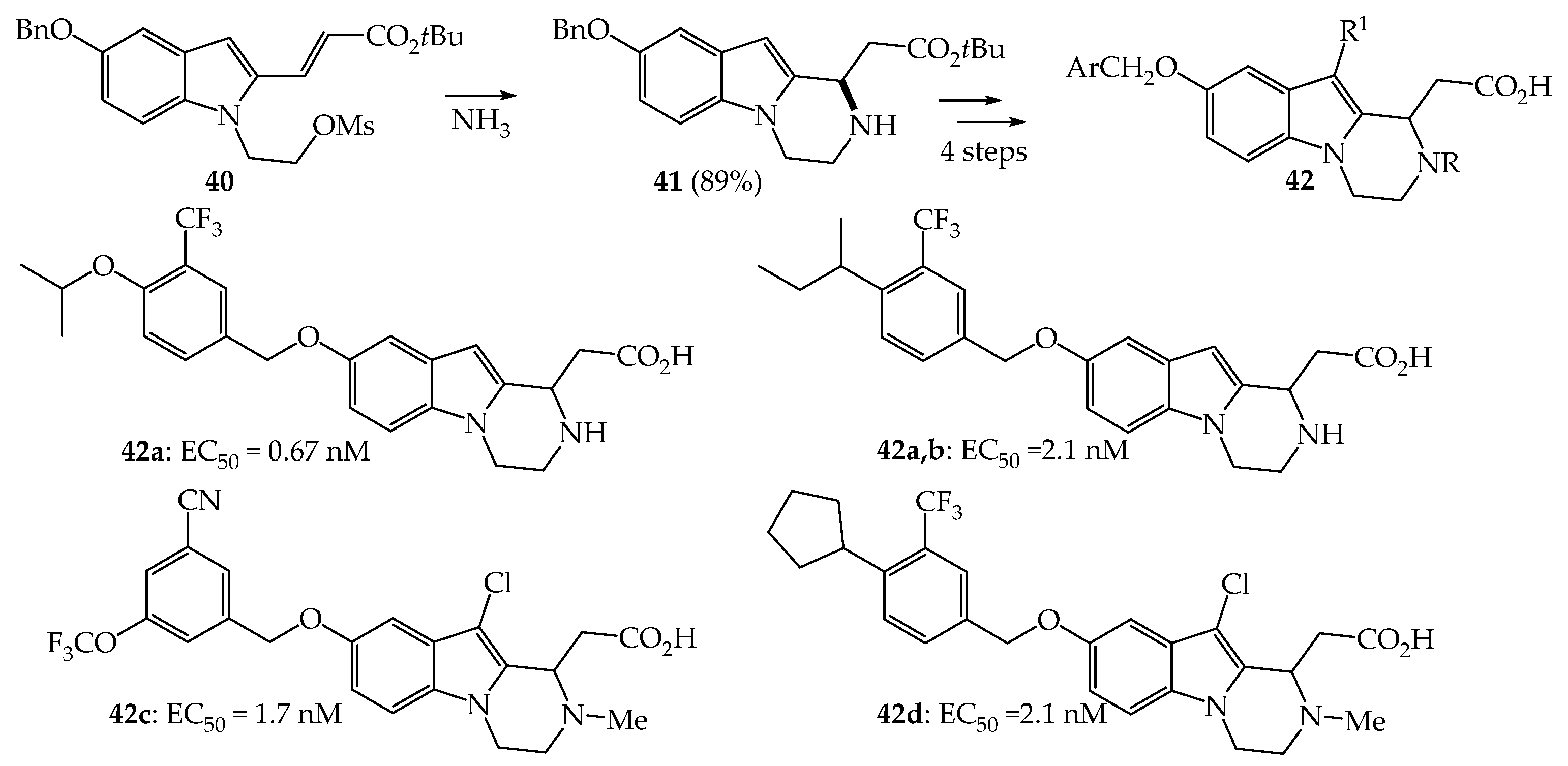{kind=link}
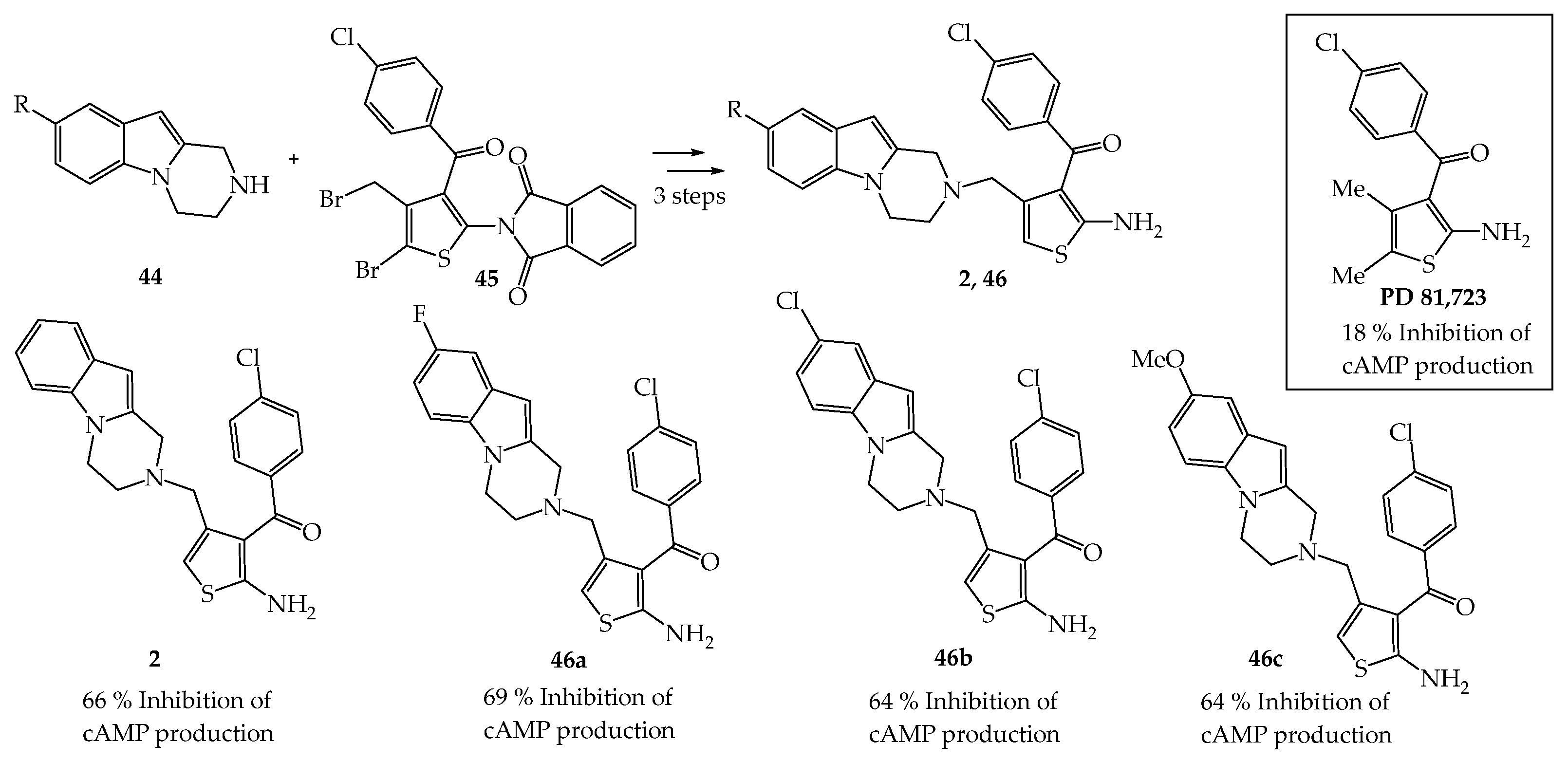{kind=link}
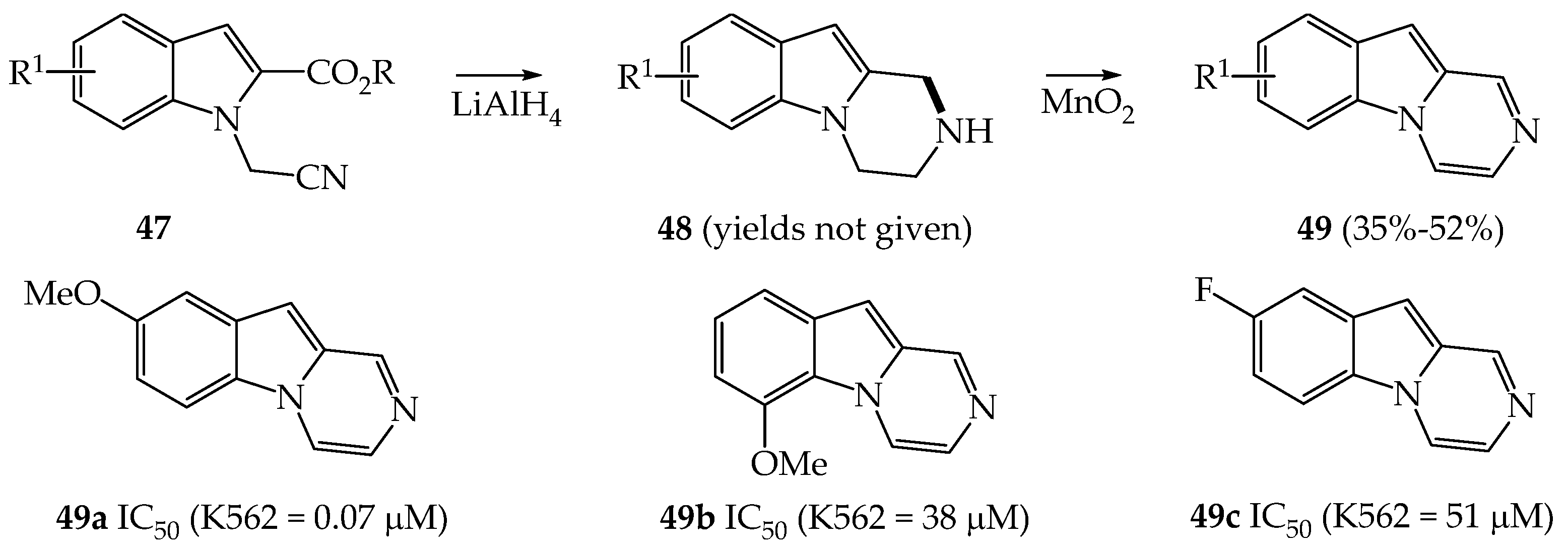{kind=link}
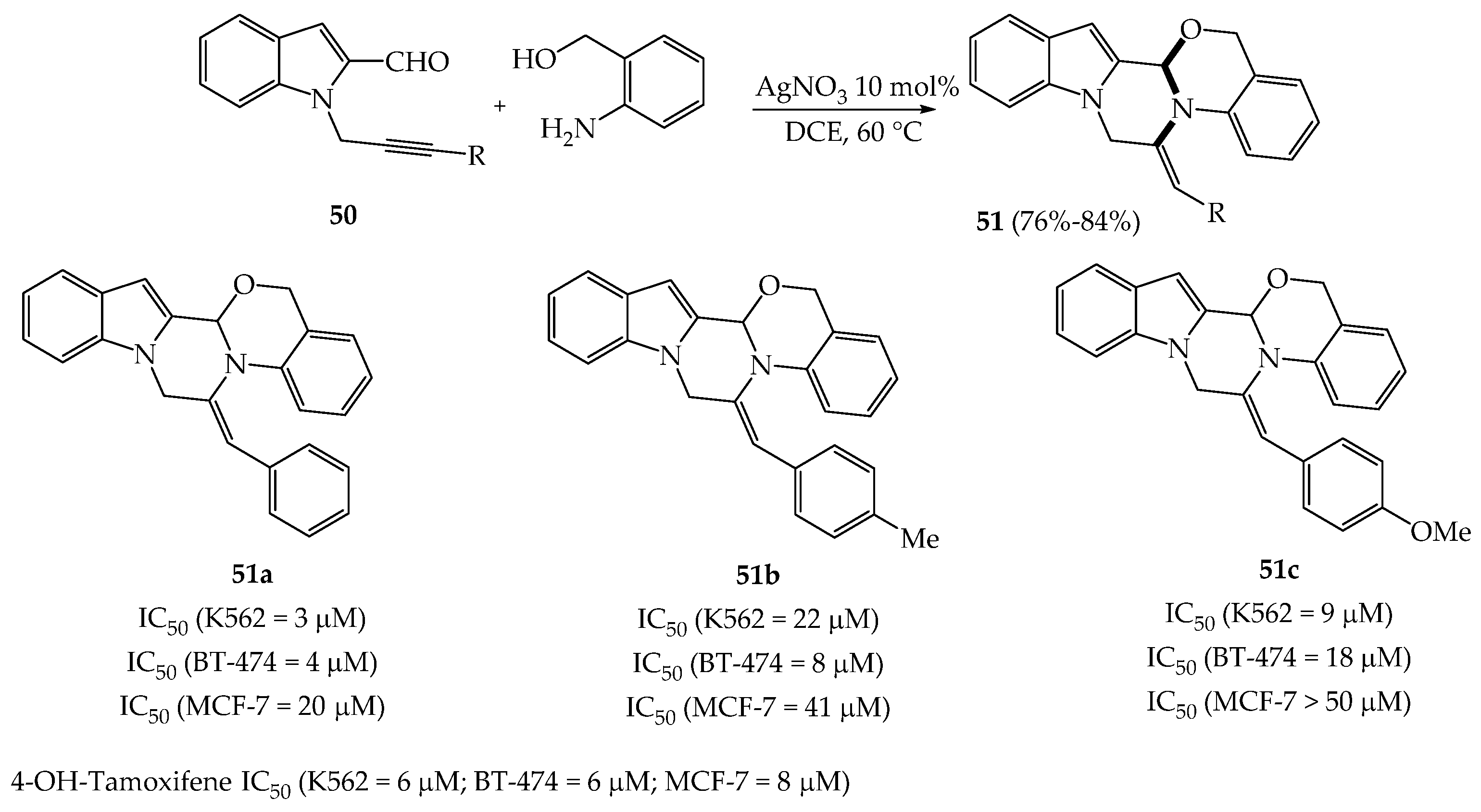{kind=link}
{kind=link}
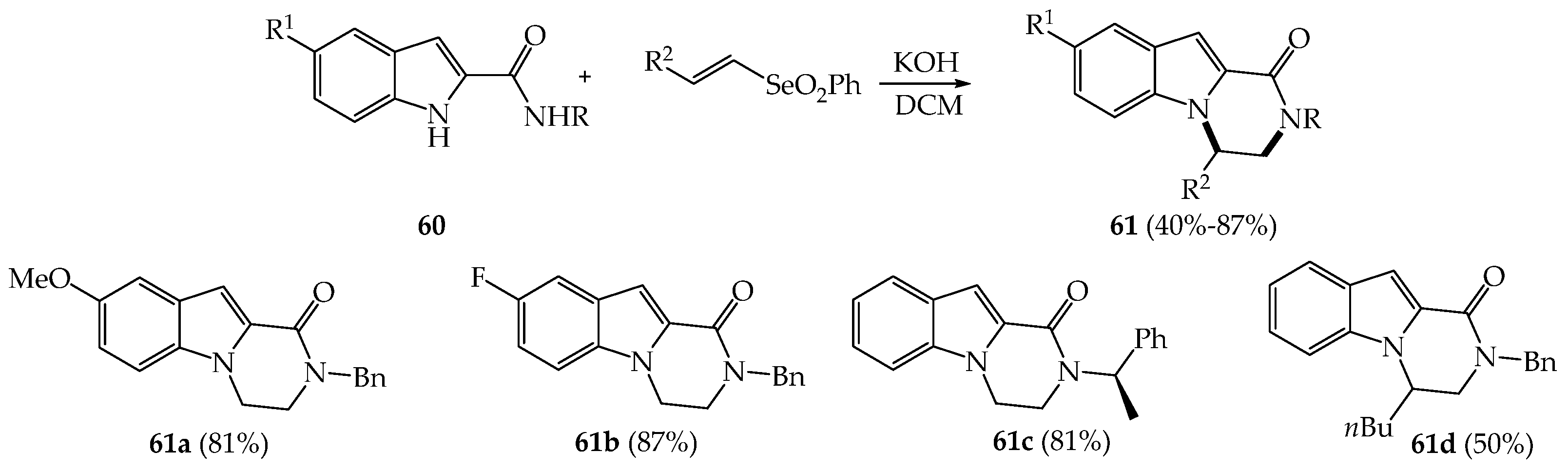{kind=link}
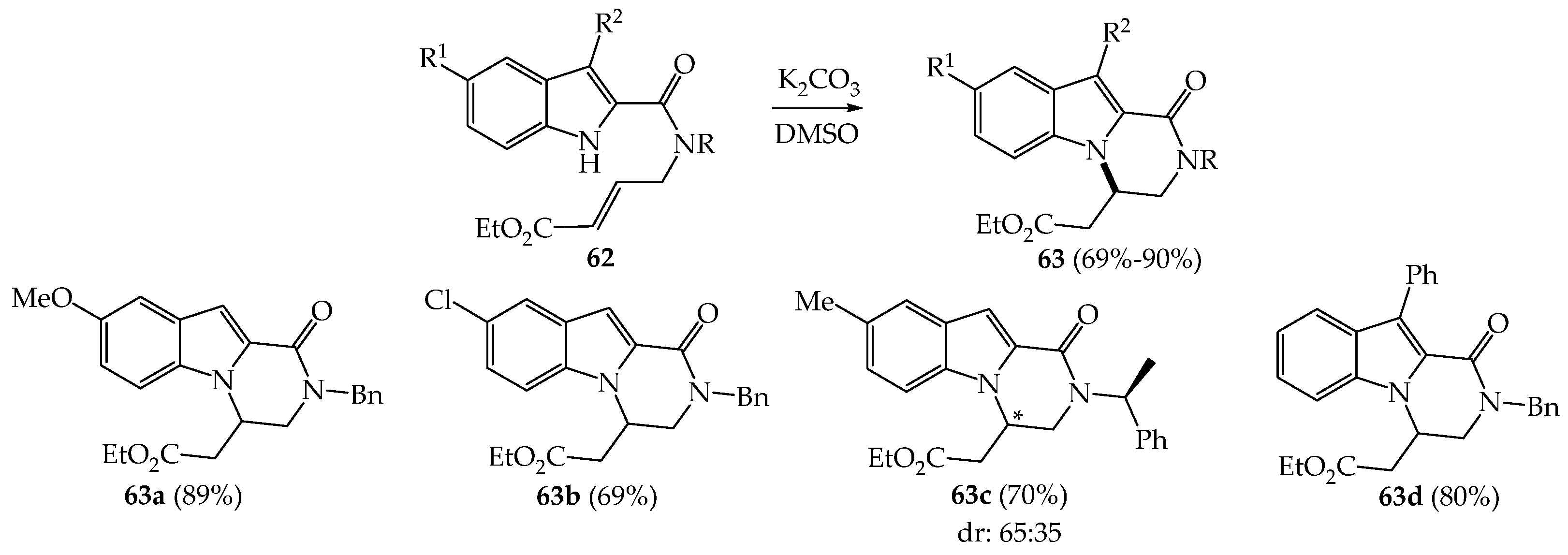{kind=link}
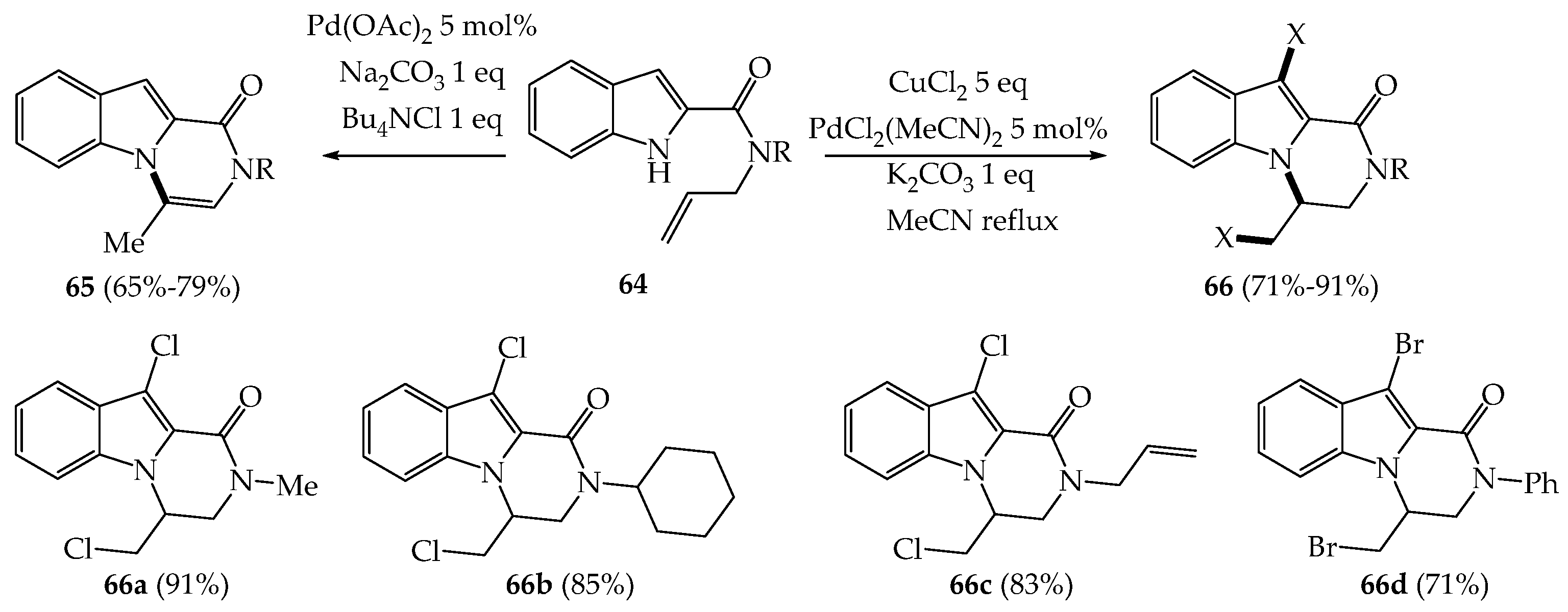{kind=link}
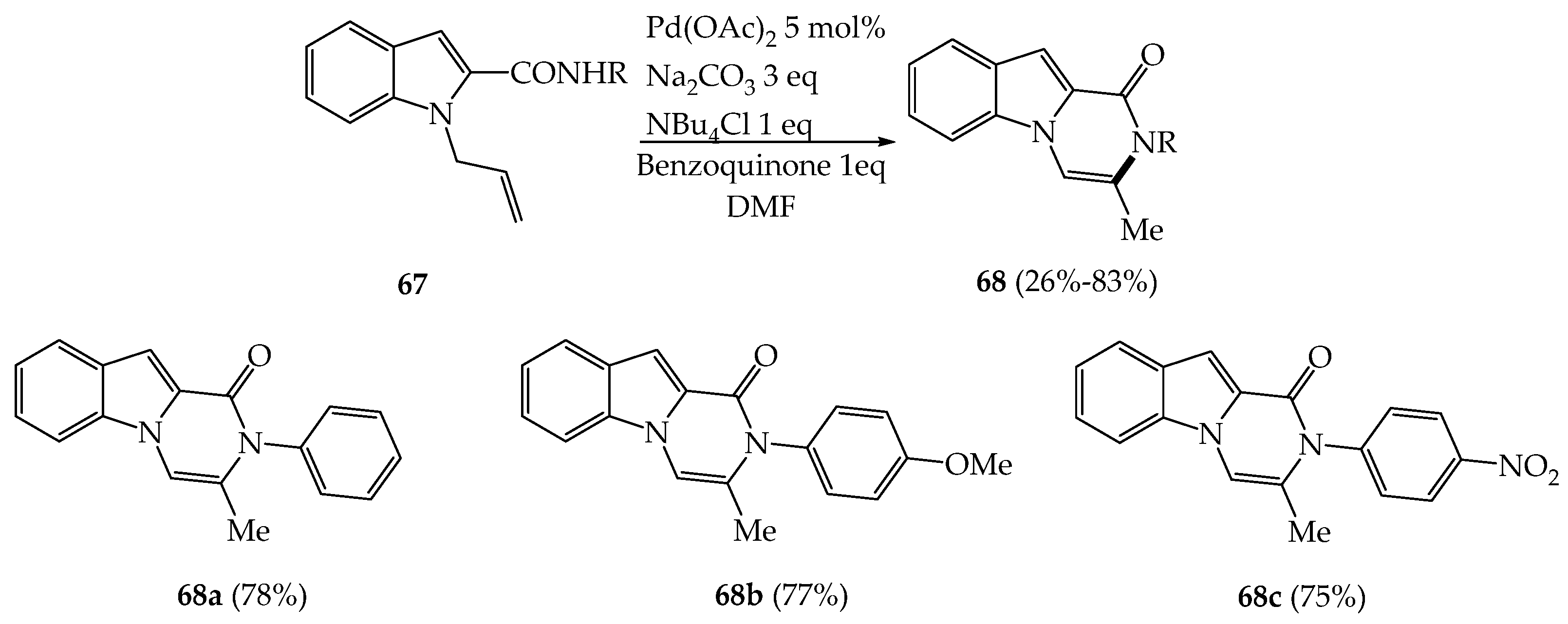{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
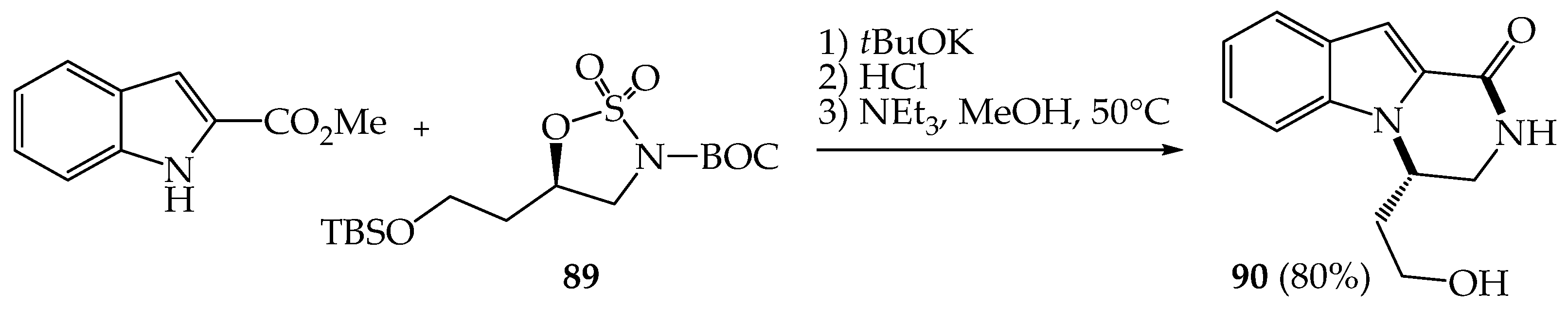{kind=link}
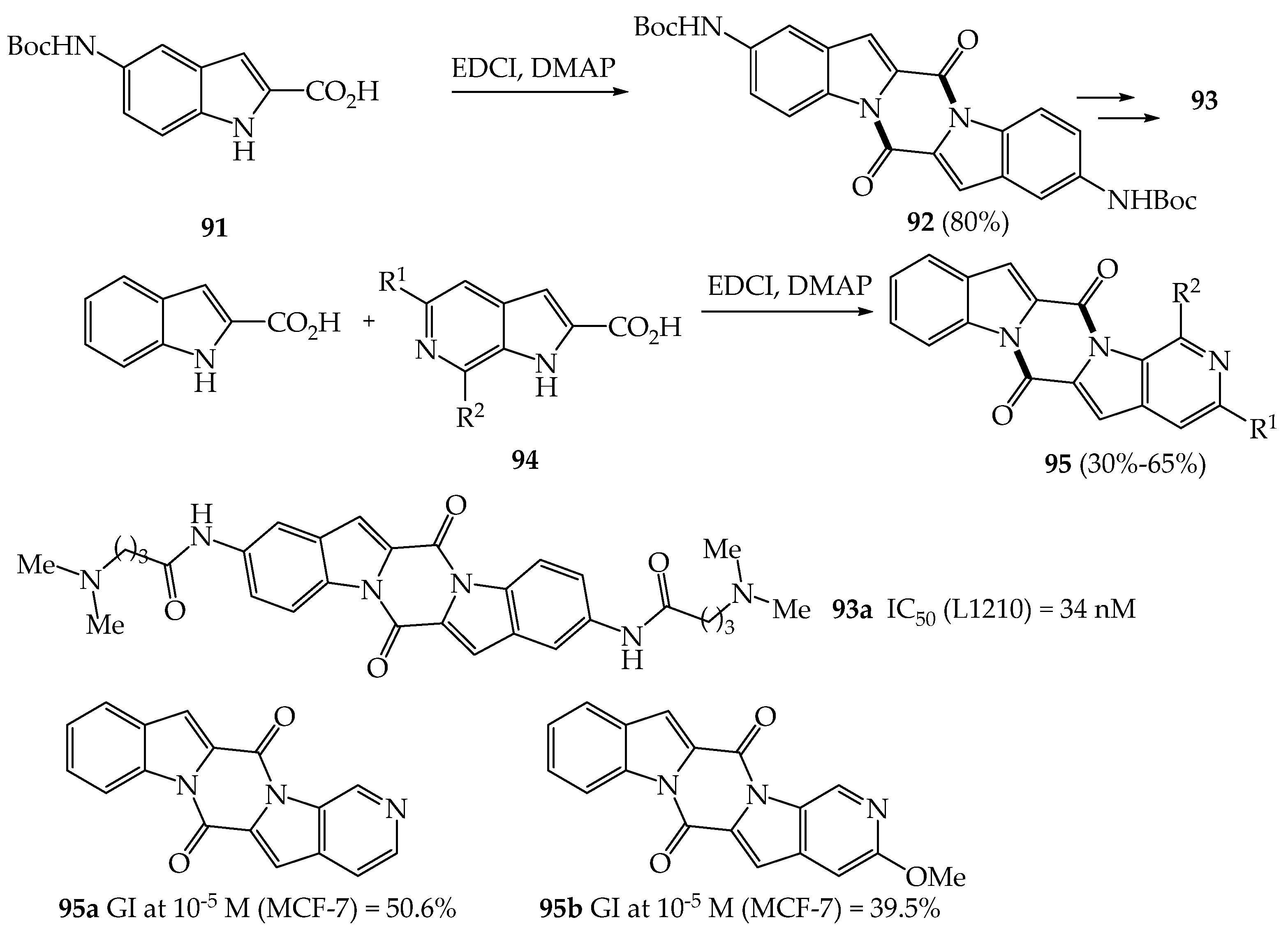{kind=link}
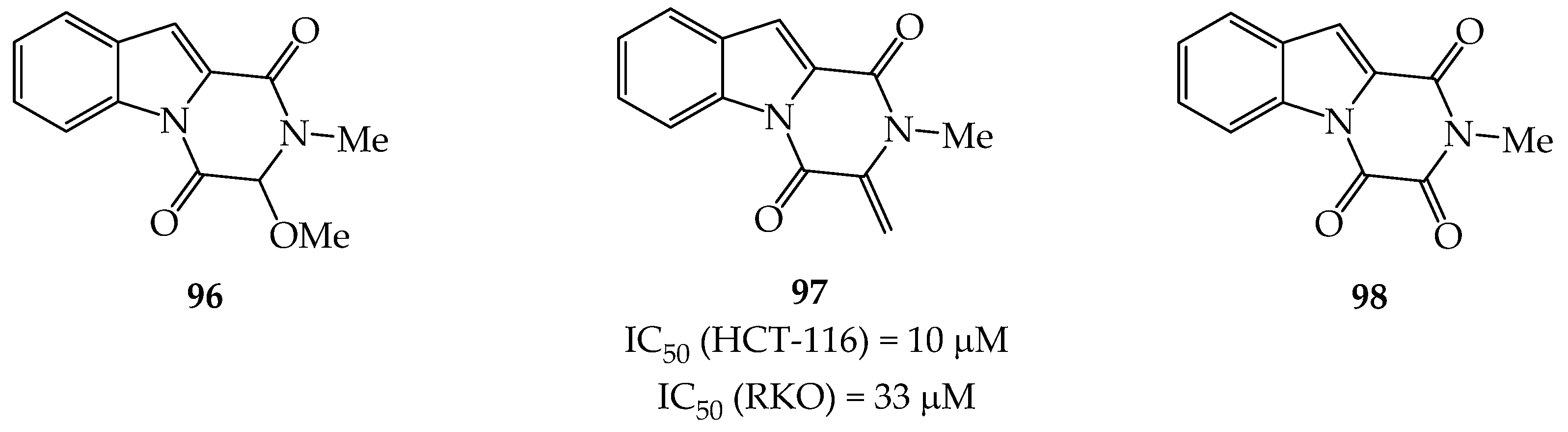{kind=link}
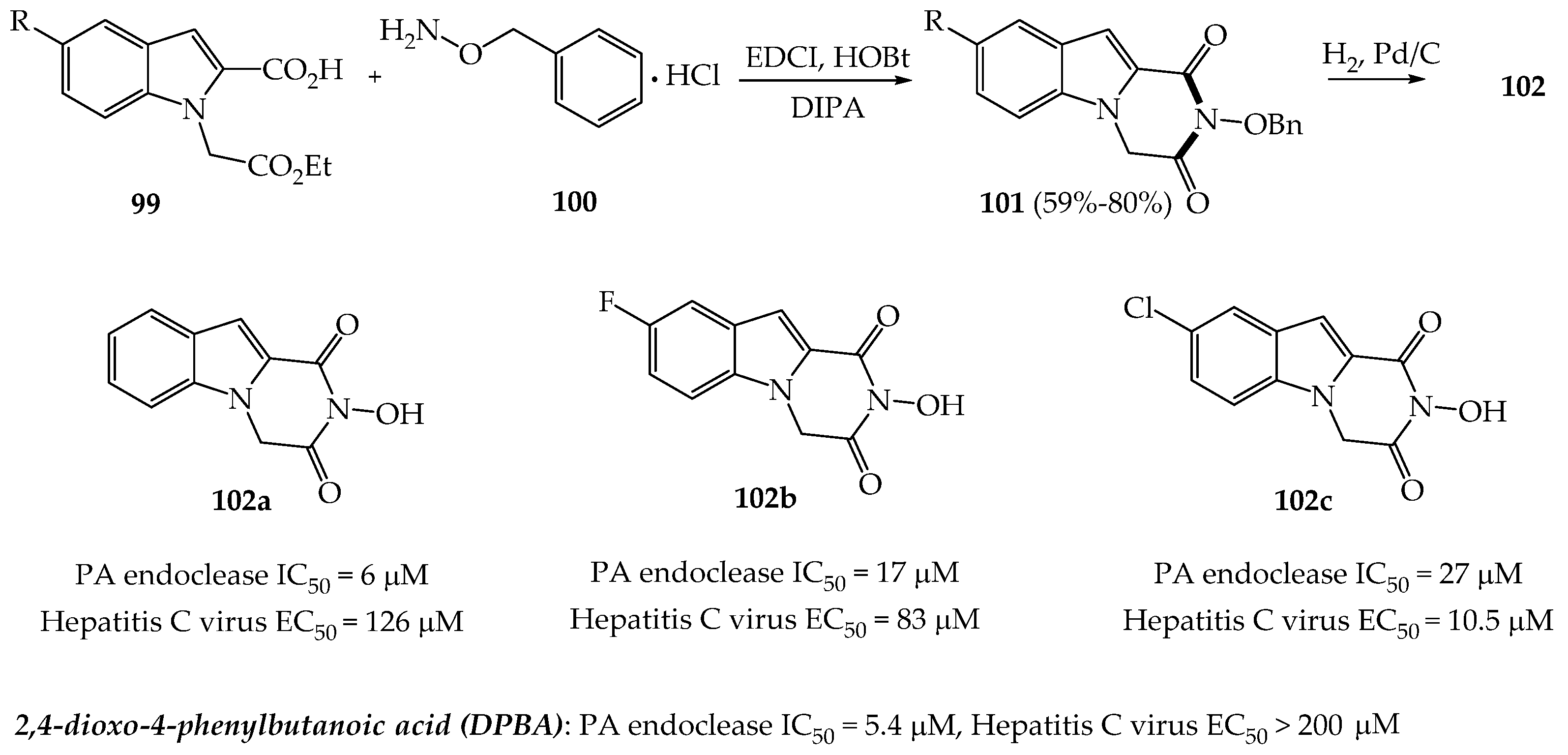{kind=link}
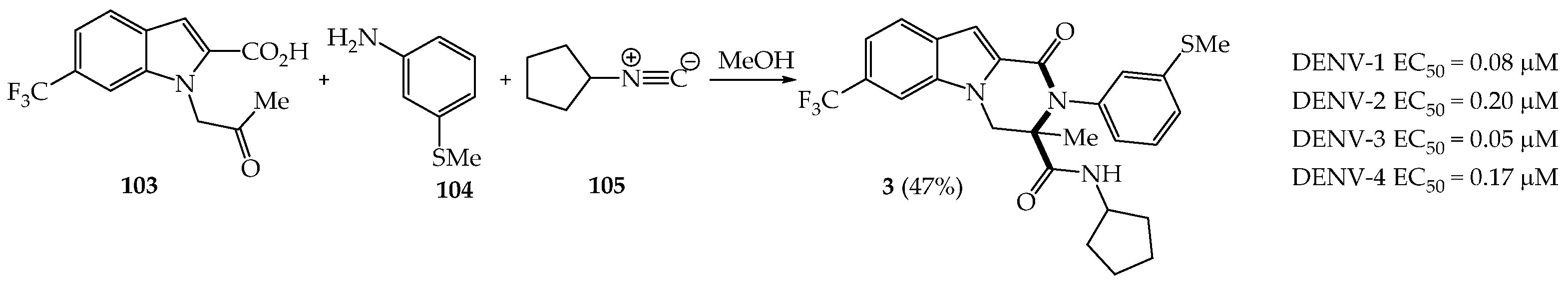{kind=link}
{kind=link}
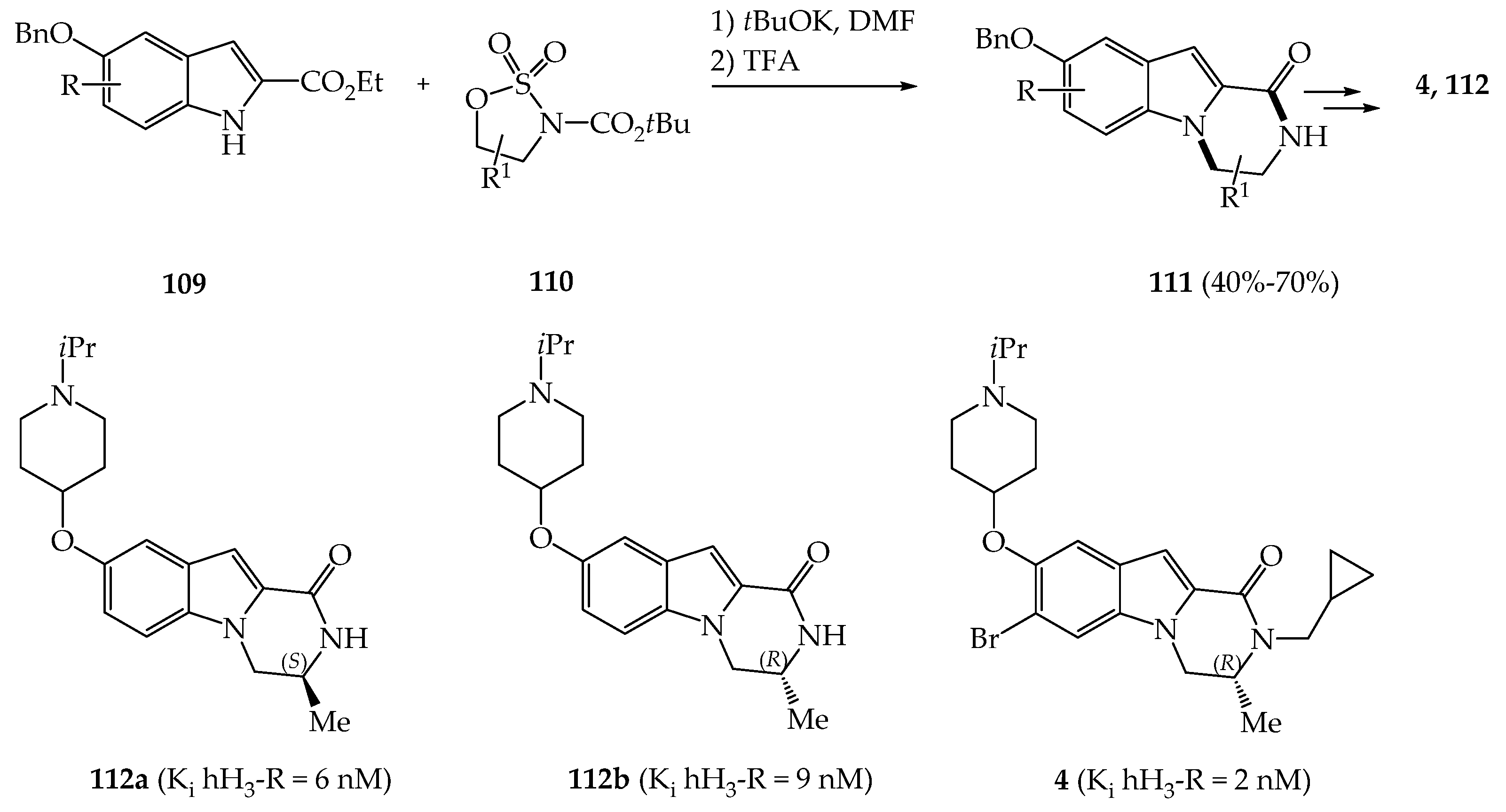{kind=link}

| Cpnd | DDA Minimum Inhibitory Concentrations (μg/disc) | |||
|---|---|---|---|---|
| S. aureus | S. typhi | P. aeruginosa | S. thermonitrificans | |
| 43a | - | - | 3.75 | - |
| 43b | - | - | 15 | 3.75 |
| 43c | 30 | 30 | 30 | 7.5 |
| 43d | 15 | 60 | 60 | 60 |
| 43e | 15 | 30 | 60 | 30 |
| 43f | - | - | 60 | 60 |
| Gentamycin | 1 | 1 | 0.5 | 1 |

| Cpnd | DDA Minimum Inhibitory Concentrations (μg/disc) | |||
|---|---|---|---|---|
| A. flavus | A. fumigatus | A. niger | C. albicans | |
| 43g | 11.7 | 5.8 | 11.7 | 15.6 |
| 43h | 47 | 23 | 47 | 62.5 |
| 43i | 187 | 94 | 187 | 125 |
| 43c | 47 | 47 | 47 | 125 |
| Gentamycin | 1 | 1 | 0.5 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, K.; Tran, C.; Alami, M.; Hamze, A.; Provot, O. Synthesis and Biological Activities of Pyrazino[1,2-a]indole and Pyrazino[1,2-a]indol-1-one Derivatives. Pharmaceuticals 2021, 14, 779. https://doi.org/10.3390/ph14080779
Zhang K, Tran C, Alami M, Hamze A, Provot O. Synthesis and Biological Activities of Pyrazino[1,2-a]indole and Pyrazino[1,2-a]indol-1-one Derivatives. Pharmaceuticals. 2021; 14(8):779. https://doi.org/10.3390/ph14080779
Chicago/Turabian StyleZhang, Kena, Christine Tran, Mouad Alami, Abdallah Hamze, and Olivier Provot. 2021. "Synthesis and Biological Activities of Pyrazino[1,2-a]indole and Pyrazino[1,2-a]indol-1-one Derivatives" Pharmaceuticals 14, no. 8: 779. https://doi.org/10.3390/ph14080779
APA StyleZhang, K., Tran, C., Alami, M., Hamze, A., & Provot, O. (2021). Synthesis and Biological Activities of Pyrazino[1,2-a]indole and Pyrazino[1,2-a]indol-1-one Derivatives. Pharmaceuticals, 14(8), 779. https://doi.org/10.3390/ph14080779