
Determination of Oxaliplatin by a UHPLC-MS/MS Method: Application to Pharmacokinetics and Tongue Tissue Distribution Studies in Rats


 ,
,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Method Development
2.1.1. Mass Spectrometry
2.1.2. Chromatography Separation
2.2. Method Validation
2.2.1. Selectivity and Specificity
2.2.2. Sensitivity and Linearity
2.2.3. Accuracy and Precision
2.2.4. Dilution Integrity
2.2.5. Matrix Effect and Extraction Recovery
2.2.6. Stability
2.3. Pharmacokinetics and Tongue Distribution Study
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Animal Purchase and Procedures
3.3. Instruments and Conditions
3.3.1. Ultra High-Performance Liquid Chromatography (UHPLC) Conditions
3.3.2. Tandem Mass Spectrometry (MS/MS) Conditions
3.4. Preparation of Stock Solutions, Calibration Standards and Quality Controls
3.5. Extraction of OXP from Plasma and Tongue Samples
3.6. Method Validation
3.6.1. Selectivity and Specificity
3.6.2. Sensitivity and Linearity
3.6.3. Carryover
3.6.4. Accuracy and Precision
3.6.5. Dilution Integrity
3.6.6. Extraction Recovery and Matrix Effect
3.6.7. Stability
3.7. Patch Formulation
3.8. Pharmacokinetics and Tongue Distribution Studies
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AUC0→90min | plasma concentration–time curve during the period of observation |
| BW | body weight |
| CAD | collision gas |
| CE | collision energy |
| CL | clearance |
| CMC | carboxymethyl cellulose |
| CPC | cancerostatic platinum compounds |
| CV% | coefficient of variation |
| CXP | cell exit potential |
| DACH | diaminocyclohexane |
| DOC | dihydrated OXP complex |
| DOPA | 3,4-dihydroxyphenylalanine |
| DP | declustering potential |
| DQC | diluted quality control |
| EMA | European Medicines Agency |
| ESI | electrospray ionization |
| FAAS | flame atomic absorption spectroscopy |
| FDA | Food and Drug Administration |
| FOLFOX | chemotherapy regimen combined 5-fluorouracil, folic acid, and oxaliplatin |
| GI | gastrointestinal |
| HILIC | hydrophilic interaction liquid chromatography |
| HPLC | high-performance liquid chromatography |
| ICP-MS | Inductively Coupled Plasma Mass Spectrometry |
| ICP-QMS | ICP quadrupole-based instrument |
| ICP-SFMS | ICP-sector field MS |
| IS | internal standard |
| LC-MS/MS | liquid chromatography with tandem mass spectrometry |
| LLOQ | lower limit of quantification |
| LOD | lower limit of detection |
| MRM | multiple reaction monitoring |
| MRT | mean residence time |
| OSCC-25 | oral squamous cell carcinoma |
| OXP | oxaliplatin |
| PAA | polyacrylic acid-974 |
| PK | pharmacokinetics |
| PUF | plasma ultrafiltrates |
| QC | quality control |
| RE% | relative error |
| SD | standard deviation |
| T1/2 | half-life |
| Vd | volume of distribution |
References
- Oun, R.; Moussa, Y.E.; Wheate, N.J. The side effects of platinum-based chemotherapy drugs: A review for chemists. Dalton Trans. 2018, 47, 6645–6653. [Google Scholar] [CrossRef]
- Dilruba, S.; Kalayda, G.V. Platinum-based drugs: Past, present and future. Cancer Chemother. Pharmacol. 2016, 77, 1103–1124. [Google Scholar] [CrossRef]
- Lin, T.; Meng, L.; Tsai, R.Y. GTP depletion synergizes the anti-proliferative activity of chemotherapeutic agents in a cell type-dependent manner. Biochem. Biophys. Res. Commun. 2011, 414, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Tsai, R.Y.L.; Ma, J.; Bhupal, P.K.; Liu, X.; Liang, D.; Xie, H. Determination and validation of mycophenolic acid by a UPLC-MS/MS method: Applications to pharmacokinetics and tongue tissue distribution studies in rats. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2020, 1136, 121930. [Google Scholar] [CrossRef]
- De Jong, L.A.W.; Elekonawo, F.M.K.; de Reuver, P.R.; Bremers, A.J.A.; de Wilt, J.H.W.; Jansman, F.G.A.; Heine, R.T.; van Erp, N.P. Hyperthermic intraperitoneal chemotherapy with oxaliplatin for peritoneal carcinomatosis: A clinical pharmacological perspective on a surgical procedure. Br. J. Clin. Pharmacol. 2019, 85, 47–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, C.H.; Khwaounjoo, P.; Hill, A.G.; Miskelly, G.M.; McKeage, M.J. Predicting effects on oxaliplatin clearance: In vitro, kinetic and clinical studies of calcium- and magnesium-mediated oxaliplatin degradation. Sci. Rep. 2017, 7, 4073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, F.R.; Yen, T.Y.; Wyrick, S.D.; Chaney, S.G. High-performance liquid chromatographic separation of the biotransformation products of oxaliplatin. J. Chromatogr. B Biomed. Sci. Appl. 1999, 724, 345–356. [Google Scholar] [CrossRef]
- Allain, P.; Heudi, O.; Cailleux, A.; Le Bouil, A.; Larra, F.; Boisdron-Celle, M.; Gamelin, E. Early biotransformations of oxaliplatin after its intravenous administration to cancer patients. Drug Metab. Dispos. 2000, 28, 1379–1384. [Google Scholar]
- Luo, F.R.; Wyrick, S.D.; Chaney, S.G. Biotransformations of oxaliplatin in rat blood in vitro. J. Biochem. Mol. Toxicol. 1999, 13, 159–169. [Google Scholar] [CrossRef]
- Jerremalm, E.; Wallin, I.; Ehrsson, H. New insights into the biotransformation and pharmacokinetics of oxaliplatin. J. Pharm. Sci. 2009, 98, 3879–3885. [Google Scholar] [CrossRef]
- Graham, M.A.; Lockwood, G.F.; Greenslade, D.; Brienza, S.; Bayssas, M.; Gamelin, E. Clinical pharmacokinetics of oxaliplatin: A critical review. Clin. Cancer Res. 2000, 6, 1205–1218. [Google Scholar]
- Reardon, J.T.; Vaisman, A.; Chaney, S.G.; Sancar, A. Efficient nucleotide excision repair of cisplatin, oxaliplatin, and Bis-aceto-ammine-dichloro-cyclohexylamine-platinum(IV) (JM216) platinum intrastrand DNA diadducts. Cancer Res. 1999, 59, 3968–3971. [Google Scholar]
- Starobova, H.; Vetter, I. Pathophysiology of Chemotherapy-Induced Peripheral Neuropathy. Front. Mol. Neurosci. 2017, 10, 174. [Google Scholar] [CrossRef] [PubMed]
- Shirao, K.; Matsumura, Y.; Yamada, Y.; Muro, K.; Gotoh, M.; Boku, N.; Ohtsu, A.; Nagashima, F.; Sano, Y.; Mutoh, M.; et al. Phase I study of single-dose oxaliplatin in Japanese patients with malignant tumors. Jpn. J. Clin. Oncol. 2006, 36, 295–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kern, W.; Braess, J.; Böttger, B.; Kaufmann, C.C.; Hiddemann, W.; Schleyer, E. Oxaliplatin pharmacokinetics during a four-hour infusion. Clin. Cancer Res. 1999, 5, 761–765. [Google Scholar] [PubMed]
- Kweekel, D.M.; Gelderblom, H.; Guchelaar, H.J. Pharmacology of oxaliplatin and the use of pharmacogenomics to individualize therapy. Cancer Treat. Rev. 2005, 31, 90–105. [Google Scholar] [CrossRef]
- Shord, S.S.; Bernard, S.A.; Lindley, C.; Blodgett, A.; Mehta, V.; Churchel, M.A.; Poole, M.; Pescatore, S.L.; Luo, F.R.; Chaney, S.G. Oxaliplatin biotransformation and pharmacokinetics: A pilot study to determine the possible relationship to neurotoxicity. Anticancer Res. 2002, 22, 2301–2309. [Google Scholar]
- Falta, T.; Koellensperger, G.; Standler, A.; Buchberger, W.; Mader, R.M.; Hann, S. Quantification of cisplatin, carboplatin, and oxaliplatin in spiked human plasma samples by ICP-SFMS and hydrophilic interaction liquid chromatography (HILIC) combined with ICP-MS detector. J. Anal. At. Spectrom. 2009, 24, 1336–1342. [Google Scholar] [CrossRef]
- Hearnden, V.; Sankar, V.; Hull, K.; Juras, D.V.; Greenberg, M.; Kerr, A.R.; Lockhart, P.B.; Patton, L.L.; Porter, S.; Thornhill, M.H. New developments and opportunities in oral mucosal drug delivery for local and systemic disease. Adv. Drug Deliv. Rev. 2012, 64, 16–28. [Google Scholar] [CrossRef]
- Kulkarni, U.; Mahalingam, R.; Pather, S.I.; Li, X.; Jasti, B. Porcine buccal mucosa as an in vitro model: Relative contribution of epithelium and connective tissue as permeability barriers. J. Pharm. Sci. 2009, 98, 471–483. [Google Scholar] [CrossRef]
- Matos, B.N.; Pereira, M.N.; Bravo, M.O.; Cunha-Filho, M.; Saldanha-Araújo, F.; Gratieri, T.; Gelfuso, G.M. Chitosan nanoparticles loading oxaliplatin as a mucoadhesive topical treatment of oral tumors: Iontophoresis further enhances drug delivery ex vivo. Int. J. Biol. Macromol. 2020, 154, 1265–1275. [Google Scholar] [CrossRef]
- Ito, H.; Yamaguchi, H.; Fujikawa, A.; Tanaka, N.; Furugen, A.; Miyamori, K.; Takahashi, N.; Ogura, J.; Kobayashi, M.; Yamada, T.; et al. A full validated hydrophilic interaction liquid chromatography-tandem mass spectrometric method for the quantification of oxaliplatin in human plasma ultrafiltrates. J. Pharm. Biomed. Anal. 2012, 71, 99–103. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Seymour, L.; Chen, E.X. Determination of intact oxaliplatin in human plasma using high performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2008, 876, 277–282. [Google Scholar] [CrossRef]
- FDA. Draft Guidance for Industry: Bioanalytical Method Validation; US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER): Washington, DC, USA, 2018. [Google Scholar]
- Bastian, G.; Barrail, A.; Urien, S. Population pharmacokinetics of oxaliplatin in patients with metastatic cancer. Anticancer Drugs 2003, 14, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Nikanjam, M.; Stewart, C.F.; Takimoto, C.H.; Synold, T.W.; Beaty, O.; Fouladi, M.; Capparelli, E.V. Population pharmacokinetic analysis of oxaliplatin in adults and children identifies important covariates for dosing. Cancer Chemother. Pharmacol. 2015, 75, 495–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, Y.; Kobuchi, S.; Shimizu, R.; Katsuyama, Y. Pharmacokinetic and toxicodynamic evaluation of oxaliplatin-induced neuropathy and hematological toxicity in rats. Cancer Chemother. Pharmacol. 2018, 81, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Hanada, K.; Suda, M.; Kanai, N.; Ogata, H. Pharmacokinetics and toxicodynamics of oxaliplatin in rats: Application of a toxicity factor to explain differences in the nephrotoxicity and myelosuppression induced by oxaliplatin and the other platinum antitumor derivatives. Pharm. Res. 2010, 27, 1893–1899. [Google Scholar] [CrossRef]
- Gao, X.; Wu, L.; Tsai, R.Y.L.; Ma, J.; Liu, X.; Chow, D.S.; Liang, D.; Xie, H. Pharmacokinetic Model Analysis of Supralingual, Oral and Intravenous Deliveries of Mycophenolic Acid. Pharmaceutics 2021, 13, 574. [Google Scholar] [CrossRef]
- De Araujo, D.R.; Padula, C.; Cereda, C.M.S.; Tofoli, G.R.; Brito, R.B.; de Paula, E.; Nicoli, S.; Santi, P. Bioadhesive Films Containing Benzocaine: Correlation Between In Vitro Permeation and In Vivo Local Anesthetic Effect. Pharm. Res. 2010, 27, 1677–1686. [Google Scholar] [CrossRef]
- Donnelly, R.F.; McCarron, P.A.; Lightowler, J.M.; Woolfson, A.D. Bioadhesive patch-based delivery of 5-aminolevulinic acid to the nail for photodynamic therapy of onychomycosis. J. Control. Release 2005, 103, 381–392. [Google Scholar] [CrossRef]
- Donnelly, R.F.; Ma, L.W.; Juzenas, P.; Iani, V.; McCarron, P.A.; Woolfson, A.D.; Moan, J. Topical bioadhesive patch systems enhance selectivity of protoporphyrin IX accumulation. Photochem. Photobiol. 2006, 82, 670–675. [Google Scholar] [CrossRef]
- Nussinovitch, A.; Gal, A.; Padula, C.; Santi, P. Physical characterization of a new skin bioadhesive film. Aaps Pharmscitech 2008, 9, 458–463. [Google Scholar] [CrossRef] [Green Version]
- Peppas, N.A.; Sahlin, J.J. Hydrogels as mucoadhesive and bioadhesive materials: A review. Biomaterials 1996, 17, 1553–1561. [Google Scholar] [CrossRef]
- Vasir, J. Bioadhesive microspheres as a controlled drug delivery system. Int. J. Pharm. 2003, 255, 13–32. [Google Scholar] [CrossRef]
- Salman, S.; Bendel, D.; Lee, T.C.; Templeton, D.; Davis, T.M. Pharmacokinetics of a novel sublingual spray formulation of the antimalarial drug artemether in healthy adults. Antimicrob. Agents Chemother. 2015, 59, 3197–3207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parikh, N.; Goskonda, V.; Chavan, A.; Dillaha, L. Single-dose pharmacokinetics of fentanyl sublingual spray and oral transmucosal fentanyl citrate in healthy volunteers: A randomized crossover study. Clin. Ther. 2013, 35, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Boddupalli, B.M.; Mohammed, Z.N.; Nath, R.A.; Banji, D. Mucoadhesive drug delivery system: An overview. J. Adv. Pharm. Technol. Res. 2010, 1, 381–387. [Google Scholar] [CrossRef] [Green Version]
- Paderni, C.; Compilato, D.; Giannola, L.I.; Campisi, G. Oral local drug delivery and new perspectives in oral drug formulation. Oral. Surg. Oral. Med. Oral. Pathol. Oral. Radiol. 2012, 114, e25–e34. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.F.; Liu, F.; Brown, M.B. Advances in oral transmucosal drug delivery. J. Control. Release 2011, 153, 106–116. [Google Scholar] [CrossRef] [Green Version]
- Palmer, D.; Levina, M.; Nokhodchi, A.; Douroumis, D.; Farrell, T.; Rajabi-Siahboomi, A. The influence of sodium carboxymethylcellulose on drug release from polyethylene oxide extended release matrices. AAPS PharmSciTech 2011, 12, 862–871. [Google Scholar] [CrossRef]
- EMA. Guideline on Bioanalytical Method Validation; European Medicines Agency: Amsterdam, The Netherlands, 2012; pp. 1–23.
- Gabrielsson, J.; Weiner, D. Non-compartmental analysis. Methods Mol. Biol. 2012, 929, 377–389. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
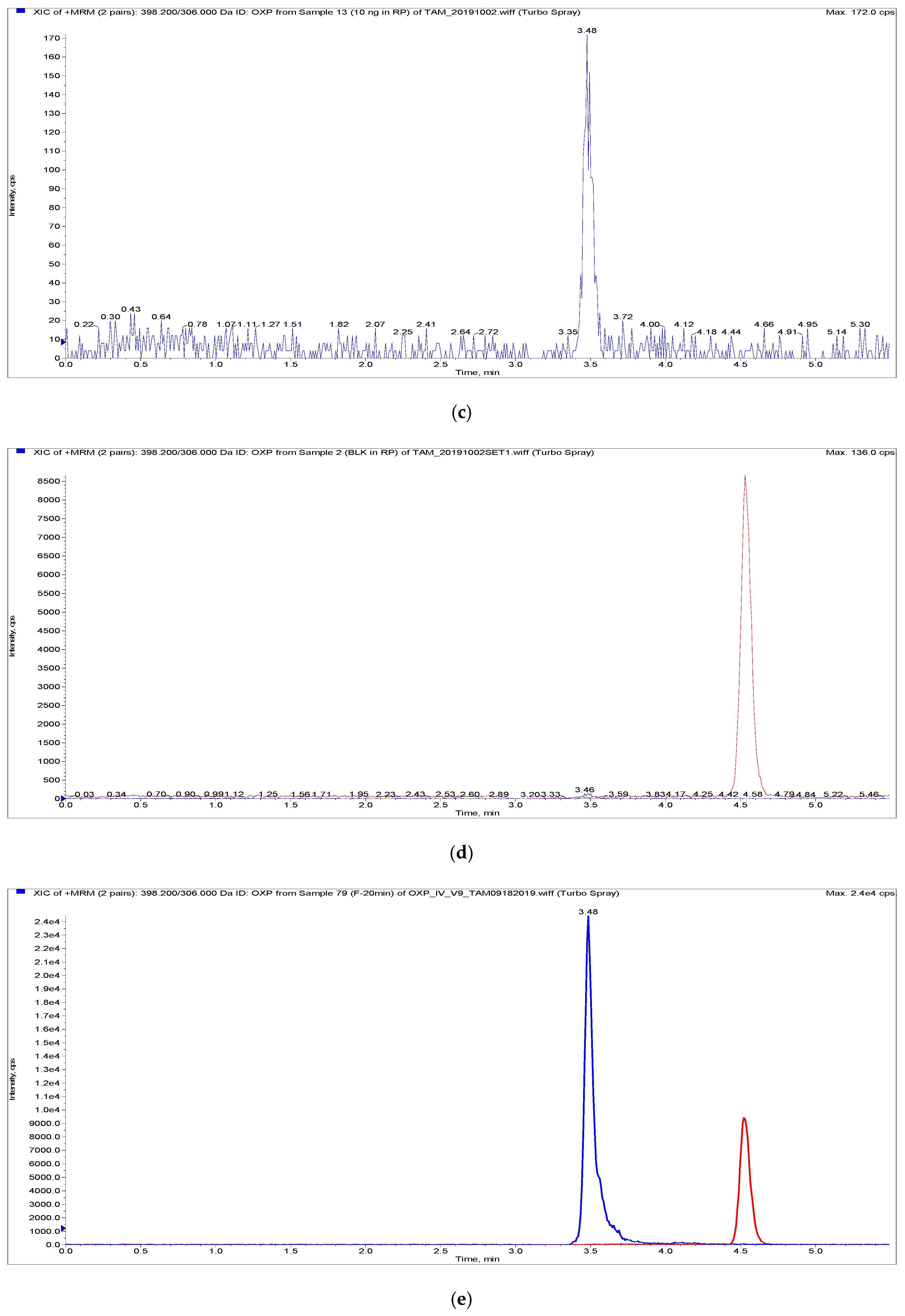{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| QCs (ng/mL) | Intra-Day (n = 5) | Inter-Day (n = 15) | |||||
|---|---|---|---|---|---|---|---|
| Observed Concentration (Mean ± SD) | RE% | CV% | Observed Concentration (Mean ± SD) | RE% | CV% | ||
| Plasma | 10 | 9.94 ± 0.85 | −0.63 | 8.54 | 9.84 ± 0.42 | −1.68 | 4.32 |
| 25 | 24.86 ± 2.15 | −0.58 | 8.67 | 26.10 ± 1.09 | 4.21 | 4.19 | |
| 2000 | 1874.44 ± 87.40 | −6.70 | 4.66 | 1903.33 ± 70.05 | −5.08 | 3.68 | |
| 4000 | 3985.00 ± 249.55 | −0.38 | 6.26 | 4065.00 ± 120.12 | 1.60 | 2.96 | |
| Tongue | 10 | 10.43 ± 0.44 | 4.15 | 4.19 | 10.45 ± 0.54 | 4.48 | 5.17 |
| 25 | 23.88 ± 0.95 | −4.68 | 3.97 | 24.95 ± 1.97 | −0.20 | 7.88 | |
| 800 | 783.33 ± 52.95 | −2.13 | 6.76 | 798.22 ± 49.90 | −0.22 | 6.25 | |
| 2000 | 2000 ± 47.33 | 0.00 | 2.37 | 2059.44 ± 109.52 | 2.97 | 5.32 | |
| Biological Samples | Nominal Concentration (ng/mL) | Matrix Effect (%) | Recovery (%) |
|---|---|---|---|
| (n = 6) | (n = 6) | ||
| Plasma | 10 | 47.02 ± 2.32 | 51.89 ± 1.50 |
| 25 | 37.17 ± 3.09 | 53.44 ± 3.98 | |
| 2000 | 46.61 ± 2.06 | 55.60 ± 2.24 | |
| 4000 | 43.24 ± 3.14 | 52.80 ± 6.85 | |
| Tongue | 10 | 12.43 ± 6.55 | 86.36 ± 7.00 |
| 25 | 1.26 ± 9.25 | 80.12 ± 7.38 | |
| 800 | 0.18 ± 6.36 | 82.30 ± 5.99 | |
| 2000 | 2.71 ± 6.71 | 84.32 ± 2.81 |
| Nominal Concentration (ng/mL) | Calculated Concentration (ng/mL) | Precision | Accuracy | |
|---|---|---|---|---|
| Mean ± SD | CV% | RE% | ||
| auto-sampler (6 h) 15 °C | 10 | 9.58 ± 0.48 | 4.98% | −4.25% |
| 25 | 25.15 ± 2.40 | 9.56% | 0.60% | |
| 2000 | 1937.50 ± 90.69 | 4.68% | −3.13% | |
| 4000 | 3487.50 ± 81.80 | 2.35% | −12.81% | |
| short-term (6 h) 4 °C | 10 | 9.91 ± 0.97 | 9.77% | −0.95% |
| 25 | 23.70 ± 1.61 | 6.81% | −5.20% | |
| 2000 | 2052.50 ± 226.62 | 11.04% | 2.63% | |
| 4000 | 3577.50 ± 102.10 | 2.85% | −10.56% | |
| 1-cycle-freeze thaw −80 °C to RT | 10 | 9.46 ± 0.69 | 7.28% | −5.43% |
| 25 | 23.45 ± 1.25 | 5.33% | −6.20% | |
| 2000 | 1950.00 ± 133.42 | 6.84% | −2.50% | |
| 4000 | 3985.00 ± 270.62 | 6.79% | −0.38% | |
| 2-cycle-freeze thaw −80 °C to RT | 10 | 8.84 ± 0.15 | 1.70% | −11.63% |
| 25 | 24.00 ± 0.94 | 3.91% | −4.00% | |
| 2000 | 1827.50 ± 126.06 | 6.90% | −8.63% | |
| 4000 | 3760.00 ± 194.42 | 5.17% | −6.00% | |
| 3-cycle-freeze thaw −80 °C to RT | 10 | 8.93 ± 1.06 | 11.90% | −10.73% |
| 25 | 17.55 ± 4.70 | 26.81% | −29.80% | |
| 2000 | 1272.50 ± 250.65 | 16.16% | −36.38% | |
| 4000 | 2630.00 ± 351.66 | 13.37% | −34.25% | |
| Long-term (one month) −80 °C | 10 | 7.185 ± 0.50 | 6.90% | −28.15% |
| 25 | 18.775 ± 0.92 | 4.89% | −24.90% | |
| 2000 | 1490 ± 73.94 | 4.96% | −25.50% | |
| 4000 | 2990 ± 47.61 | 1.59% | −25.25% | |
| Long-term (one week) −80 °C | 10 | 9.74 ± 0.21 | 2.19% | −2.60% |
| 25 | 26.9 ± 0.48 | 1.77% | 7.60% | |
| 2000 | 2127.5 ± 113.25 | 5.32% | 6.38% | |
| 4000 | 4387.5 ± 195.34 | 4.45% | 9.69% |
| Nominal Concentration (ng/mL) | Calculated Concentration (ng/mL) | Precision | Accuracy | |
|---|---|---|---|---|
| Mean ± SD | CV (%) | RE (%) | ||
| auto-sampler (6 h) 15 °C | 10 | 9.02 ± 0.72 | 8.02% | −9.80% |
| 25 | 22.40 ± 0.42 | 1.89% | −10.40% | |
| 800 | 733.25 ±37.43 | 5.10% | −8.34% | |
| 2000 | 1930.00 ± 29.16 | 2.03% | −3.50% | |
| short-term (6 h) 4 °C | 10 | 10.23 ± 1.43 | 13.97% | 2.25% |
| 25 | 26.80 ± 0.85 | 3.17% | 7.20% | |
| 800 | 858.00 ± 62.06 | 7.23% | 7.25% | |
| 2000 | 2055.00 ± 59.72 | 2.91% | 2.75% | |
| 1-cycle-freeze thaw −80 °C to RT | 10 | 10.875 ± 0.26 | 2.42% | 8.75% |
| 25 | 26.6 ± 0.28 | 1.06% | 6.40% | |
| 800 | 812.5 ± 79.33 | 9.76% | 1.56% | |
| 2000 | 1970 ± 212.60 | 10.79% | −1.50% | |
| 2-cycle-freeze thaw −80 °C to RT | 10 | 11.025 ± 0.59 | 5.31% | 10.25% |
| 25 | 25.05 ± 0.78 | 3.11% | 0.20% | |
| 800 | 696.00 ± 13.66 | 1.96% | −13.00% | |
| 2000 | 1855.00 ± 59.16 | 3.19% | −7.25% | |
| 3-cycle-freeze thaw −80 °C to RT | 10 | 6.57 ± 0.29 | 4.43% | −34.35% |
| 25 | 15.70 ± 0.99 | 6.31% | −37.20% | |
| 800 | 437.25 ± 67.07 | 15.34% | −45.34% | |
| 2000 | 1097.50 ± 20.62 | 1.88% | −45.13% | |
| Long-term (one month) −80 °C | 10 | 9.96 ± 0.75 | 7.49% | −0.45% |
| 25 | 20.60 ± 1.27 | 6.18% | −17.60% | |
| 800 | 507.75 ± 16.01 | 3.15% | −36.53% | |
| 2000 | 1232.50 ± 90.32 | 7.33% | −38.38% | |
| Long-term (one week) −80 °C | 10 | 9.50 ± 0.35 | 3.71% | −5.05% |
| 25 | 25.25 ± 3.46 | 13.72% | 1.00% | |
| 800 | 731.25 ± 12.84 | 1.76% | −8.59% | |
| 2000 | 2045.00 ± 186.28 | 9.11% | 2.25% |
| Rat A | Rat B | Rat C | Mean | SD | CV% | ||
|---|---|---|---|---|---|---|---|
| Parameter | Units | Estimate | |||||
| AUC0→90min | min.μg/mL | 159.40 | 217.62 | 224.09 | 200.37 | 35.63 | 17.8 |
| CL | mL/min/kg | 156.84 | 114.88 | 111.56 | 127.76 | 25.24 | 19.8 |
| T1/2 | min | 16.85 | 13.50 | 18.27 | 16.21 | 2.45 | 15.1 |
| MRT | min | 6.06 | 7.38 | 7.20 | 6.88 | 0.72 | 10.4 |
| Vd | mL/kg | 949.91 | 848.24 | 802.80 | 866.98 | 75.32 | 8.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, X.; Tsai, R.Y.L.; Ma, J.; Wang, Y.; Liu, X.; Liang, D.; Xie, H. Determination of Oxaliplatin by a UHPLC-MS/MS Method: Application to Pharmacokinetics and Tongue Tissue Distribution Studies in Rats. Pharmaceuticals 2022, 15, 52. https://doi.org/10.3390/ph15010052
Gao X, Tsai RYL, Ma J, Wang Y, Liu X, Liang D, Xie H. Determination of Oxaliplatin by a UHPLC-MS/MS Method: Application to Pharmacokinetics and Tongue Tissue Distribution Studies in Rats. Pharmaceuticals. 2022; 15(1):52. https://doi.org/10.3390/ph15010052
Chicago/Turabian StyleGao, Xiuqing, Robert Y. L. Tsai, Jing Ma, Yang Wang, Xiaohua Liu, Dong Liang, and Huan Xie. 2022. "Determination of Oxaliplatin by a UHPLC-MS/MS Method: Application to Pharmacokinetics and Tongue Tissue Distribution Studies in Rats" Pharmaceuticals 15, no. 1: 52. https://doi.org/10.3390/ph15010052
APA StyleGao, X., Tsai, R. Y. L., Ma, J., Wang, Y., Liu, X., Liang, D., & Xie, H. (2022). Determination of Oxaliplatin by a UHPLC-MS/MS Method: Application to Pharmacokinetics and Tongue Tissue Distribution Studies in Rats. Pharmaceuticals, 15(1), 52. https://doi.org/10.3390/ph15010052