
Pharmacological Modulation and (Patho)Physiological Roles of TRPM4 Channel—Part 1: Modulation of TRPM4

 and
and
Abstract
:1. Introduction
2. Activation of TRPM4
2.1. Ca2+
2.2. Phosphatidylinositol 4,5-Bisphosphate (PIP2)
2.3. Calmodulin
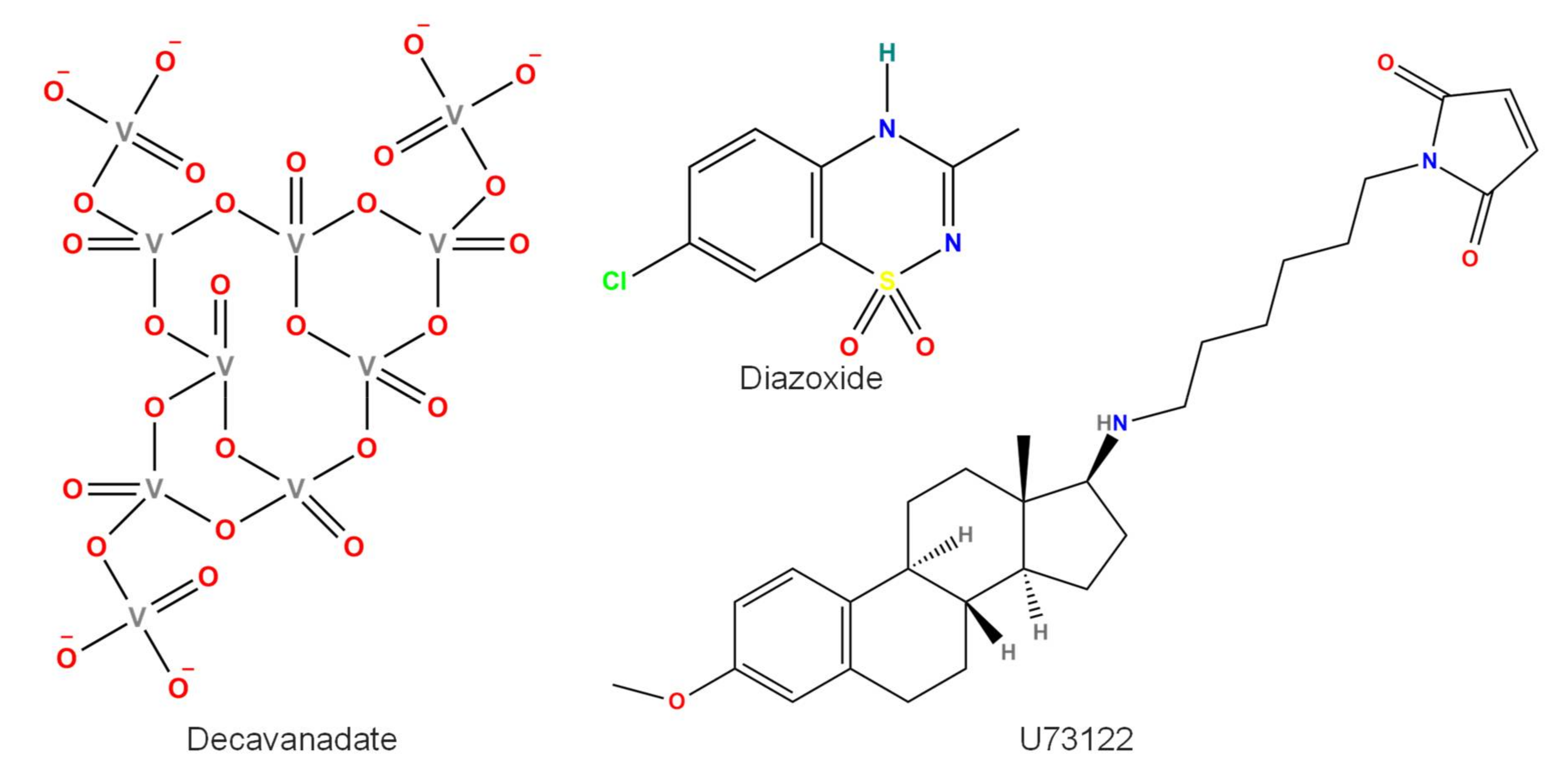
2.4. Decavanadate
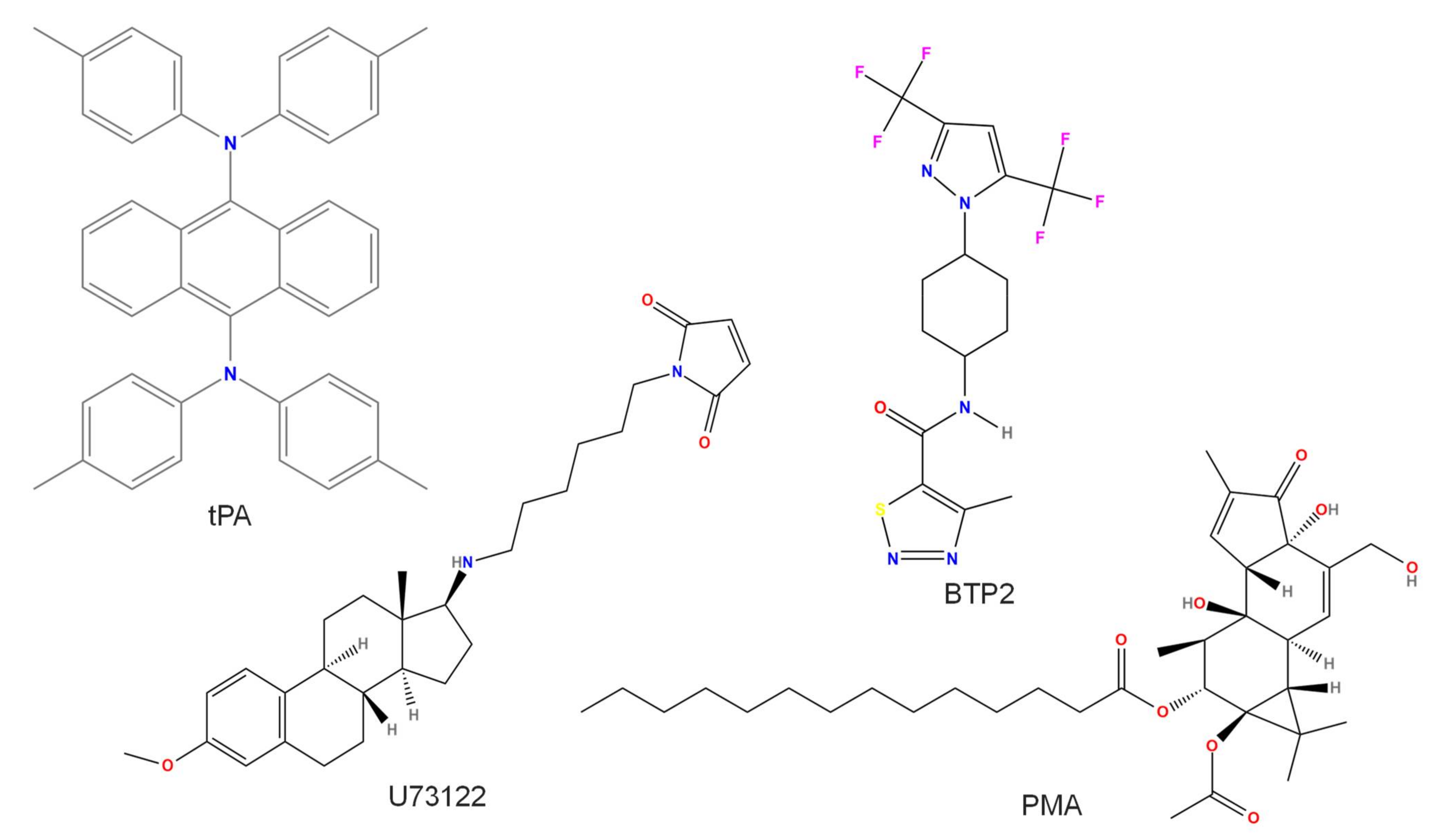
2.5. BTP2 or YM-58483
2.6. H2O2
2.7. Tissue Plasminogen Activator (tPA)
2.8. U73122
2.9. Diazoxide
2.10. PKC-Mediated Phosphorylation
3. Inhibition of TRPM4
3.1. Adenosine Triphosphate (ATP)
3.2. Nitric Oxide (NO)
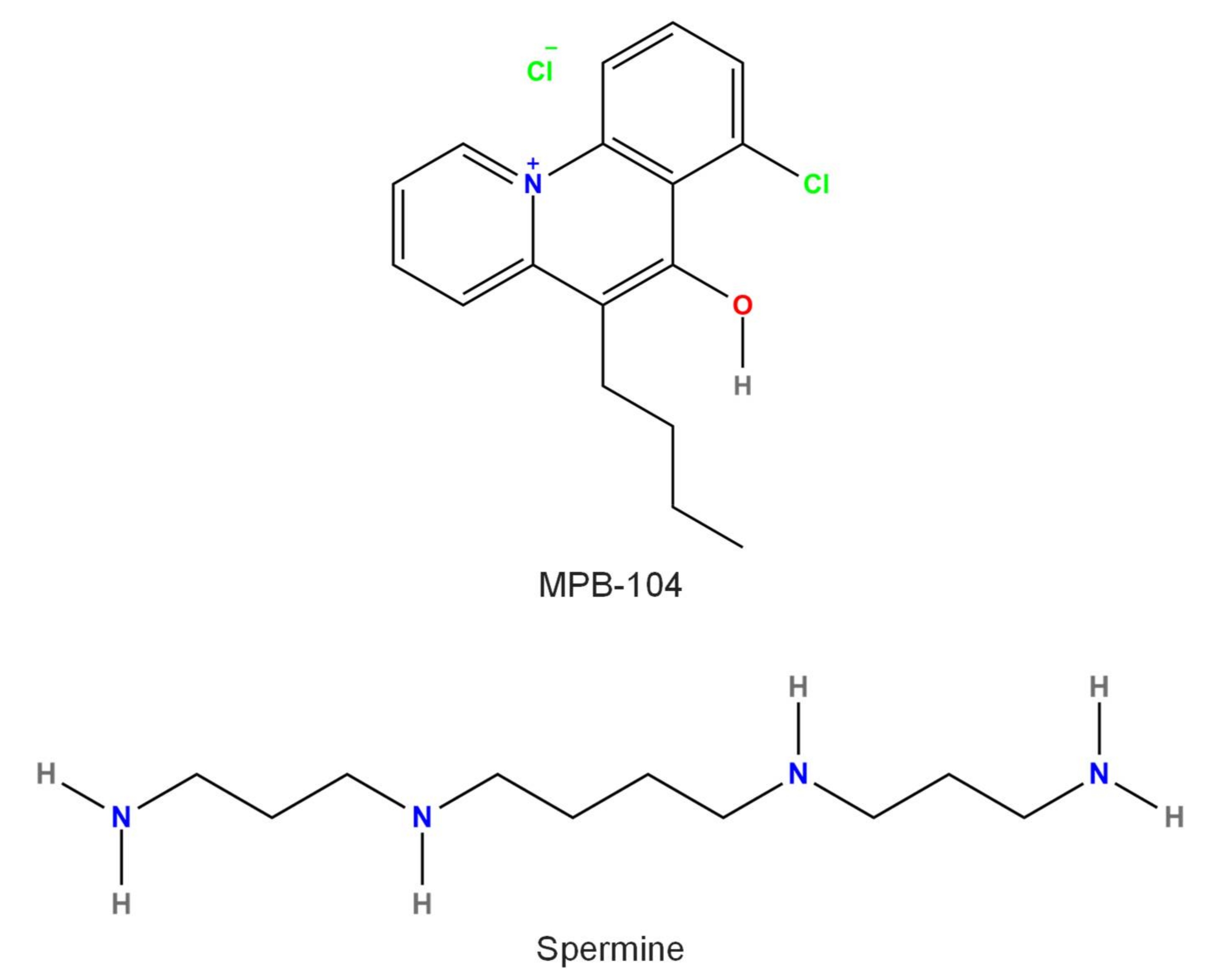
3.3. Spermine
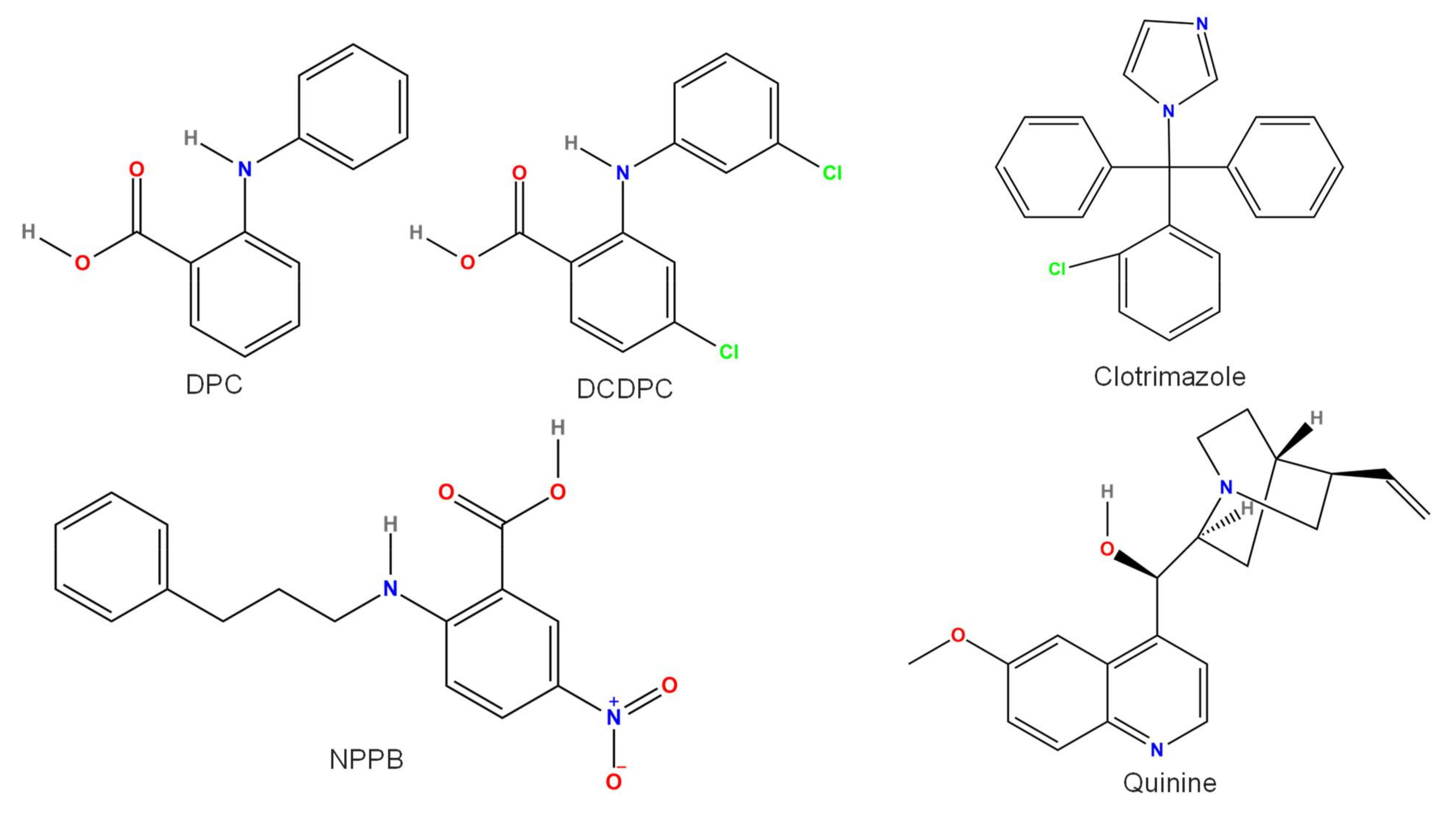
3.4. Quinine
3.5. MPB-104
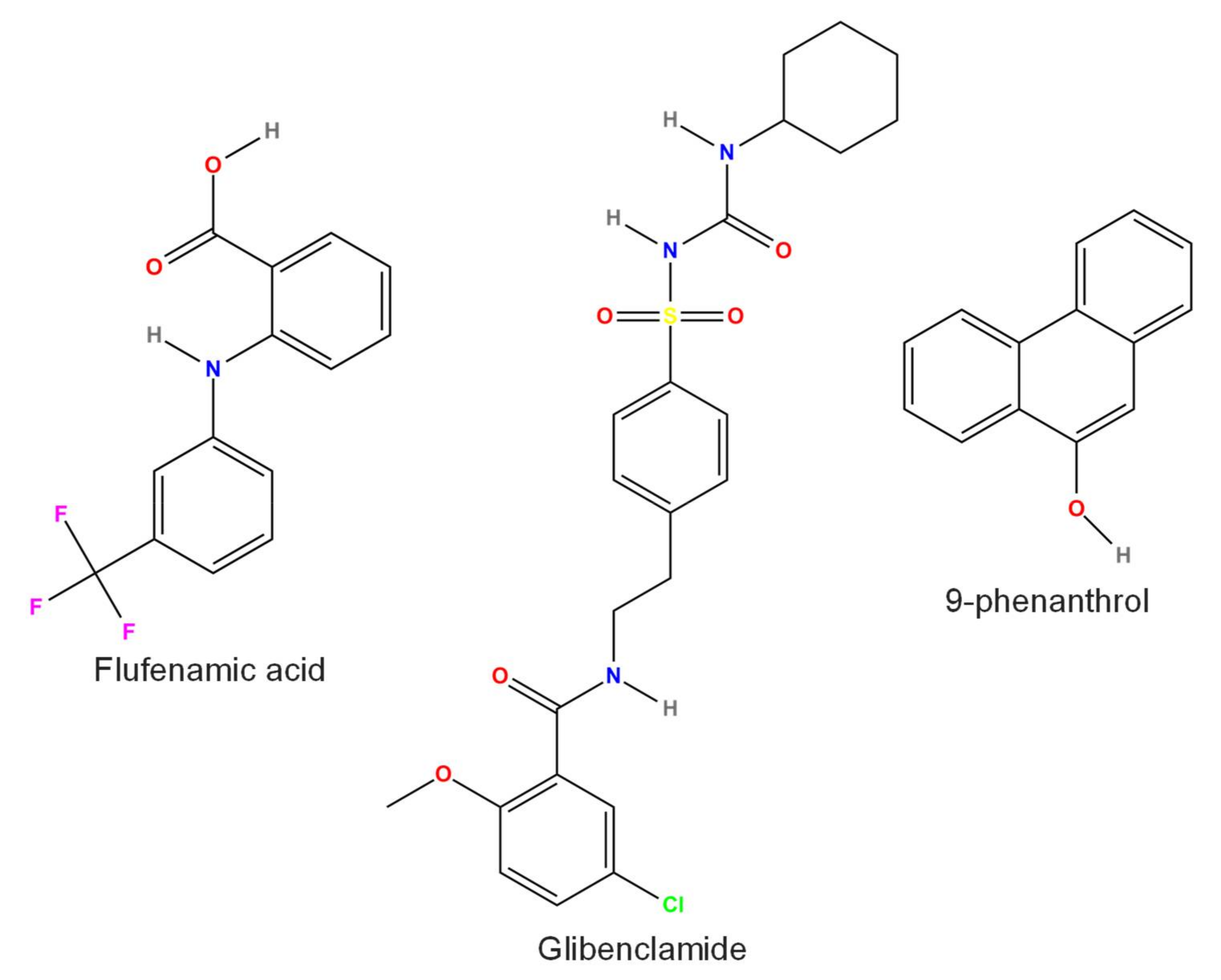
3.6. Flufenamic Acid (FFA)
3.7. Glibenclamide
3.8. Clotrimazole
3.9. DPC, DCDPC, and NPPB
3.10. 9-Phenanthrol
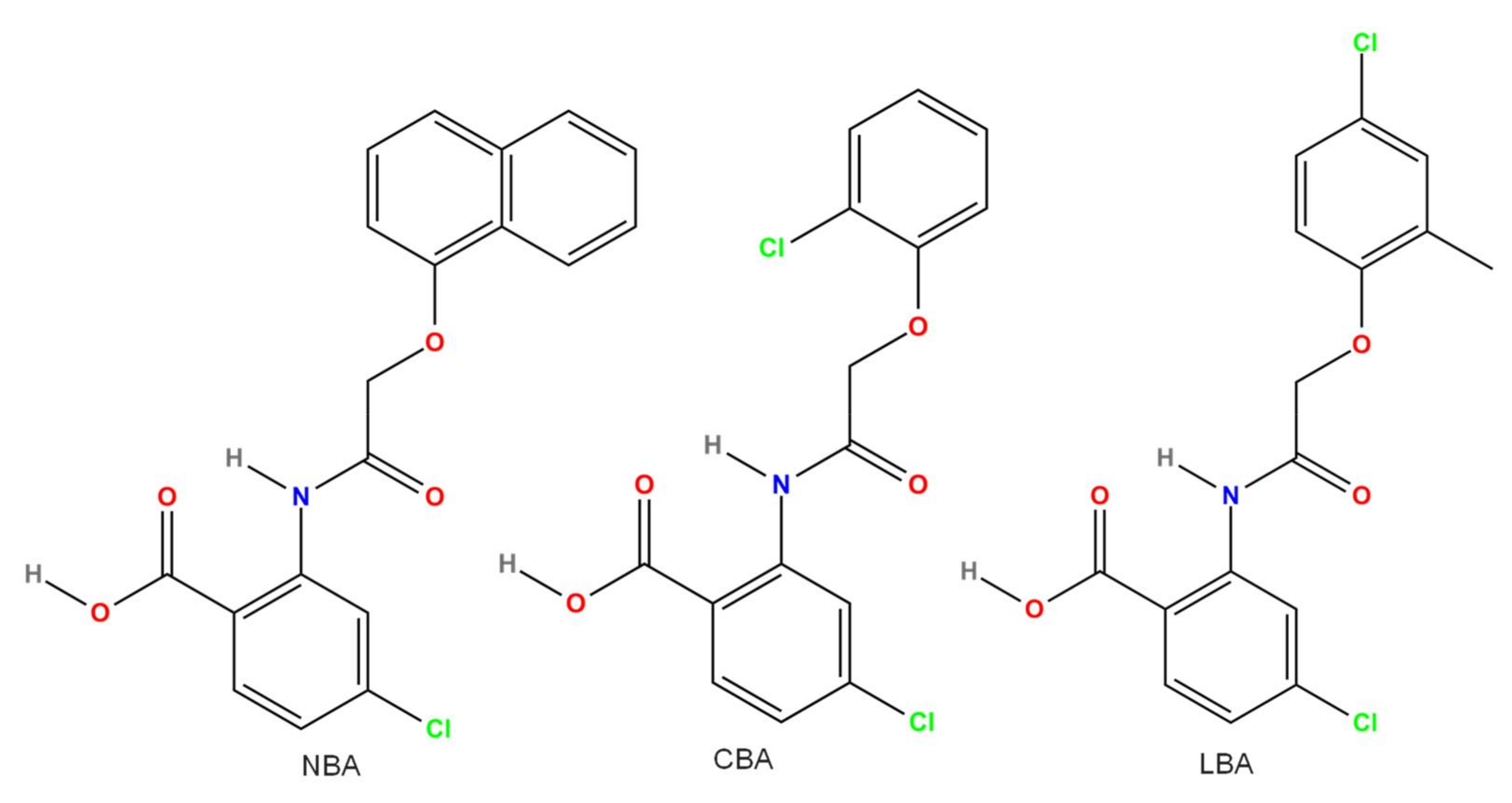
3.11. CBA and Other Related Compounds (LBA and NBA)
3.12. M4P, M4M, and M4M1 Anti-TRPM4 Antibodies
3.13. siRNA Approach
3.14. Dominant-Negative Splice Variants
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AP | action potential |
| ATP | adenosine triphosphate |
| BTP2 | 3,5-bis(trifluoromethyl)pyrazole derivative |
| CBA | 4-chloro-2-[[2-(2-chlorophenoxy)acetyl]amino]benzoic acid |
| CFTR | cystic fibrosis transmembrane conductance regulator |
| CHO | Chinese hamster ovary |
| DCDPC | 3′,5-dichlorodiphenylamine-2-carboxylic acid |
| DPC | diphenylamine-2-carboxylic acid |
| DV | decavanadate |
| EC50 | half effective activator concentration |
| FFA | flufenamic acid |
| HEK | human embryonic kidney |
| HUVEC | human umbilical vein endothelial cells |
| IC50 | half inhibitory concentration |
| IP3 | inositol 1,4,5-trisphosphate |
| I(NSCCa) | Ca2+-activated nonspecific cationic current |
| KATP | adenosine triphosphate-dependent K+ |
| KCax.x | Ca2+-activated K+ channels type x.x |
| Kirx.x | inward rectifier K+ channel type x.x |
| KD | knock-down |
| KO | knock-out |
| LBA | 4-chloro-2-(2-(4-chloro-2-methylphenoxy)propanamido) benzoic acid |
| LNCaP | lymph node carcinoma of the prostate |
| NBA | 4-chloro-2-(1-naphthyloxyacetamido)benzoic acid |
| NMDA | N-methyl-d-aspartic acid |
| NO | nitric oxide |
| NPPB | 5-nitro-2-(3-phenylpropylamino)benzoic acid |
| NSCCa | Ca2+-activated nonspecific cationic channel |
| PIP2 | phosphatidylinositol 4,5-bisphosphate |
| PKC | protein kinase C |
| PLC | phospholipase C |
| PMA | phorbol 12-myristate 13-acetate |
| tPA | tissue plasminogen activator |
| siRNA | small interfering RNA |
| SUR1 | sulfonylurea receptor 1 |
| TRP | transient receptor potential |
| TRPA | transient receptor potential ankyrin |
| TRPC | transient receptor potential canonical |
| TRPM | transient receptor potential melastatin |
| TRPML | transient receptor potential mucolipin |
| TRPP | transient receptor potential polycystin |
| TRPV | transient receptor potential vanilloid |
References
- Montell, C.; Rubin, G.M. Molecular characterization of the Drosophila trp locus: A putative integral membrane protein required for phototransduction. Neuron 1989, 2, 1313–1323. [Google Scholar] [CrossRef]
- Chang, Y.; Schlenstedt, G.; Flockerzi, V.; Beck, A. Properties of the intracellular transient receptor potential (TRP) channel in yeast, Yvc1. FEBS Lett. 2010, 584, 2028–2032. [Google Scholar] [CrossRef] [Green Version]
- Jimenez, I.; Prado, Y.; Marchant, F.; Otero, C.; Eltit, F.; Cabello-Verrugio, C.; Cerda, O.; Simon, F. TRPM Channels in Human Diseases. Cells 2020, 9, 2604. [Google Scholar] [CrossRef] [PubMed]
- Guinamard, R.; Chatelier, A.; Demion, M.; Potreau, D.; Patri, S.; Rahmati, M.; Bois, P. Functional characterization of a Ca2+-activated non-selective cation channel in human atrial cardiomyocytes. J. Physiol. 2004, 558, 75–83. [Google Scholar] [CrossRef]
- Guinamard, R.; Sallé, L.; Simard, C. The non-selective monovalent cationic channels TRPM4 and TRPM5. Adv. Exp. Med. Biol. 2011, 704, 147–171. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.Z.; Moebius, F.; Gill, D.L.; Montell, C. Regulation of melastatin, a TRP-related protein, through interaction with a cytoplasmic isoform. Proc. Natl. Acad. Sci. USA 2001, 98, 10692–10697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Launay, P.; Fleig, A.; Perraud, A.L.; Scharenberg, A.M.; Penner, R.; Kinet, J.P. TRPM4 is a Ca2+-activated nonselective cation channel mediating cell membrane depolarization. Cell 2002, 109, 397–407. [Google Scholar] [CrossRef] [Green Version]
- Ullrich, N.D.; Voets, T.; Prenen, J.; Vennekens, R.; Talavera, K.; Droogmans, G.; Nilius, B. Comparison of functional properties of the Ca2+-activated cation channels TRPM4 and TRPM5 from mice. Cell Calcium 2005, 37, 267–278. [Google Scholar] [CrossRef]
- Demion, M.; Bois, P.; Launay, P.; Guinamard, R. TRPM4, a Ca2+-activated nonselective cation channel in mouse sino-atrial node cells. Cardiovasc. Res. 2007, 73, 531–538. [Google Scholar] [CrossRef] [Green Version]
- Demion, M.; Thireau, J.; Gueffier, M.; Finan, A.; Khoueiry, Z.; Cassan, C.; Serafini, N.; Aimond, F.; Granier, M.; Pasquié, J.L.; et al. Trpm4 gene invalidation leads to cardiac hypertrophy and electrophysiological alterations. PLoS ONE 2014, 9, e115256. [Google Scholar] [CrossRef] [Green Version]
- Nilius, B.; Prenen, J.; Tang, J.; Wang, C.; Owsianik, G.; Janssens, A.; Voets, T.; Zhu, M.X. Regulation of the Ca2+ sensitivity of the nonselective cation channel TRPM4. J. Biol. Chem. 2005, 280, 6423–6433. [Google Scholar] [CrossRef] [Green Version]
- Bouchghoul, H.; Bouyer, J.; Senat, M.V.; Mandelbrot, L.; Letourneau, A.; Bourcigaux, N.; Becquemont, L.; Verstuyft, C. Hypoglycemia and glycemic control with glyburide in women with gestational diabetes and genetic variants of cytochrome P450 2C9 and/or OATP1B3. Clin. Pharmacol. Ther. 2020, 110, 141–148. [Google Scholar] [CrossRef]
- Medert, R.; Pironet, A.; Bacmeister, L.; Segin, S.; Londono, J.E.C.; Vennekens, R.; Freichel, M. Genetic background influences expression and function of the cation channel TRPM4 in the mouse heart. Basic Res. Cardiol. 2020, 115, 70. [Google Scholar] [CrossRef]
- Medert, R.; Jungmann, A.; Hildebrand, S.; Busch, M.; Grimm, D.; Flockerzi, V.; Müller, O.J.; Most, P.; Schumacher, D.; Freichel, M. Development of an AAV9-RNAi-mediated silencing strategy to abrogate TRPM4 expression in the adult heart. Pflugers Arch. 2021, 473, 533–546. [Google Scholar] [CrossRef]
- Nilius, B.; Mahieu, F.; Prenen, J.; Janssens, A.; Owsianik, G.; Vennekens, R.; Voets, T. The Ca2+-activated cation channel TRPM4 is regulated by phosphatidylinositol 4,5-biphosphate. EMBO J. 2006, 25, 467–478. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Okawa, H.; Wang, Y.; Liman, E.R. Phosphatidylinositol 4,5-bisphosphate rescues TRPM4 channels from desensitization. J. Biol. Chem. 2005, 280, 39185–39192. [Google Scholar] [CrossRef] [Green Version]
- Woo, S.K.; Kwon, M.S.; Ivanov, A.; Gerzanich, V.; Simard, J.M. The sulfonylurea receptor 1 (Sur1)-transient receptor potential melastatin 4 (Trpm4) channel. J. Biol. Chem. 2013, 288, 3655–3667. [Google Scholar] [CrossRef] [Green Version]
- Nilius, B.; Prenen, J.; Janssens, A.; Voets, T.; Droogmans, G. Decavanadate modulates gating of TRPM4 cation channels. J. Physiol. 2004, 560, 753–765. [Google Scholar] [CrossRef] [Green Version]
- Takezawa, R.; Cheng, H.; Beck, A.; Ishikawa, J.; Launay, P.; Kubota, H.; Kinet, J.P.; Fleig, A.; Yamada, T.; Penner, R. A pyrazole derivative potently inhibits lymphocyte Ca2+ influx and cytokine production by facilitating transient receptor potential melastatin 4 channel activity. Mol. Pharmacol. 2006, 69, 1413–1420. [Google Scholar] [CrossRef]
- Simon, F.; Leiva-Salcedo, E.; Armisén, R.; Riveros, A.; Cerda, O.; Varela, D.; Eguiguren, A.L.; Olivero, P.; Stutzin, A. Hydrogen peroxide removes TRPM4 current desensitization conferring increased vulnerability to necrotic cell death. J. Biol. Chem. 2010, 285, 37150–37158. [Google Scholar] [CrossRef] [Green Version]
- Gerzanich, V.; Kwon, M.S.; Woo, S.K.; Ivanov, A.; Simard, J.M. SUR1-TRPM4 channel activation and phasic secretion of MMP-9 induced by tPA in brain endothelial cells. PLoS ONE 2018, 13, e0195526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leitner, M.G.; Michel, N.; Behrendt, M.; Dierich, M.; Dembla, S.; Wilke, B.U.; Konrad, M.; Lindner, M.; Oberwinkler, J.; Oliver, D. Direct modulation of TRPM4 and TRPM3 channels by the phospholipase C inhibitor U73122. Br. J. Pharmacol. 2016, 173, 2555–2569. [Google Scholar] [CrossRef] [PubMed]
- Ngoc Tran, T.D.; Stovall, K.E.; Suantawee, T.; Hu, Y.; Yao, S.; Yang, L.J.; Adisakwattana, S.; Cheng, H. Transient receptor potential melastatin 4 channel is required for rat dental pulp stem cell proliferation and survival. Cell Prolif. 2017, 50. [Google Scholar] [CrossRef]
- Kappel, S.; Stokłosa, P.; Hauert, B.; Ross-Kaschitza, D.; Borgström, A.; Baur, R.; Galván, J.A.; Zlobec, I.; Peinelt, C. TRPM4 is highly expressed in human colorectal tumor buds and contributes to proliferation, cell cycle, and invasion of colorectal cancer cells. Mol. Oncol. 2019, 13, 2393–2405. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Beck, A.; Launay, P.; Gross, S.A.; Stokes, A.J.; Kinet, J.P.; Fleig, A.; Penner, R. TRPM4 controls insulin secretion in pancreatic beta-cells. Cell Calcium 2007, 41, 51–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzales, A.L.; Earley, S. Endogenous cytosolic Ca2+ buffering is necessary for TRPM4 activity in cerebral artery smooth muscle cells. Cell Calcium 2012, 51, 82–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guinamard, R.; Demion, M.; Magaud, C.; Potreau, D.; Bois, P. Functional expression of the TRPM4 cationic current in ventricular cardiomyocytes from spontaneously hypertensive rats. Hypertension 2006, 48, 587–594. [Google Scholar] [CrossRef] [Green Version]
- Yarishkin, O.V.; Hwang, E.M.; Park, J.Y.; Kang, D.; Han, J.; Hong, S.G. Endogenous TRPM4-like channel in Chinese hamster ovary (CHO) cells. Biochem. Biophys. Res. Commun. 2008, 369, 712–717. [Google Scholar] [CrossRef]
- Autzen, H.E.; Myasnikov, A.G.; Campbell, M.G.; Asarnow, D.; Julius, D.; Cheng, Y. Structure of the human TRPM4 ion channel in a lipid nanodisc. Science 2018, 359, 228–232. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Fliegert, R.; Guse, A.H.; Lü, W.; Du, J. A structural overview of the ion channels of the TRPM family. Cell Calcium 2020, 85, 102111. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Tanimoto, A.; Iwasa, S.; Otsuguro, K. TRPM4 and TRPM5 Channels Share Crucial Amino Acid Residues for Ca. Int. J. Mol. Sci. 2019, 20, 2012. [Google Scholar] [CrossRef] [Green Version]
- Constantine, M.; Liew, C.K.; Lo, V.; Macmillan, A.; Cranfield, C.G.; Sunde, M.; Whan, R.; Graham, R.M.; Martinac, B. Heterologously-expressed and Liposome-reconstituted Human Transient Receptor Potential Melastatin 4 Channel (TRPM4) is a Functional Tetramer. Sci. Rep. 2016, 6, 19352. [Google Scholar] [CrossRef] [Green Version]
- Rivas, J.; Díaz, N.; Silva, I.; Morales, D.; Lavanderos, B.; Álvarez, A.; Saldías, M.P.; Pulgar, E.; Cruz, P.; Maureira, D.; et al. KCTD5, a novel TRPM4-regulatory protein required for cell migration as a new predictor for breast cancer prognosis. FASEB J. 2020, 34, 7847–7865. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Brayden, J.E. Rho kinase activity governs arteriolar myogenic depolarization. J. Cereb. Blood Flow Metab. 2017, 37, 140–152. [Google Scholar] [CrossRef]
- Flannery, R.J.; Kleene, N.K.; Kleene, S.J. A TRPM4-dependent current in murine renal primary cilia. Am. J. Physiol. Renal Physiol. 2015, 309, F697–F707. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.X.; Zhang, B.L.; Yu, M.; Yang, Y.C.; Ao, X.; Zhu, D.; Wang, Q.S.; Lou, J.; Liang, C.; Tang, L.L.; et al. Cholesterol Stimulates the Transient Receptor Potential Melastatin 4 Channel in mpkCCD c14 Cells. Front. Pharmacol. 2021, 12, 627875. [Google Scholar] [CrossRef]
- Bousova, K.; Jirku, M.; Bumba, L.; Bednarova, L.; Sulc, M.; Franek, M.; Vyklicky, L.; Vondrasek, J.; Teisinger, J. PIP2 and PIP3 interact with N-terminus region of TRPM4 channel. Biophys. Chem. 2015, 205, 24–32. [Google Scholar] [CrossRef]
- Bousova, K.; Barvik, I.; Herman, P.; Hofbauerová, K.; Monincova, L.; Majer, P.; Zouharova, M.; Vetyskova, V.; Postulkova, K.; Vondrasek, J. Mapping of CaM, S100A1 and PIP2-Binding Epitopes in the Intracellular N- and C-Termini of TRPM4. Int. J. Mol. Sci. 2020, 21, 4323. [Google Scholar] [CrossRef]
- Chuang, H.H.; Prescott, E.D.; Kong, H.; Shields, S.; Jordt, S.E.; Basbaum, A.I.; Chao, M.V.; Julius, D. Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature 2001, 411, 957–962. [Google Scholar] [CrossRef]
- Liu, D.; Liman, E.R. Intracellular Ca2+ and the phospholipid PIP2 regulate the taste transduction ion channel TRPM5. Proc. Natl. Acad. Sci. USA 2003, 100, 15160–15165. [Google Scholar] [CrossRef] [Green Version]
- Runnels, L.W.; Yue, L.; Clapham, D.E. The TRPM7 channel is inactivated by PIP(2) hydrolysis. Nat. Cell Biol. 2002, 4, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Qin, F. Functional control of cold- and menthol-sensitive TRPM8 ion channels by phosphatidylinositol 4,5-bisphosphate. J. Neurosci. 2005, 25, 1674–1681. [Google Scholar] [CrossRef] [PubMed]
- Rohács, T.; Lopes, C.M.; Michailidis, I.; Logothetis, D.E. PI(4,5)P2 regulates the activation and desensitization of TRPM8 channels through the TRP domain. Nat. Neurosci. 2005, 8, 626–634. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Cha, S.K.; Sun, T.J.; Huang, C.L. PIP2 activates TRPV5 and releases its inhibition by intracellular Mg2+. J. Gen. Physiol. 2005, 126, 439–451. [Google Scholar] [CrossRef] [Green Version]
- Lopes, C.M.; Rohács, T.; Czirják, G.; Balla, T.; Enyedi, P.; Logothetis, D.E. PIP2 hydrolysis underlies agonist-induced inhibition and regulates voltage gating of two-pore domain K+ channels. J. Physiol. 2005, 564, 117–129. [Google Scholar] [CrossRef]
- Wu, L.; Bauer, C.S.; Zhen, X.G.; Xie, C.; Yang, J. Dual regulation of voltage-gated calcium channels by PtdIns(4,5)P2. Nature 2002, 419, 947–952. [Google Scholar] [CrossRef]
- Gamper, N.; Reznikov, V.; Yamada, Y.; Yang, J.; Shapiro, M.S. Phosphatidylinositol [correction] 4,5-bisphosphate signals underlie receptor-specific Gq/11-mediated modulation of N-type Ca2+ channels. J. Neurosci. 2004, 24, 10980–10992. [Google Scholar] [CrossRef] [Green Version]
- Hille, B.; Dickson, E.J.; Kruse, M.; Vivas, O.; Suh, B.C. Phosphoinositides regulate ion channels. Biochim. Biophys. Acta 2015, 1851, 844–856. [Google Scholar] [CrossRef]
- Winkler, P.A.; Huang, Y.; Sun, W.; Du, J.; Lü, W. Electron cryo-microscopy structure of a human TRPM4 channel. Nature 2017, 552, 200–204. [Google Scholar] [CrossRef]
- Michel, A.D.; Xing, M.; Thompson, K.M.; Jones, C.A.; Humphrey, P.P. Decavanadate, a P2X receptor antagonist, and its use to study ligand interactions with P2X7 receptors. Eur. J. Pharmacol. 2006, 534, 19–29. [Google Scholar] [CrossRef]
- Föhr, K.J.; Wahl, Y.; Engling, R.; Kemmer, T.P.; Gratzl, M. Decavanadate displaces inositol 1,4,5-trisphosphate (IP3) from its receptor and inhibits IP3 induced Ca2+ release in permeabilized pancreatic acinar cells. Cell Calcium 1991, 12, 735–742. [Google Scholar] [CrossRef] [Green Version]
- Aureliano, M. Vanadate oligomer inhibition of passive and active Ca2+ translocation by the Ca2+ pump of sarcoplasmic reticulum. J. Inorg. Biochem. 2000, 80, 145–147. [Google Scholar] [CrossRef]
- Zhang, X.; Xin, P.; Yoast, R.E.; Emrich, S.M.; Johnson, M.T.; Pathak, T.; Benson, J.C.; Azimi, I.; Gill, D.L.; Monteith, G.R.; et al. Distinct pharmacological profiles of ORAI1, ORAI2, and ORAI3 channels. Cell Calcium 2020, 91, 102281. [Google Scholar] [CrossRef]
- He, L.P.; Hewavitharana, T.; Soboloff, J.; Spassova, M.A.; Gill, D.L. A functional link between store-operated and TRPC channels revealed by the 3,5-bis(trifluoromethyl)pyrazole derivative, BTP2. J. Biol. Chem. 2005, 280, 10997–11006. [Google Scholar] [CrossRef] [Green Version]
- Piao, H.; Takahashi, K.; Yamaguchi, Y.; Wang, C.; Liu, K.; Naruse, K. Transient receptor potential melastatin-4 is involved in hypoxia-reoxygenation injury in the cardiomyocytes. PLoS ONE 2015, 10, e0121703. [Google Scholar] [CrossRef]
- Sarmiento, D.; Montorfano, I.; Cerda, O.; Cáceres, M.; Becerra, A.; Cabello-Verrugio, C.; Elorza, A.A.; Riedel, C.; Tapia, P.; Velásquez, L.A.; et al. Increases in reactive oxygen species enhance vascular endothelial cell migration through a mechanism dependent on the transient receptor potential melastatin 4 ion channel. Microvasc. Res. 2015, 98, 187–196. [Google Scholar] [CrossRef]
- Ding, X.Q.; Ban, T.; Liu, Z.Y.; Lou, J.; Tang, L.L.; Wang, J.X.; Chu, W.F.; Zhao, D.; Song, B.L.; Zhang, Z.R. Transient Receptor Potential Melastatin 4 (TRPM4) Contributes to High Salt Diet-Mediated Early-Stage Endothelial Injury. Cell Physiol. Biochem. 2017, 41, 835–848. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.M.; Zhai, Y.J.; Li, Y.X.; Hu, Q.Q.; Wang, Z.R.; Wei, S.P.; Zou, L.; Alli, A.A.; Thai, T.L.; Zhang, Z.R.; et al. Hydrogen peroxide suppresses TRPM4 trafficking to the apical membrane in mouse cortical collecting duct principal cells. Am. J. Physiol. Renal Physiol. 2016, 311, F1360–F1368. [Google Scholar] [CrossRef]
- Keckeis, S.; Wernecke, L.; Salchow, D.J.; Reichhart, N.; Strauß, O. Activation of a Ca. Exp. Eye Res. 2017, 161, 61–70. [Google Scholar] [CrossRef]
- Sakaguchi, R.; Mori, Y. Transient receptor potential (TRP) channels: Biosensors for redox environmental stimuli and cellular status. Free Radic. Biol. Med. 2020, 146, 36–44. [Google Scholar] [CrossRef]
- Das, L.; Azmoon, P.; Banki, M.A.; Mantuano, E.; Gonias, S.L. Tissue-type plasminogen activator selectively inhibits multiple toll-like receptors in CSF-1-differentiated macrophages. PLoS ONE 2019, 14, e0224738. [Google Scholar] [CrossRef] [PubMed]
- Gonias, S.L.; Banki, M.A.; Gilder, A.S.; Azmoon, P.; Campana, W.M.; Mantuano, E. PAI1 blocks NMDA receptor-mediated effects of tissue-type plasminogen activator on cell signaling and physiology. J. Cell Sci. 2018, 131, jcs217083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.; Youm, J.B.; Ryu, S.Y.; Earm, Y.E.; Ho, W.K. Inhibition of acetylcholine-activated K+ currents by U73122 is mediated by the inhibition of PIP(2)-channel interaction. Br. J. Pharmacol. 2001, 134, 1066–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klose, A.; Huth, T.; Alzheimer, C. 1-[6-[[(17beta)-3-methoxyestra-1,3,5(10)-trien-17-yl]amino]hexyl]-1H-pyrrole-2,5-dione (U73122) selectively inhibits Kir3 and BK channels in a phospholipase C-independent fashion. Mol. Pharmacol. 2008, 74, 1203–1214. [Google Scholar] [CrossRef] [Green Version]
- Sickmann, T.; Klose, A.; Huth, T.; Alzheimer, C. Unexpected suppression of neuronal G protein-activated, inwardly rectifying K+ current by common phospholipase C inhibitor. Neurosci. Lett. 2008, 436, 102–106. [Google Scholar] [CrossRef]
- Mogami, H.; Lloyd Mills, C.; Gallacher, D.V. Phospholipase C inhibitor, U73122, releases intracellular Ca2+, potentiates Ins(1,4,5)P3-mediated Ca2+ release and directly activates ion channels in mouse pancreatic acinar cells. Biochem. J. 1997, 324 Pt 2, 645–651. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Feng, L.; Yao, H.; Yang, L.; Qin, Y. Efficacy and safety of diazoxide for treating hyperinsulinemic hypoglycemia: A systematic review and meta-analysis. PLoS ONE 2021, 16, e0246463. [Google Scholar] [CrossRef]
- Pipatpolkai, T.; Usher, S.; Stansfeld, P.J.; Ashcroft, F.M. New insights into K. Nat. Rev. Endocrinol. 2020, 16, 378–393. [Google Scholar] [CrossRef]
- Hansen, J.B. Towards selective Kir6.2/SUR1 potassium channel openers, medicinal chemistry and therapeutic perspectives. Curr. Med. Chem. 2006, 13, 361–376. [Google Scholar] [CrossRef]
- Guinamard, R.; Rahmati, M.; Lenfant, J.; Bois, P. Characterization of a Ca2+-activated nonselective cation channel during dedifferentiation of cultured rat ventricular cardiomyocytes. J. Membr. Biol. 2002, 188, 127–135. [Google Scholar] [CrossRef]
- Nilius, B.; Prenen, J.; Voets, T.; Droogmans, G. Intracellular nucleotides and polyamines inhibit the Ca2+-activated cation channel TRPM4b. Pflugers Arch. 2004, 448, 70–75. [Google Scholar] [CrossRef]
- Suh, S.H.; Watanabe, H.; Droogmans, G.; Nilius, B. ATP and nitric oxide modulate a Ca2+-activated non-selective cation current in macrovascular endothelial cells. Pflugers Arch. 2002, 444, 438–445. [Google Scholar] [CrossRef]
- Talavera, K.; Yasumatsu, K.; Yoshida, R.; Margolskee, R.F.; Voets, T.; Ninomiya, Y.; Nilius, B. The taste transduction channel TRPM5 is a locus for bitter-sweet taste interactions. FASEB J. 2008, 22, 1343–1355. [Google Scholar] [CrossRef]
- Grand, T.; Demion, M.; Norez, C.; Mettey, Y.; Launay, P.; Becq, F.; Bois, P.; Guinamard, R. 9-phenanthrol inhibits human TRPM4 but not TRPM5 cationic channels. Br. J. Pharmacol. 2008, 153, 1697–1705. [Google Scholar] [CrossRef] [Green Version]
- Guinamard, R.; Simard, C.; Del Negro, C. Flufenamic acid as an ion channel modulator. Pharmacol. Ther. 2013, 138, 272–284. [Google Scholar] [CrossRef] [Green Version]
- Vennekens, R.; Nilius, B. Insights into TRPM4 function, regulation and physiological role. Handb. Exp. Pharmacol. 2007, 269–285. [Google Scholar] [CrossRef]
- Guinamard, R.; Hof, T.; Del Negro, C.A. The TRPM4 channel inhibitor 9-phenanthrol. Br. J. Pharmacol. 2014, 171, 1600–1613. [Google Scholar] [CrossRef] [Green Version]
- Ozhathil, L.C.; Delalande, C.; Bianchi, B.; Nemeth, G.; Kappel, S.; Thomet, U.; Ross-Kaschitza, D.; Simonin, C.; Rubin, M.; Gertsch, J.; et al. Identification of potent and selective small molecule inhibitors of the cation channel TRPM4. Br. J. Pharmacol. 2018, 175, 2504–2519. [Google Scholar] [CrossRef] [Green Version]
- Delalande, C.; Awale, M.; Rubin, M.; Probst, D.; Ozhathil, L.C.; Gertsch, J.; Abriel, H.; Reymond, J.L. Optimizing TRPM4 inhibitors in the MHFP6 chemical space. Eur. J. Med. Chem. 2019, 166, 167–177. [Google Scholar] [CrossRef]
- Chen, B.; Gao, Y.; Wei, S.; Low, S.W.; Ng, G.; Yu, D.; Tu, T.M.; Soong, T.W.; Nilius, B.; Liao, P. TRPM4-specific blocking antibody attenuates reperfusion injury in a rat model of stroke. Pflugers Arch. 2019, 471, 1455–1466. [Google Scholar] [CrossRef] [Green Version]
- Low, S.W.; Gao, Y.; Wei, S.; Chen, B.; Nilius, B.; Liao, P. Development and characterization of a monoclonal antibody blocking human TRPM4 channel. Sci. Rep. 2021, 11, 10411. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Ng, G.; Gao, Y.; Low, S.W.; Sandanaraj, E.; Ramasamy, B.; Sekar, S.; Bhakoo, K.; Soong, T.W.; Nilius, B.; et al. Non-Invasive Multimodality Imaging Directly Shows TRPM4 Inhibition Ameliorates Stroke Reperfusion Injury. Transl. Stroke Res. 2019, 10, 91–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balla, T. Pharmacology of phosphoinositides, regulators of multiple cellular functions. Curr. Pharm. Des. 2001, 7, 475–507. [Google Scholar] [CrossRef] [PubMed]
- Nichols, C.G. KATP channels as molecular sensors of cellular metabolism. Nature 2006, 440, 470–476. [Google Scholar] [CrossRef]
- Liman, E.R. Regulation by voltage and adenine nucleotides of a Ca2+-activated cation channel from hamster vomeronasal sensory neurons. J. Physiol. 2003, 548, 777–787. [Google Scholar] [CrossRef]
- Guo, J.; She, J.; Zeng, W.; Chen, Q.; Bai, X.C.; Jiang, Y. Structures of the calcium-activated, non-selective cation channel TRPM4. Nature 2017, 552, 205–209. [Google Scholar] [CrossRef]
- Chen, M.; Simard, J.M. Cell swelling and a nonselective cation channel regulated by internal Ca2+ and ATP in native reactive astrocytes from adult rat brain. J. Neurosci. 2001, 21, 6512–6521. [Google Scholar] [CrossRef] [Green Version]
- Ali, S.; Solano, A.S.; Gonzales, A.L.; Thakore, P.; Krishnan, V.; Yamasaki, E.; Earley, S. Nitric Oxide Signals Through IRAG to Inhibit TRPM4 Channels and Dilate Cerebral Arteries. Function 2021, 2, zqab051. [Google Scholar] [CrossRef]
- Krippeit-Drews, P.; Kröncke, K.D.; Welker, S.; Zempel, G.; Roenfeldt, M.; Ammon, H.P.; Lang, F.; Drews, G. The effects of nitric oxide on the membrane potential and ionic currents of mouse pancreatic B cells. Endocrinology 1995, 136, 5363–5369. [Google Scholar] [CrossRef]
- Miyoshi, H.; Nakaya, Y.; Moritoki, H. Nonendothelial-derived nitric oxide activates the ATP-sensitive K+ channel of vascular smooth muscle cells. FEBS Lett. 1994, 345, 47–49. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.B.; Tenneti, L.; Le, D.A.; Ortiz, J.; Bai, G.; Chen, H.S.; Lipton, S.A. Molecular basis of NMDA receptor-coupled ion channel modulation by S-nitrosylation. Nat. Neurosci. 2000, 3, 15–21. [Google Scholar] [CrossRef]
- Matsumoto, S.; Takahashi, T.; Ikeda, M.; Nishikawa, T.; Yoshida, S.; Kawase, T. Effects of N(G)-monomethyl-l-arginine on Ca2+ current and nitric-oxide synthase in rat ventricular myocytes. J. Pharmacol. Exp. Ther. 2000, 294, 216–223. [Google Scholar]
- Koh, S.D.; Campbell, J.D.; Carl, A.; Sanders, K.M. Nitric oxide activates multiple potassium channels in canine colonic smooth muscle. J. Physiol. 1995, 489 Pt 3, 735–743. [Google Scholar] [CrossRef]
- Pegg, A.E. Functions of Polyamines in Mammals. J. Biol. Chem. 2016, 291, 14904–14912. [Google Scholar] [CrossRef] [Green Version]
- Lopatin, A.N.; Nichols, C.G. K+ dependence of polyamine-induced rectification in inward rectifier potassium channels (IRK1, Kir2.1). J. Gen. Physiol. 1996, 108, 105–113. [Google Scholar] [CrossRef] [Green Version]
- Mott, D.D.; Washburn, M.S.; Zhang, S.; Dingledine, R.J. Subunit-dependent modulation of kainate receptors by extracellular protons and polyamines. J. Neurosci. 2003, 23, 1179–1188. [Google Scholar] [CrossRef] [Green Version]
- Washburn, M.S.; Dingledine, R. Block of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors by polyamines and polyamine toxins. J. Pharmacol. Exp. Ther. 1996, 278, 669–678. [Google Scholar]
- Kerschbaum, H.H.; Kozak, J.A.; Cahalan, M.D. Polyvalent cations as permeant probes of MIC and TRPM7 pores. Biophys. J. 2003, 84, 2293–2305. [Google Scholar] [CrossRef] [Green Version]
- Chubanov, V.; Mederos y Schnitzler, M.; Meißner, M.; Schäfer, S.; Abstiens, K.; Hofmann, T.; Gudermann, T. Natural and synthetic modulators of SK (K(ca)2) potassium channels inhibit magnesium-dependent activity of the kinase-coupled cation channel TRPM7. Br. J. Pharmacol. 2012, 166, 1357–1376. [Google Scholar] [CrossRef] [Green Version]
- Marivingt-Mounir, C.; Norez, C.; Dérand, R.; Bulteau-Pignoux, L.; Nguyen-Huy, D.; Viossat, B.; Morgant, G.; Becq, F.; Vierfond, J.M.; Mettey, Y. Synthesis, SAR, crystal structure, and biological evaluation of benzoquinoliziniums as activators of wild-type and mutant cystic fibrosis transmembrane conductance regulator channels. J. Med. Chem. 2004, 47, 962–972. [Google Scholar] [CrossRef]
- Norez, C.; Bilan, F.; Kitzis, A.; Mettey, Y.; Becq, F. Proteasome-dependent pharmacological rescue of cystic fibrosis transmembrane conductance regulator revealed by mutation of glycine 622. J. Pharmacol. Exp. Ther. 2008, 325, 89–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodward, R.M.; Polenzani, L.; Miledi, R. Effects of fenamates and other nonsteroidal anti-inflammatory drugs on rat brain GABAA receptors expressed in Xenopus oocytes. J. Pharmacol. Exp. Ther. 1994, 268, 806–817. [Google Scholar] [PubMed]
- Liantonio, A.; Giannuzzi, V.; Picollo, A.; Babini, E.; Pusch, M.; Conte Camerino, D. Niflumic acid inhibits chloride conductance of rat skeletal muscle by directly inhibiting the CLC-1 channel and by increasing intracellular calcium. Br. J. Pharmacol. 2007, 150, 235–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amarouch, M.Y.; Syam, N.; Abriel, H. Biochemical, single-channel, whole-cell patch clamp, and pharmacological analyses of endogenous TRPM4 channels in HEK293 cells. Neurosci. Lett. 2013, 541, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Burt, R.; Graves, B.M.; Gao, M.; Li, C.; Williams, D.L.; Fregoso, S.P.; Hoover, D.B.; Li, Y.; Wright, G.L.; Wondergem, R. 9-Phenanthrol and flufenamic acid inhibit calcium oscillations in HL-1 mouse cardiomyocytes. Cell Calcium 2013, 54, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Buntschu, S.; Tscherter, A.; Heidemann, M.; Streit, J. Critical Components for Spontaneous Activity and Rhythm Generation in Spinal Cord Circuits in Culture. Front. Cell Neurosci. 2020, 14, 81. [Google Scholar] [CrossRef]
- Yao, Y.; Xu, J.; Yu, T.; Chen, Z.; Xiao, Z.; Wang, J.; Hu, Y.; Wu, Y.; Zhu, D. Flufenamic acid inhibits secondary hemorrhage and BSCB disruption after spinal cord injury. Theranostics 2018, 8, 4181–4198. [Google Scholar] [CrossRef]
- Malysz, J.; Maxwell, S.E.; Yarotskyy, V.; Petkov, G.V. TRPM4 channel inhibitors 9-phenanthrol and glibenclamide differentially decrease guinea pig detrusor smooth muscle whole-cell cation currents and phasic contractions. Am. J. Physiol. Cell Physiol. 2020, 318, C406–C421. [Google Scholar] [CrossRef]
- Hernandez-Benito, M.J.; Macianskiene, R.; Sipido, K.R.; Flameng, W.; Mubagwa, K. Suppression of transient outward potassium currents in mouse ventricular myocytes by imidazole antimycotics and by glybenclamide. J. Pharmacol. Exp. Ther. 2001, 298, 598–606. [Google Scholar]
- Melin, P.; Hosy, E.; Vivaudou, M.; Becq, F. CFTR inhibition by glibenclamide requires a positive charge in cytoplasmic loop three. Biochim. Biophys. Acta 2007, 1768, 2438–2446. [Google Scholar] [CrossRef]
- Schaffer, P.; Pelzmann, B.; Bernhart, E.; Lang, P.; Mächler, H.; Rigler, B.; Koidl, B. The sulphonylurea glibenclamide inhibits voltage dependent potassium currents in human atrial and ventricular myocytes. Br. J. Pharmacol. 1999, 128, 1175–1180. [Google Scholar] [CrossRef]
- Jiang, B.; Zhang, Y.; Wang, Y.; Li, Z.; Chen, Q.; Tang, J.; Zhu, G. Glibenclamide Attenuates Neuroinflammation and Promotes Neurological Recovery After Intracerebral Hemorrhage in Aged Rats. Front. Aging Neurosci. 2021, 13, 729652. [Google Scholar] [CrossRef]
- Takahashi, K.; Sakamoto, K.; Kimura, J. Hypoxic stress induces transient receptor potential melastatin 2 (TRPM2) channel expression in adult rat cardiac fibroblasts. J. Pharmacol. Sci. 2012, 118, 186–197. [Google Scholar] [CrossRef] [Green Version]
- Hill, K.; McNulty, S.; Randall, A.D. Inhibition of TRPM2 channels by the antifungal agents clotrimazole and econazole. Naunyn Schmiedebergs Arch. Pharmacol. 2004, 370, 227–237. [Google Scholar] [CrossRef]
- Jensen, B.S.; Strobaek, D.; Christophersen, P.; Jorgensen, T.D.; Hansen, C.; Silahtaroglu, A.; Olesen, S.P.; Ahring, P.K. Characterization of the cloned human intermediate-conductance Ca2+-activated K+ channel. Am. J. Physiol. 1998, 275, C848–C856. [Google Scholar] [CrossRef]
- Thomas, G.P.; Karmazyn, M.; Zygmunt, A.C.; Antzelevitch, C.; Narayanan, N. The antifungal antibiotic clotrimazole potently inhibits L-type calcium current in guinea-pig ventricular myocytes. Br. J. Pharmacol. 1999, 126, 1531–1533. [Google Scholar] [CrossRef] [Green Version]
- Fearon, I.M.; Ball, S.G.; Peers, C. Clotrimazole inhibits the recombinant human cardiac L-type Ca2+ channel alpha 1C subunit. Br. J. Pharmacol. 2000, 129, 547–554. [Google Scholar] [CrossRef] [Green Version]
- Gögelein, H.; Pfannmüller, B. The nonselective cation channel in the basolateral membrane of rat exocrine pancreas. Inhibition by 3′,5-dichlorodiphenylamine-2-carboxylic acid (DCDPC) and activation by stilbene disulfonates. Pflugers Arch. 1989, 413, 287–298. [Google Scholar] [CrossRef]
- Van den Abbeele, T.; Tran Ba Huy, P.; Teulon, J. A calcium-activated nonselective cationic channel in the basolateral membrane of outer hair cells of the guinea-pig cochlea. Pflugers Arch. 1994, 427, 56–63. [Google Scholar] [CrossRef]
- Chraïbi, A.; Van den Abbeele, T.; Guinamard, R.; Teulon, J. A ubiquitous non-selective cation channel in the mouse renal tubule with variable sensitivity to calcium. Pflugers Arch. 1994, 429, 90–97. [Google Scholar] [CrossRef]
- Tran, T.D.; Zolochevska, O.; Figueiredo, M.L.; Wang, H.; Yang, L.J.; Gimble, J.M.; Yao, S.; Cheng, H. Histamine-induced Ca2+ signalling is mediated by TRPM4 channels in human adipose-derived stem cells. Biochem. J. 2014, 463, 123–134. [Google Scholar] [CrossRef]
- Simard, C.; Sallé, L.; Rouet, R.; Guinamard, R. Transient receptor potential melastatin 4 inhibitor 9-phenanthrol abolishes arrhythmias induced by hypoxia and re-oxygenation in mouse ventricle. Br. J. Pharmacol. 2012, 165, 2354–2364. [Google Scholar] [CrossRef] [Green Version]
- Simard, C.; Hof, T.; Keddache, Z.; Launay, P.; Guinamard, R. The TRPM4 non-selective cation channel contributes to the mammalian atrial action potential. J. Mol. Cell Cardiol. 2013, 59, 11–19. [Google Scholar] [CrossRef]
- Gonzales, A.L.; Garcia, Z.I.; Amberg, G.C.; Earley, S. Pharmacological inhibition of TRPM4 hyperpolarizes vascular smooth muscle. Am. J. Physiol. Cell Physiol. 2010, 299, C1195–C1202. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.H.; Ternai, B.; Polya, G.M. Specific inhibition of cyclic AMP-dependent protein kinase by the antimalarial halofantrine and by related phenanthrenes. Biol. Chem. Hoppe Seyler 1994, 375, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Veress, R.; Baranyai, D.; Hegyi, B.; Kistamás, K.; Dienes, C.; Magyar, J.; Bányász, T.; Nánási, P.P.; Szentandrássy, N.; Horváth, B. Transient receptor potential melastatin 4 channel inhibitor 9-phenanthrol inhibits K. Can. J. Physiol. Pharmacol. 2018, 96, 1022–1029. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.W.; Fei, Y.D.; Li, W.; Chen, Y.H.; Wang, Q.; Xiao, Y.; Wang, Y.P.; Li, Y.G. The transient receptor potential melastatin 4 channel inhibitor 9-phenanthrol modulates cardiac sodium channel. Br. J. Pharmacol. 2018, 175, 4325–4337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garland, C.J.; Smirnov, S.V.; Bagher, P.; Lim, C.S.; Huang, C.Y.; Mitchell, R.; Stanley, C.; Pinkney, A.; Dora, K.A. TRPM4 inhibitor 9-phenanthrol activates endothelial cell intermediate conductance calcium-activated potassium channels in rat isolated mesenteric artery. Br. J. Pharmacol. 2015, 172, 1114–1123. [Google Scholar] [CrossRef] [Green Version]
- Burris, S.K.; Wang, Q.; Bulley, S.; Neeb, Z.P.; Jaggar, J.H. 9-Phenanthrol inhibits recombinant and arterial myocyte TMEM16A channels. Br. J. Pharmacol. 2015, 172, 2459–2468. [Google Scholar] [CrossRef] [Green Version]
- Arullampalam, P.; Preti, B.; Ross-Kaschitza, D.; Lochner, M.; Rougier, J.S.; Abriel, H. Species-Specific Effects of Cation Channel TRPM4 Small-Molecule Inhibitors. Front. Pharmacol. 2021, 12, 712354. [Google Scholar] [CrossRef]
- Dienes, C.; Hézső, T.; Kiss, D.Z.; Baranyai, D.; Kovács, Z.M.; Szabó, L.; Magyar, J.; Bányász, T.; Nánási, P.P.; Horváth, B.; et al. Electrophysiological Effects of the Transient Receptor Potential Melastatin 4 Channel Inhibitor (4-Chloro-2-(2-chlorophenoxy)acetamido) Benzoic Acid (CBA) in Canine Left Ventricular Cardiomyocytes. Int. J. Mol. Sci 2021, 22, 9499. [Google Scholar] [CrossRef]
- Borgström, A.; Hauert, B.; Kappel, S.; Zoni, E.; Kiener, M.; Stokłosa, P.; Baur, R.; Spahn, M.; Kruithof-de Julio, M.; Peinelt, C. Small Molecular Inhibitors Block TRPM4 Currents in Prostate Cancer Cells, with Limited Impact on Cancer Hallmark Functions. J. Mol. Biol. 2020, 433, 166665. [Google Scholar] [CrossRef]
- Stokłosa, P.; Borgström, A.; Hauert, B.; Baur, R.; Peinelt, C. Investigation of Novel Small Molecular TRPM4 Inhibitors in Colorectal Cancer Cells. Cancers 2021, 13, 5400. [Google Scholar] [CrossRef]
- Bianchi, B.; Smith, P.A.; Abriel, H. The ion channel TRPM4 in murine experimental autoimmune encephalomyelitis and in a model of glutamate-induced neuronal degeneration. Mol. Brain 2018, 11, 41. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Abbott, S.B.G.; Shi, Y.; Eggan, P.; Gonye, E.C.; Bayliss, D.A. TRPM4 mediates a subthreshold membrane potential oscillation in respiratory chemoreceptor neurons that drives pacemaker firing and breathing. Cell Rep. 2021, 34, 108714. [Google Scholar] [CrossRef]
- Wei, S.; Low, S.W.; Poore, C.P.; Chen, B.; Gao, Y.; Nilius, B.; Liao, P. Comparison of Anti-oncotic Effect of TRPM4 Blocking Antibody in Neuron, Astrocyte and Vascular Endothelial Cell Under Hypoxia. Front. Cell Dev. Biol. 2020, 8, 562584. [Google Scholar] [CrossRef]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef]
- Gönczi, M.; Szentandrássy, N.; Johnson, I.T.; Heagerty, A.M.; Weston, A.H. Investigation of the role of TASK-2 channels in rat pulmonary arteries; pharmacological and functional studies following RNA interference procedures. Br. J. Pharmacol. 2006, 147, 496–505. [Google Scholar] [CrossRef] [Green Version]
- Gonzales, A.L.; Amberg, G.C.; Earley, S. Ca2+ release from the sarcoplasmic reticulum is required for sustained TRPM4 activity in cerebral artery smooth muscle cells. Am. J. Physiol. Cell Physiol. 2010, 299, C279–C288. [Google Scholar] [CrossRef] [Green Version]
- Becerra, A.; Echeverría, C.; Varela, D.; Sarmiento, D.; Armisén, R.; Nuñez-Villena, F.; Montecinos, M.; Simon, F. Transient receptor potential melastatin 4 inhibition prevents lipopolysaccharide-induced endothelial cell death. Cardiovasc. Res. 2011, 91, 677–684. [Google Scholar] [CrossRef] [Green Version]
- Loh, K.P.; Ng, G.; Yu, C.Y.; Fhu, C.K.; Yu, D.; Vennekens, R.; Nilius, B.; Soong, T.W.; Liao, P. TRPM4 inhibition promotes angiogenesis after ischemic stroke. Pflugers Arch. 2014, 466, 563–576. [Google Scholar] [CrossRef] [PubMed]
- Hazalin, N.A.M.N.; Liao, P.; Hassan, Z. TRPM4 inhibition improves spatial memory impairment and hippocampal long-term potentiation deficit in chronic cerebral hypoperfused rats. Behav. Brain Res. 2020, 393, 112781. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.C.; Choi, S.K.; Lim, M.; Yeon, S.I.; Lee, Y.H. Role of endogenous ENaC and TRP channels in the myogenic response of rat posterior cerebral arteries. PLoS ONE 2013, 8, e84194. [Google Scholar] [CrossRef] [PubMed]
- Holzmann, C.; Kappel, S.; Kilch, T.; Jochum, M.M.; Urban, S.K.; Jung, V.; Stöckle, M.; Rother, K.; Greiner, M.; Peinelt, C. Transient receptor potential melastatin 4 channel contributes to migration of androgen-insensitive prostate cancer cells. Oncotarget 2015, 6, 41783–41793. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.X.; Zhang, Y.Y.; Wu, X.Y.; Tang, H.X.; Liang, X.Q.; Xue, Z.M.; Xue, Y.D.; Li, J.; Zhu, H.; Huo, R.; et al. Transient receptor potential melastatin 4 contributes to early-stage endothelial injury induced by arsenic trioxide. Toxicol. Lett. 2019, 312, 98–108. [Google Scholar] [CrossRef]
- Wang, F.; Wu, P.; Gong, S.; Chen, Y.; Gao, J.; Wang, S.; Shen, Q.; Tao, H.; Hua, F.; Zhou, Z.; et al. Aberrant TRPM4 expression in MLL-rearranged acute myeloid leukemia and its blockade induces cell cycle arrest via AKT/GLI1/Cyclin D1 pathway. Cell Signal. 2020, 72, 109643. [Google Scholar] [CrossRef]
- Launay, P.; Cheng, H.; Srivatsan, S.; Penner, R.; Fleig, A.; Kinet, J.P. TRPM4 regulates calcium oscillations after T cell activation. Science 2004, 306, 1374–1377. [Google Scholar] [CrossRef] [Green Version]
- Nilius, B.; Prenen, J.; Janssens, A.; Owsianik, G.; Wang, C.; Zhu, M.X.; Voets, T. The selectivity filter of the cation channel TRPM4. J. Biol. Chem. 2005, 280, 22899–22906. [Google Scholar] [CrossRef] [Green Version]
- Marigo, V.; Courville, K.; Hsu, W.H.; Feng, J.M.; Cheng, H. TRPM4 impacts on Ca2+ signals during agonist-induced insulin secretion in pancreatic beta-cells. Mol. Cell Endocrinol. 2009, 299, 194–203. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Studied Preparation | Results | Conclusion | Reference |
|---|---|---|---|
| Rat cerebral arteries | 80% reduction in TRPM4 mRNA | TRPM4 channels are regulated by Ca2+ release from IP3 receptor | [139] |
| Rat posterior cerebral artery segments | 70% reduction in TRPM4 mRNA | Epithelial sodium channels and TRPM4 interact and contribute to pressure-induced vasoconstriction | [143] |
| Prostate cancer cell lines | 50–75% reduction in TRPM4 mRNA | TRPM4 contributes to cancer cell migration | [144] |
| Human umbilical vein endothelial cells (HUVEC) | 75% reduction in TRPM4 mRNA and ~50% reduction in TRPM4 protein | TRPM4 is involved in endothelial injury induced by arsenic trioxide | [145] |
| Leukemia cell lines with the MLL gene rearrangement | 75% reduction in TRPM4 mRNA and ~50% reduction in TRPM4 protein | TRPM4 may be involved in the pathogenesis of MLL-rearranged leukemia | [146] |
| Permanent middle cerebral artery of rat | Prevented the expression of TRPM4 | TRPM4 upregulation contributes to cerebral damage in acute phase of stroke | [141] |
| Bilateral common carotid arteries occlusion rat models | Prevented the expression of TRPM4 | TRPM4 mediates cognitive deficits and LTP impairment and reduces the expression of synaptic proteins | [142] |
| HUVEC | At least 90% reduction in TRPM4 mRNA and protein | TRPM4 is involved in lipopolysaccharide-induced endothelial cell death | [140] |
| Jurkat cells | Some reduction in TRPM4 mRNA and protein | TRPM4-mediated depolarization modulates Ca2+ oscillations | [147] |
| Type of Variant | Studied Preparation | Results | Conclusion | Reference |
|---|---|---|---|---|
| Single amino acid modification (D984A) | HEK cells | Nonconducting TRPM4 channels | Information about the selectivity filter of TRPM4 | [148] |
| D984A variant | Colorectal cancer cell line HCT116 | Complete inhibition of the current without the reduction in TRPM4 protein expression | Ion conduction of TRPM4 plays a versatile role in cancer cell proliferation, cell cycle, and invasion | [24] |
| D984A variant | Human prostate cancer cells line | Nonconducting TRPM4 channels | TRPM4 is involved in cancer hallmark functions (cell viability, proliferation, migration, and cell cycle shift) | [132] |
| Deletion of first 177 amino acids in the N-terminus (ΔN-TRPM4) | Jurkat cells | Hardly conducts any current | TRPM4-mediated depolarization modulates Ca2+ oscillations | [147] |
| ΔN-TRPM4 | HUVEC | Suppression of TRPM4 activity | TRPM4 contributes to lipopolysaccharide-induced endothelial cell death | [140] |
| ΔN-TRPM4 | Rat insulinoma cell line INS-1 | Reduced TRPM4-mediated current | TRPM4 is involved in glucose- or arginine-vasopressin-induced insulin secretion | [25] |
| ΔN-TRPM4 | Rat insulinoma cell line INS-1 | Reduced TRPM4-mediated current | TRPM4 contributes to calcium signals and insulin secretion | [149] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovács, Z.M.; Dienes, C.; Hézső, T.; Almássy, J.; Magyar, J.; Bányász, T.; Nánási, P.P.; Horváth, B.; Szentandrássy, N. Pharmacological Modulation and (Patho)Physiological Roles of TRPM4 Channel—Part 1: Modulation of TRPM4. Pharmaceuticals 2022, 15, 81. https://doi.org/10.3390/ph15010081
Kovács ZM, Dienes C, Hézső T, Almássy J, Magyar J, Bányász T, Nánási PP, Horváth B, Szentandrássy N. Pharmacological Modulation and (Patho)Physiological Roles of TRPM4 Channel—Part 1: Modulation of TRPM4. Pharmaceuticals. 2022; 15(1):81. https://doi.org/10.3390/ph15010081
Chicago/Turabian StyleKovács, Zsigmond Máté, Csaba Dienes, Tamás Hézső, János Almássy, János Magyar, Tamás Bányász, Péter P. Nánási, Balázs Horváth, and Norbert Szentandrássy. 2022. "Pharmacological Modulation and (Patho)Physiological Roles of TRPM4 Channel—Part 1: Modulation of TRPM4" Pharmaceuticals 15, no. 1: 81. https://doi.org/10.3390/ph15010081
APA StyleKovács, Z. M., Dienes, C., Hézső, T., Almássy, J., Magyar, J., Bányász, T., Nánási, P. P., Horváth, B., & Szentandrássy, N. (2022). Pharmacological Modulation and (Patho)Physiological Roles of TRPM4 Channel—Part 1: Modulation of TRPM4. Pharmaceuticals, 15(1), 81. https://doi.org/10.3390/ph15010081