
Recent Advances in the Treatment of Pulmonary Arterial Hypertension
 , and
, and
Abstract
:1. Introduction
2. Definition of Pulmonary Hypertension
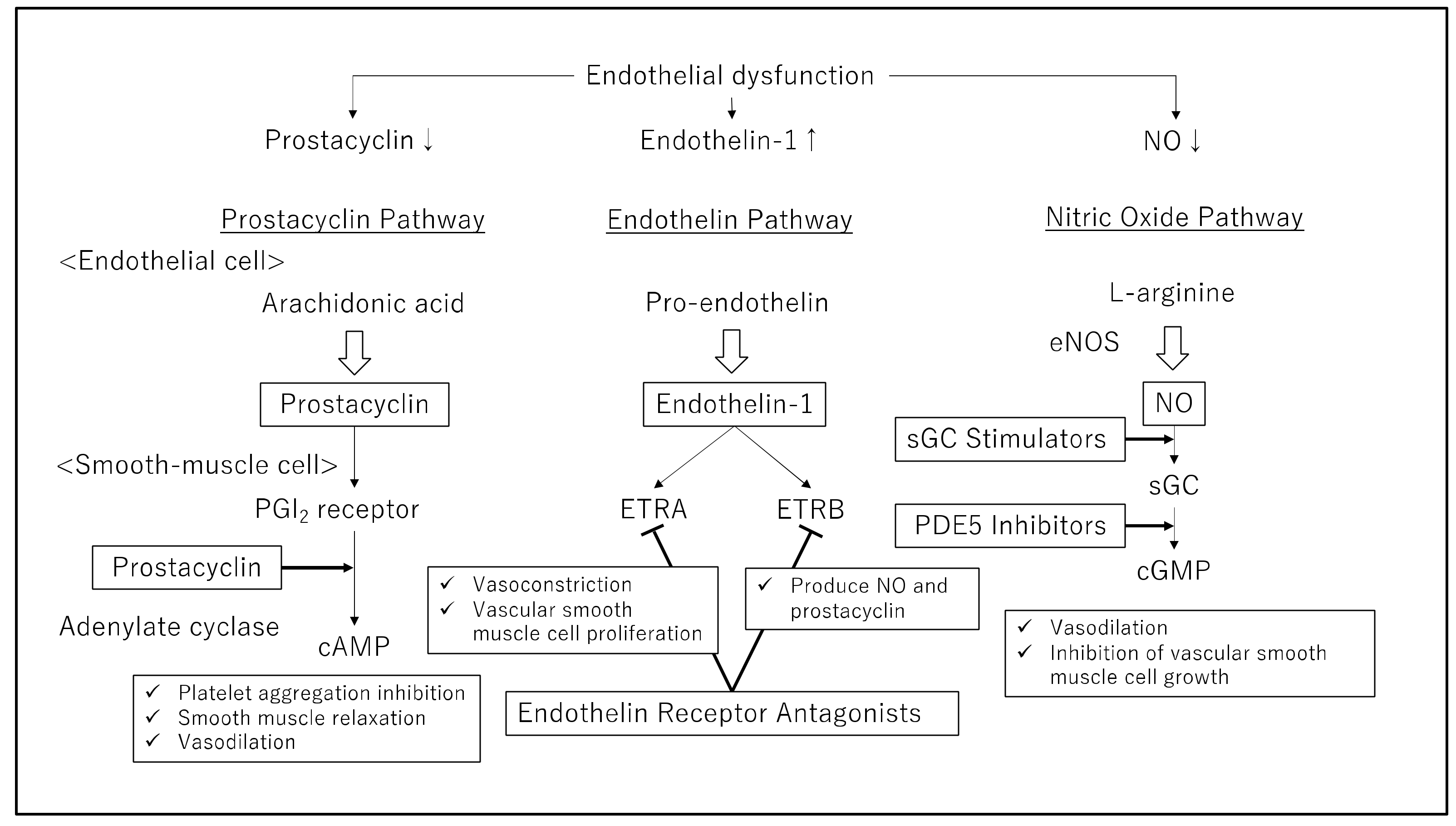
3. Current PAH Medication Therapies
4. Prostacyclin Therapy (for Targeting the Prostacyclin Pathway)
4.1. Epoprostenol
4.2. Treprostinil
4.3. Iloprost
4.4. Beraprost
4.5. Selexipag
5. Endothelin Receptor Antagonists (for Targeting the Endothelin Pathway)
5.1. Bosentan
5.2. Ambrisentan
5.3. Macitentan
5.4. Sitaxsentan
6. Phosphodiesterase Type 5 Inhibitors and Soluble Guanylate Cyclase Stimulators (Targeting the Nitric Oxide Pathway)
6.1. Sildenafil
6.2. Tadalafil
6.3. Vardenafil
6.4. Riociguat
7. Initial Dual Oral Combination Therapy
8. Development of New Therapeutic Agents
8.1. Drugs for Already-Known Targets
8.1.1. Tyrosine Kinase Inhibitors
8.1.2. Serotonin Receptor Antagonists
8.1.3. Rho-Kinase Inhibitors
8.1.4. Ranolazine
8.1.5. Tocilizumab
8.2. Drugs with New Targets
Sotatercept
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Runo, J.R.; Loyd, J.E. Primary pulmonary hypertension. Lancet 2003, 361, 1533–1544. [Google Scholar] [CrossRef] [Green Version]
- Rich, S.; Kaufmann, E.; Levy, P.S. The effect of high doses of calcium-channel blockers on survival in primary pulmonary hypertension. N. Engl. J. Med. 1992, 327, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Sitbon, O.; Chaouat, A.; Bertocchi, M.; Habib, G.; Gressin, V.; Yaïci, A.; Weitzenblum, E.; Cordier, J.F.; Chabot, F.; et al. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation 2010, 122, 156–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Alonzo, G.E.; Barst, R.J.; Ayres, S.M.; Bergofsky, E.H.; Brundage, B.H.; Detre, K.M.; Fishman, A.P.; Goldring, R.M.; Groves, B.M.; Kernis, J.T.; et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann. Intern. Med. 1991, 115, 343–349. [Google Scholar] [CrossRef]
- Hatano, S.; Strasser, T. Primary pulmonary hypertension. In Report on a WHO Meeting; World Health Organization: Geneva, Switzerland, 1975. [Google Scholar]
- Kovacs, G.; Berghold, A.; Scheidl, S.; Olschewski, H. Pulmonary arterial pressure during rest and exercise in healthy subjects: A systematic review. Eur. Respir. J. 2009, 34, 888–894. [Google Scholar] [CrossRef] [Green Version]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef]
- Galiè, N.; Hoeper, M.M.; Humbert, M.; Torbicki, A.; Vachiery, J.L.; Barbera, J.A.; Beghetti, M.; Corris, P.; Gaine, S.; Gibbs, J.S.; et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: The Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur. Heart J. 2009, 30, 2493–2537. [Google Scholar] [CrossRef]
- Sitbon, O.; Jaïs, X.; Savale, L.; Cottin, V.; Bergot, E.; Macari, E.A.; Bouvaist, H.; Dauphin, C.; Picard, F.; Bulifon, S.; et al. Upfront triple combination therapy in pulmonary arterial hypertension: A pilot study. Eur. Respir. J. 2014, 43, 1691–1697. [Google Scholar] [CrossRef] [Green Version]
- Moncada, S.; Gryglewski, R.; Bunting, S.; Vane, J.R. An enzyme isolated from arteries transforms prostaglandin endoperoxides to an unstable substance that inhibits platelet aggregation. Nature 1976, 263, 663–665. [Google Scholar] [CrossRef]
- Christman, B.W.; McPherson, C.D.; Newman, J.H.; King, G.A.; Bernard, G.R.; Groves, B.M.; Loyd, J.E. An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N. Engl. J. Med. 1992, 327, 70–75. [Google Scholar] [CrossRef]
- Tuder, R.M.; Cool, C.D.; Geraci, M.W.; Wang, J.; Abman, S.H.; Wright, L.; Badesch, D.; Voelkel, N.F. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am. J. Respir. Crit. Care Med. 1999, 159, 1925–1932. [Google Scholar] [CrossRef]
- Ruan, C.H.; Dixon, R.A.; Willerson, J.T.; Ruan, K.H. Prostacyclin therapy for pulmonary arterial hypertension. Tex. Heart Inst. J. 2010, 37, 391–399. [Google Scholar]
- Sitbon, O.; Humbert, M.; Nunes, H.; Parent, F.; Garcia, G.; Hervé, P.; Rainisio, M.; Simonneau, G. Long-term intravenous epoprostenol infusion in primary pulmonary hypertension: Prognostic factors and survival. J. Am. Coll. Cardiol. 2002, 40, 780–788. [Google Scholar] [CrossRef] [Green Version]
- Montani, D.; Günther, S.; Dorfmüller, P.; Perros, F.; Girerd, B.; Garcia, G.; Jaïs, X.; Savale, L.; Artaud-Macari, E.; Price, L.C.; et al. Pulmonary arterial hypertension. Orphanet. J. Rare Dis. 2013, 8, 97. [Google Scholar] [CrossRef] [Green Version]
- Barst, R.J.; Rubin, L.J.; Long, W.A.; McGoon, M.D.; Rich, S.; Badesch, D.B.; Groves, B.M.; Tapson, V.F.; Bourge, R.C.; Brundage, B.H.; et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N. Engl. J. Med. 1996, 334, 296–301. [Google Scholar] [CrossRef]
- McLaughlin, V.V.; Shillington, A.; Rich, S. Survival in primary pulmonary hypertension: The impact of epoprostenol therapy. Circulation 2002, 106, 1477–1482. [Google Scholar] [CrossRef] [Green Version]
- Laliberte, K.; Arneson, C.; Jeffs, R.; Hunt, T.; Wade, M. Pharmacokinetics and steady-state bioequivalence of treprostinil sodium (Remodulin) administered by the intravenous and subcutaneous route to normal volunteers. J. Cardiovasc. Pharmacol. 2004, 44, 209–214. [Google Scholar] [CrossRef]
- Simonneau, G.; Barst, R.J.; Galie, N.; Naeije, R.; Rich, S.; Bourge, R.C.; Keogh, A.; Oudiz, R.; Frost, A.; Blackburn, S.D.; et al. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: A double-blind, randomized, placebo-controlled trial. Am. J. Respir. Crit. Care Med. 2002, 165, 800–804. [Google Scholar] [CrossRef]
- McLaughlin, V.V.; Benza, R.L.; Rubin, L.J.; Channick, R.N.; Voswinckel, R.; Tapson, V.F.; Robbins, I.M.; Olschewski, H.; Rubenfire, M.; Seeger, W. Addition of inhaled treprostinil to oral therapy for pulmonary arterial hypertension: A randomized controlled clinical trial. J. Am. Coll. Cardiol. 2010, 55, 1915–1922. [Google Scholar] [CrossRef] [Green Version]
- White, R.J.; Torres, F.; Allen, R.; Jerjes, C.; Pulido, T.; Yehle, D.; Howell, M.; Laliberte, K.; Marier, J.F.; Tapson, V.F. Pharmacokinetics of oral treprostinil sustained release tablets during chronic administration to patients with pulmonary arterial hypertension. J. Cardiovasc. Pharmacol. 2013, 61, 474–481. [Google Scholar] [CrossRef]
- Olschewski, H.; Simonneau, G.; Galiè, N.; Higenbottam, T.; Naeije, R.; Rubin, L.J.; Nikkho, S.; Speich, R.; Hoeper, M.M.; Behr, J.; et al. Inhaled iloprost for severe pulmonary hypertension. N. Engl. J. Med. 2002, 347, 322–329. [Google Scholar] [CrossRef]
- Hoeper, M.M.; Schwarze, M.; Ehlerding, S.; Adler-Schuermeyer, A.; Spiekerkoetter, E.; Niedermeyer, J.; Hamm, M.; Fabel, H. Long-term treatment of primary pulmonary hypertension with aerosolized iloprost, a prostacyclin analogue. N. Engl. J. Med. 2000, 342, 1866–1870. [Google Scholar] [CrossRef]
- Nagaya, N.; Uematsu, M.; Okano, Y.; Satoh, T.; Kyotani, S.; Sakamaki, F.; Nakanishi, N.; Miyatake, K.; Kunieda, T. Effect of orally active prostacyclin analogue on survival of outpatients with primary pulmonary hypertension. J. Am. Coll. Cardiol. 1999, 34, 1188–1192. [Google Scholar] [CrossRef] [Green Version]
- Galiè, N.; Humbert, M.; Vachiéry, J.L.; Vizza, C.D.; Kneussl, M.; Manes, A.; Sitbon, O.; Torbicki, A.; Delcroix, M.; Naeije, R.; et al. Effects of beraprost sodium, an oral prostacyclin analogue, in patients with pulmonary arterial hypertension: A randomized, double-blind, placebo-controlled trial. J. Am. Coll. Cardiol. 2002, 39, 1496–1502. [Google Scholar] [CrossRef]
- Kunieda, T.; Nakanishi, N.; Matsubara, H.; Ohe, T.; Okano, Y.; Kondo, H.; Nishimura, M.; Shirato, K.; Tanabe, N.; Homma, S.; et al. Effects of long-acting beraprost sodium (TRK-100STP) in Japanese patients with pulmonary arterial hypertension. Int. Heart J. 2009, 50, 513–529. [Google Scholar] [CrossRef] [Green Version]
- Kuwano, K.; Hashino, A.; Noda, K.; Kosugi, K.; Kuwabara, K. A long-acting and highly selective prostacyclin receptor agonist prodrug, 2-{4-[(5,6-diphenylpyrazin-2-yl)(isopropyl)amino]butoxy}-N-(methylsulfonyl)acetamide (NS-304), ameliorates rat pulmonary hypertension with unique relaxant responses of its active form, {4-[(5,6-diphenylpyrazin-2-yl)(isopropyl)amino]butoxy}acetic acid (MRE-269), on rat pulmonary artery. J. Pharmacol. Exp. Ther. 2008, 326, 691–699. [Google Scholar] [CrossRef]
- Sitbon, O.; Channick, R.; Chin, K.M.; Frey, A.; Gaine, S.; Galiè, N.; Ghofrani, H.A.; Hoeper, M.M.; Lang, I.M.; Preiss, R.; et al. Selexipag for the treatment of pulmonary arterial hypertension. N. Engl. J. Med. 2015, 373, 252–2533. [Google Scholar] [CrossRef] [Green Version]
- Yanagisawa, M.; Kurihara, H.; Kimura, S.; Tomobe, Y.; Kobayashi, M.; Mitsui, Y.; Yazaki, Y.; Goto, K.; Masaki, T. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 1988, 332, 411–415. [Google Scholar] [CrossRef] [Green Version]
- Inoue, A.; Yanagisawa, M.; Kimura, S.; Kasuya, Y.; Miyauchi, T.; Goto, K.; Masaki, T. The human endothelin family: Three structurally and pharmacologically distinct isopeptides predicted by three separate genes. Proc. Natl. Acad. Sci. USA 1989, 86, 2863–2867. [Google Scholar] [CrossRef] [Green Version]
- Miyagawa, K.; Emoto, N. Current state of endothelin receptor antagonism in hypertension and pulmonary hypertension. Ther. Adv. Cardiovasc. Dis. 2014, 8, 202–216. [Google Scholar] [CrossRef]
- Kedzierski, R.M.; Yanagisawa, M. Endothelin system: The double-edged sword in health and disease. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 851–876. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T.; Yanagisawa, M.; Takuwa, Y.; Miyazaki, H.; Kimura, S.; Goto, K.; Masaki, T. Cloning of a cDNA encoding a non-isopeptide-selective subtype of the endothelin receptor. Nature 1990, 348, 732–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benigni, A.; Remuzzi, G. Endothelin antagonists. Lancet 1999, 353, 133–138. [Google Scholar] [CrossRef]
- Channick, R.N.; Simonneau, G.; Sitbon, O.; Robbins, I.M.; Frost, A.; Tapson, V.F.; Badesch, D.B.; Roux, S.; Rainisio, M.; Bodin, F.; et al. Effects of the dual endothelin-receptor antagonist bosentan in patients with pulmonary hypertension: A randomised placebo-controlled study. Lancet 2001, 358, 1119–1123. [Google Scholar] [CrossRef]
- Rubin, L.J.; Badesch, D.B.; Barst, R.J.; Galie, N.; Black, C.M.; Keogh, A.; Pulido, T.; Frost, A.; Roux, S.; Leconte, I.; et al. Bosentan therapy for pulmonary arterial hypertension. N. Engl. J. Med. 2002, 346, 896–903. [Google Scholar] [CrossRef]
- Humbert, M.; Barst, R.J.; Robbins, I.M.; Channick, R.N.; Galiè, N.; Boonstra, A.; Rubin, L.J.; Horn, E.M.; Manes, A.; Simonneau, G. Combination of bosentan with epoprostenol in pulmonary arterial hypertension: BREATHE-2. Eur. Respir. J. 2004, 24, 353–359. [Google Scholar] [CrossRef] [Green Version]
- Galiè, N.; Beghetti, M.; Gatzoulis, M.A.; Granton, J.; Berger, R.M.; Lauer, A.; Chiossi, E.; Landzberg, M.; Bosentan Randomized Trial of Endothelin Antagonist Therapy-5 (BREATHE-5) Investigators. Bosentan therapy in patients with Eisenmenger syndrome: A multicenter, double-blind, randomized, placebo-controlled study. Circulation 2006, 114, 48–54. [Google Scholar] [CrossRef] [Green Version]
- Galiè, N.; Rubin, L.J.; Hoeper, M.; Jansa, P.; Al-Hiti, H.; Meyer, G.; Chiossi, E.; Kusic-Pajic, A.; Simonneau, G. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): A double-blind, randomised controlled trial. Lancet 2008, 371, 2093–2100. [Google Scholar] [CrossRef]
- Galié, N.; Badesch, D.; Oudiz, R.; Simonneau, G.; McGoon, M.D.; Keogh, A.M.; Frost, A.E.; Zwicke, D.; Naeije, R.; Shapiro, S.; et al. Ambrisentan therapy for pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2005, 46, 529–535. [Google Scholar] [CrossRef]
- Galiè, N.; Olschewski, H.; Oudiz, R.J.; Torres, F.; Frost, A.; Ghofrani, H.A.; Badesch, D.B.; McGoon, M.D.; McLaughlin, V.V.; Roecker, E.B.; et al. Ambrisentan for the treatment of pulmonary arterial hypertension: Results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation 2008, 117, 3010–3019. [Google Scholar] [CrossRef] [Green Version]
- Galiè, N.; Corris, P.A.; Frost, A.; Girgis, R.E.; Granton, J.; Jing, Z.C.; Klepetko, W.; McGoon, M.D.; McLaughlin, V.V.; Preston, I.R.; et al. Updated treatment algorithm of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2013, 62 (Suppl. 25), D60–D72. [Google Scholar] [CrossRef]
- Pulido, T.; Adzerikho, I.; Channick, R.N.; Delcroix, M.; Galiè, N.; Ghofrani, H.A.; Jansa, P.; Jing, Z.C.; Le Brun, F.O.; Mehta, S.; et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N. Engl. J. Med. 2013, 369, 809–818. [Google Scholar] [CrossRef] [Green Version]
- Barst, R.J.; Rich, S.; Widlitz, A.; Horn, E.M.; McLaughlin, V.; McFarlin, J. Clinical efficacy of sitaxsentan, an endothelin-A receptor antagonist, in patients with pulmonary arterial hypertension: Open-label pilot study. Chest 2002, 121, 1860–1868. [Google Scholar] [CrossRef]
- Barst, R.J.; Langleben, D.; Badesch, D.; Frost, A.; Lawrence, E.C.; Shapiro, S.; Naeije, R.; Galie, N.; STRIDE-2 Study Group. Treatment of pulmonary arterial hypertension with the selective endothelin-A receptor antagonist sitaxsentan. J. Am. Coll. Cardiol. 2006, 47, 2049–2056. [Google Scholar] [CrossRef]
- Barst, R.J.; Langleben, D.; Frost, A.; Horn, E.M.; Oudiz, R.; Shapiro, S.; McLaughlin, V.; Hill, N.; Tapson, V.F.; Robbins, I.M.; et al. Sitaxsentan therapy for pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2004, 169, 441–447. [Google Scholar] [CrossRef]
- Lavelle, A.; Sugrue, R.; Lawler, G.; Mulligan, N.; Kelleher, B.; Murphy, D.M.; Gaine, S.P. Sitaxentan-induced hepatic failure in two patients with pulmonary arterial hypertension. Eur. Respir. J. 2009, 34, 770–771. [Google Scholar] [CrossRef]
- Watanabe, H.; Ohashi, K.; Takeuchi, K.; Yamashita, K.; Yokoyama, T.; Tran, Q.K.; Satoh, H.; Terada, H.; Ohashi, H.; Hayashi, H. Sildenafil for primary and secondary pulmonary hypertension. Clin. Pharmacol. Ther. 2002, 71, 398–402. [Google Scholar] [CrossRef]
- Galiè, N.; Ghofrani, H.A.; Torbicki, A.; Barst, R.J.; Rubin, L.J.; Badesch, D.; Fleming, T.; Parpia, T.; Burgess, G.; Branzi, A.; et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N. Engl. J. Med. 2005, 353, 2148–2157. [Google Scholar] [CrossRef] [Green Version]
- Rubin, L.J.; Badesch, D.B.; Fleming, T.R.; Galiè, N.; Simonneau, G.; Ghofrani, H.A.; Oakes, M.; Layton, G.; Serdarevic-Pehar, M.; McLaughlin, V.V.; et al. Long-term treatment with sildenafil citrate in pulmonary arterial hypertension: The SUPER-2 study. Chest 2011, 140, 1274–1283. [Google Scholar] [CrossRef]
- Galiè, N.; Brundage, B.H.; Ghofrani, H.A.; Oudiz, R.J.; Simonneau, G.; Safdar, Z.; Shapiro, S.; White, R.J.; Chan, M.; Beardsworth, A.; et al. Tadalafil therapy for pulmonary arterial hypertension. Circulation 2009, 119, 2894–2903. [Google Scholar] [CrossRef] [Green Version]
- Oudiz, R.J.; Brundage, B.H.; Galiè, N.; Ghofrani, H.A.; Simonneau, G.; Botros, F.T.; Chan, M.; Beardsworth, A.; Barst, R.J.; PHIRST Study Group. Tadalafil for the treatment of pulmonary arterial hypertension: A double-blind 52-week uncontrolled extension study. J. Am. Coll. Cardiol. 2012, 60, 768–774. [Google Scholar] [CrossRef]
- Coyle, K.; Coyle, D.; Blouin, J.; Lee, K.; Jabr, M.F.; Tran, K.; Mielniczuk, L.; Swiston, J.; Innes, M. Cost Effectiveness of First-Line Oral Therapies for Pulmonary Arterial Hypertension: A Modelling Study. Pharmacoeconomics 2016, 34, 509–520. [Google Scholar] [CrossRef] [Green Version]
- Jing, Z.C.; Yu, Z.X.; Shen, J.Y.; Wu, B.X.; Xu, K.F.; Zhu, X.Y.; Pan, L.; Zhang, Z.L.; Liu, X.Q.; Zhang, Y.S.; et al. Vardenafil in pulmonary arterial hypertension: A randomized, double-blind, placebo-controlled study. Am. J. Respir. Crit. Care Med. 2011, 183, 1723–1729. [Google Scholar] [CrossRef]
- Ghofrani, H.A.; Galiè, N.; Grimminger, F.; Grünig, E.; Humbert, M.; Jing, Z.C.; Keogh, A.M.; Langleben, D.; Kilama, M.O.; Fritsch, A.; et al. Riociguat for the treatment of pulmonary arterial hypertension. N. Engl. J. Med. 2013, 369, 330–340. [Google Scholar] [CrossRef] [Green Version]
- O’Callaghan, D.S.; Gaine, S.P. Combination therapy and new types of agents for pulmonary arterial hypertension. Clin. Chest Med. 2007, 28, 169–185. [Google Scholar] [CrossRef]
- O’Callaghan, D.S.; Savale, L.; Montani, D.; Jaïs, X.; Sitbon, O.; Simonneau, G.; Humbert, M. Treatment of pulmonary arterial hypertension with targeted therapies. Nat. Rev. Cardiol. 2011, 8, 526–538. [Google Scholar] [CrossRef]
- Galiè, N.; Barberà, J.A.; Frost, A.E.; Ghofrani, H.A.; Hoeper, M.M.; McLaughlin, V.V.; Peacock, A.J.; Simonneau, G.; Vachiery, J.L.; Grünig, E.; et al. Initial use of ambrisentan plus tadalafil in pulmonary arterial hypertension. N. Engl. J. Med. 2015, 373, 834–844. [Google Scholar] [CrossRef]
- Galiè, N.; Channick, R.N.; Frantz, R.P.; Grünig, E.; Jing, Z.C.; Moiseeva, O.; Preston, I.R.; Pulido, T.; Safdar, Z.; Tamura, Y.; et al. Risk stratification and medical therapy of pulmonary arterial hypertension. Eur. Respir. J. 2019, 53, 1801889. [Google Scholar] [CrossRef]
- Sitbon, O.; Sattler, C.; Bertoletti, L.; Savale, L.; Cottin, V.; Jaïs, X.; de Groote, P.; Chaouat, A.; Chabannes, C.; Bergot, E.; et al. Initial dual oral combination therapy in pulmonary arterial hypertension. Eur. Respir. J. 2016, 47, 1727–1736. [Google Scholar] [CrossRef] [Green Version]
- Sulica, R.; Sangli, S.; Chakravarti, A.; Steiger, D. Clinical and hemodynamic benefit of macitentan and riociguat upfront combination in patients with pulmonary arterial hypertension. Pulm. Circ. 2019, 9, 2045894019826944. [Google Scholar] [CrossRef] [Green Version]
- Coghlan, J.G.; Channick, R.; Chin, K.; Di Scala, L.; Galiè, N.; Ghofrani, H.A.; Hoeper, M.M.; Lang, I.M.; McLaughlin, V.; Preiss, R.; et al. Targeting the prostacyclin pathway with selexipag in patients with pulmonary arterial hypertension receiving double combination therapy: Insights from the randomized controlled GRIPHON Study. Am. J. Cardiovasc. Drugs 2018, 18, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Toba, M.; Alzoubi, A.; Ito, M.; Fagan, K.A.; Cool, C.D.; Voelkel, N.F.; McMurtry, I.F.; Oka, M. Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation 2010, 121, 2747–2754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, K.; Shinoda, M.; Tanaka, M.; Kuwabara, Y.; Yoshida, K.; Hirooka, Y.; McMurtry, I.F.; Oka, M.; Sunagawa, K. Haemodynamic unloading reverses occlusive vascular lesions in severe pulmonary hypertension. Cardiovasc. Res. 2016, 111, 16–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perros, F.; Montani, D.; Dorfmüller, P.; Durand-Gasselin, I.; Tcherakian, C.; Le Pavec, J.; Mazmanian, M.; Fadel, E.; Mussot, S.; Mercier, O.; et al. Platelet-derived growth factor expression and function in idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2008, 178, 81–88. [Google Scholar] [CrossRef]
- Hoeper, M.M.; Barst, R.J.; Bourge, R.C.; Feldman, J.; Frost, A.E.; Galié, N.; Gómez-Sánchez, M.A.; Grimminger, F.; Grünig, E.; Hassoun, P.M.; et al. Imatinib mesylate as add-on therapy for pulmonary arterial hypertension: Results of the randomized IMPRES study. Circulation 2013, 127, 1128–1138. [Google Scholar] [CrossRef] [Green Version]
- Weatherald, J.; Chaumais, M.C.; Montani, D. Pulmonary arterial hypertension induced by tyrosine kinase inhibitors. Curr. Opin. Pulm. Med. 2017, 23, 392–397. [Google Scholar] [CrossRef]
- Hervé, P.; Launay, J.M.; Scrobohaci, M.L.; Brenot, F.; Simonneau, G.; Petitpretz, P.; Poubeau, P.; Cerrina, J.; Duroux, P.; Drouet, L. Increased plasma serotonin in primary pulmonary hypertension. Am. J. Med. 1995, 99, 249–254. [Google Scholar] [CrossRef]
- Dempsie, Y.; MacLean, M.R. Pulmonary hypertension: Therapeutic targets within the serotonin system. Br. J. Pharmacol. 2008, 155, 455–462. [Google Scholar] [CrossRef]
- Dumitrascu, R.; Kulcke, C.; Königshoff, M.; Kouri, F.; Yang, X.; Morrell, N.; Ghofrani, H.A.; Weissmann, N.; Reiter, R.; Seeger, W.; et al. Terguride ameliorates monocrotaline-induced pulmonary hypertension in rats. Eur. Respir. J. 2011, 37, 1104–1118. [Google Scholar] [CrossRef]
- Ghofrani, H.A.; Al-Hiti, H.; Vonk-Noordegraaf, A.; Behr, J.; Neurohr, C.; Jansa, P.; Wilkens, H.; Hoeper, M.H.; Gruenig, E.; Opitz, C.; et al. Proof-of-concept study to investi- gate the efficacy, hemodynamics and tolerability of terguride vs. placebo in subjects with pulmonary arterial hypertension: Results of a double blind, randomised, prospective phase IIa study. Am. J. Respir. Crit. Care Med. 2012, 185, A2496. [Google Scholar]
- Leung, T.; Manser, E.; Tan, L.; Lim, L. A novel serine/threonine kinase binding the Ras-related RhoA GTPase which translocates the kinase to peripheral membranes. J. Biol. Chem. 1995, 270, 29051–29054. [Google Scholar] [CrossRef] [Green Version]
- Shimokawa, H. Rho-kinase as a novel therapeutic target in treatment of cardiovascular diseases. J. Cardiovasc. Pharmacol. 2002, 39, 319–327. [Google Scholar] [CrossRef]
- Odagiri, K.; Watanabe, H. Effects of the Rho-kinase inhibitor, fasudil, on pulmonary hypertension. Circ. J. 2015, 79, 1213–1214. [Google Scholar] [CrossRef] [Green Version]
- Fukumoto, Y.; Yamada, N.; Matsubara, H.; Mizoguchi, M.; Uchino, K.; Yao, A.; Kihara, Y.; Kawano, M.; Watanabe, H.; Takeda, Y.; et al. Double-blind, placebo-controlled clinical trial with a rho-kinase inhibitor in pulmonary arterial hypertension. Circ. J. 2013, 77, 2619–2625. [Google Scholar] [CrossRef] [Green Version]
- Stone, P.H. Ranolazine: New paradigm for management of myocardial ischemia, myocardial dysfunction, and arrhythmias. Cardiol. Clin. 2008, 26, 603–614. [Google Scholar] [CrossRef]
- Khan, S.S.; Cuttica, M.J.; Beussink-Nelson, L.; Kozyleva, A.; Sanchez, C.; Mkrdichian, H.; Selvaraj, S.; Dematte, J.E.; Lee, D.C.; Shah, S.J. Effects of ranolazine on exercise capacity, right ventricular indices, and hemodynamic characteristics in pulmonary arterial hypertension: A pilot study. Pulm. Circ. 2015, 5, 547–556. [Google Scholar] [CrossRef] [Green Version]
- Humbert, M.; Morrell, N.W.; Archer, S.L.; Stenmark, K.R.; MacLean, M.R.; Lang, I.M.; Christman, B.W.; Weir, E.K.; Eickelberg, O.; Voelkel, N.F.; et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2004, 43 (Suppl. S12), 13S–24S. [Google Scholar] [CrossRef] [Green Version]
- Soon, E.; Holmes, A.M.; Treacy, C.M.; Doughty, N.J.; Southgate, L.; Machado, R.D.; Trembath, R.C.; Jennings, S.; Barker, L.; Nicklin, P.; et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation 2010, 122, 920–927. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto-Kataoka, T.; Hosen, N.; Sonobe, T.; Arita, Y.; Yasui, T.; Masaki, T.; Minami, M.; Inagaki, T.; Miyagawa, S.; Sawa, Y.; et al. Interleukin-6/interleukin-21 signaling axis is critical in the pathogenesis of pulmonary arterial hypertension. Proc. Natl. Acad. Sci. USA 2015, 112, E2677–E2686. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Sánchez, J.; Harlow, L.; Church, C.; Gaine, S.; Knightbridge, E.; Bunclark, K.; Gor, D.; Bedding, A.; Morrell, N.; Corris, P.; et al. Clinical trial protocol for TRANSFORM-UK: A therapeutic open-label study of tocilizumab in the treatment of pulmonary arterial hypertension. Pulm. Circ. 2018, 8, 2045893217735820. [Google Scholar] [CrossRef] [Green Version]
- Leopold, J.A.; Maron, B.A. Molecular mechanisms of pulmonary vascular remodeling in pulmonary arterial hypertension. Int. J. Mol. Sci. 2016, 17, 761. [Google Scholar] [CrossRef] [Green Version]
- Austin, E.D.; Ma, L.; LeDuc, C.; Berman Rosenzweig, E.; Borczuk, A.; Phillips, J.A., 3rd; Palomero, T.; Sumazin, P.; Kim, H.R.; Talati, M.H.; et al. Whole exome sequencing to identify a novel gene (caveolin-1) associated with human pulmonary arterial hypertension. Circ. Cardiovasc. Genet. 2012, 5, 336–343. [Google Scholar] [CrossRef]
- Yu, Y.; Keller, S.H.; Remillard, C.V.; Safrina, O.; Nicholson, A.; Zhang, S.L.; Jiang, W.; Vangala, N.; Landsberg, J.W.; Wang, J.Y.; et al. A functional single-nucleotide polymorphism in the TRPC6 gene promoter associated with idiopathic pulmonary arterial hypertension. Circulation 2009, 119, 2313–2322. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Roman-Campos, D.; Austin, E.D.; Eyries, M.; Sampson, K.S.; Soubrier, F.; Germain, M.; Trégouët, D.A.; Borczuk, A.; Rosenzweig, E.B.; et al. A novel channelopathy in pulmonary arterial hypertension. N. Engl. J. Med. 2013, 369, 351–361. [Google Scholar] [CrossRef] [Green Version]
- Doe, Z.; Fukumoto, Y.; Takaki, A.; Tawara, S.; Ohashi, J.; Nakano, M.; Tada, T.; Saji, K.; Sugimura, K.; Fujita, H.; et al. Evidence for Rho-kinase activation in patients with pulmonary arterial hypertension. Circ. J. 2009, 73, 1731–1739. [Google Scholar] [CrossRef] [Green Version]
- Humbert, M.; McLaughlin, V.; Gibbs, J.S.R.; Gomberg-Maitland, M.; Hoeper, M.M.; Preston, I.R.; Souza, R.; Waxman, A.; Escribano Subias, P.; Feldman, J.; et al. Sotatercept for the treatment of pulmonary arterial hypertension. N. Engl. J. Med. 2021, 384, 1204–1215. [Google Scholar] [CrossRef]


{kind=link}
| Target-Based Actions | Drug Name | Dosage | Adverse Effects |
|---|---|---|---|
| Prostacyclins | Epoprostenol | Initial: 2–4 ng/kg/min IV infusion pump. Titrate by 1–2 ng/kg/min every 15 min or longer and gradually raise to 10–16 ng/kg/min according to adverse drug reactions. | >10%
|
| Treprostinil | <Injectable> Initial: 1.25 ng/kg/min continuous SC/IV infusion (0.625 ng/kg/min if not tolerated). Titrate by no more than 1.25 ng/kg/min every 1 week in first 4 weeks, then no more than 2.5 ng/kg/min every 1 week. <Extended-release tablets> Initial: 0.125 mg PO TID or 0.25 mg PO BID. Titrate by 0.125 mg TID or 0.25 or 0.5 mg BID, not more frequently than every 3–4 days. | >10%
| |
| Iloprost | Initial: 2.5 μg inhaled by a nebulizer, then 5 μg subsequent doses 6–9 times/day. | >10%
| |
| Beraprost | Initial: 20 μg PO TID, to highest tolerated dose; not to exceed 180 μg TID. |
| |
| Selexipag | Initial: 0.2 mg PO BID. Increase dose by 0.2 mg BID, at weekly intervals, to highest tolerated dose; not to exceed 1.6 mg BID. | >10%
| |
| Endothelin receptor antagonists | Bosentan | <40 kg: Maintain dose at 62.5 mg PO BID. >40 kg: 62.5 mg PO BID for 4 weeks and then increased to maintenance dosage 125 mg PO BID; not to exceed 250 mg BID. | >10%
|
| Ambrisentan | Initiate treatment at 5 mg PO QD, as needed and tolerated, not to exceed 10 mg. | >10%
| |
| Macitentan | 10 mg PO QD | >10%
| |
| Sitaxsentan | Withdrawn from the market | ||
| Phosphodiesterase type 5 inhibitors | Sildenafil | 5 mg or 20 mg PO TID; administer 4–6 h apart | >10%
|
| Tadalafil | 40 mg PO QD | >10%
| |
| Vardenafil | Vardenafil is not yet approved for PAH at present. | ||
| Soluble guanylate cyclase stimulators | Riociguat | Initial dose: 1 mg PO TID; consider 0.5 mg PO TID if patient may not tolerate hypotensive effect. If systolic blood pressure > 95 mmHg and no symptoms of hypotension, up-titrate dose by 0.5 mg PO TID, with dose increases no sooner than 2 weeks apart to highest tolerated dose. Maintenance dose: not to exceed 2.5 mg PO TID. If symptoms of hypotension occur, decrease dose by 0.5 mg TID. | >10%
|
| Target-Based Actions | Drug Name | Administration Route | Development Status | |
|---|---|---|---|---|
| Drugs for already-known targets | Tyrosine kinase inhibitors | Imatinib | Oral administration | Phase 3 (no development) |
| Serotonin receptor antagonists | Terguride | Oral administration | Phase 2 (no development) | |
| Rho-kinase inhibitors | Fasudil | Oral administration | Phase 2 | |
| Inhibition of INa and fatty acid oxidation | Ranolazine | Oral administration | Phase 3 | |
| IL6 receptor antagonists | Tocilizumab | Injection (intravenous administration) | Phase 3 | |
| Drugs in new targets | Ligand trap for TGF-β | Sotatercept | Injection (subcutaneous injection) | Phase 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Otani, N.; Tomoe, T.; Kawabe, A.; Sugiyama, T.; Horie, Y.; Sugimura, H.; Yasu, T.; Nakamoto, T. Recent Advances in the Treatment of Pulmonary Arterial Hypertension. Pharmaceuticals 2022, 15, 1277. https://doi.org/10.3390/ph15101277
Otani N, Tomoe T, Kawabe A, Sugiyama T, Horie Y, Sugimura H, Yasu T, Nakamoto T. Recent Advances in the Treatment of Pulmonary Arterial Hypertension. Pharmaceuticals. 2022; 15(10):1277. https://doi.org/10.3390/ph15101277
Chicago/Turabian StyleOtani, Naoyuki, Takashi Tomoe, Atsuhiko Kawabe, Takushi Sugiyama, Yasuto Horie, Hiroyuki Sugimura, Takanori Yasu, and Takaaki Nakamoto. 2022. "Recent Advances in the Treatment of Pulmonary Arterial Hypertension" Pharmaceuticals 15, no. 10: 1277. https://doi.org/10.3390/ph15101277
APA StyleOtani, N., Tomoe, T., Kawabe, A., Sugiyama, T., Horie, Y., Sugimura, H., Yasu, T., & Nakamoto, T. (2022). Recent Advances in the Treatment of Pulmonary Arterial Hypertension. Pharmaceuticals, 15(10), 1277. https://doi.org/10.3390/ph15101277