
Alterations of HDL’s to piHDL’s Proteome in Patients with Chronic Inflammatory Diseases, and HDL-Targeted Therapies


Abstract
:
1. Introduction
2. Chronic Inflammatory Diseases Are Associated with Increased CVD Risk
3. The Pathophysiology of Atherosclerosis
4. Cardioprotective Effect of HDL Particles
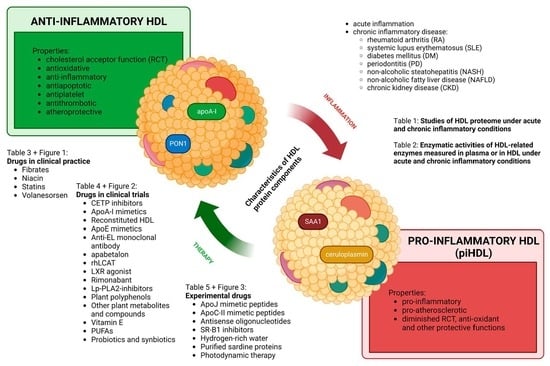
5. HDL Protein Components
6. Protein Components of HDL Relevant to Atherosclerosis
6.1. apoA-I
6.2. apoA-II
6.3. apoA-IV
6.4. apoE
6.5. apoA-V
6.6. apoC
6.7. apoJ
6.8. apoM, apoD and apoF
6.9. CETP
6.10. Lp-PLA2
6.11. PLTP
6.12. LPS-Binding Protein (LBP)
6.13. LCAT
6.14. Gpx-3
6.15. PON
6.16. Transferrin
6.17. SAA
6.18. Ceruloplasmin
6.19. Fibrinogen
6.20. Hp, Hb and Hx
6.21. AAT
6.22. α1-Acid Glycoprotein (AAG)
6.23. α2-Macroglobulin (α2M)
6.24. β2-Microglobulin (B2M)
6.25. Secretory Phospholipase A2 (sPLA2)
6.26. Complement Component 3 (C3)
7. Conversion of Anti-Inflammatory HDL to piHDL and Its Relevance to Atherosclerosis
8. Alterations of HDL Proteome under Inflammatory Conditions
9. Post-Translational Changes of HDL Proteins
9.1. Oxidation
9.2. Carbamylation
9.3. Glycation
10. Currently Available HDL-Targeted Therapies
10.1. Drugs in Clinical Practice
10.2. HDL-Affecting Drugs in Clinical Trials for Cardiovascular Application
10.3. Experimental Therapies
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wu, S.; Wu, F.; Ding, Y.; Hou, J.; Bi, J.; Zhang, Z. Association of non-alcoholic fatty liver disease with major adverse cardiovascular events: A systematic review and meta-analysis. Sci. Rep. 2016, 6, 33386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maradit-Kremers, H.; Nicola, P.J.; Crowson, C.S.; Ballman, K.V.; Gabriel, S.E. Cardiovascular death in rheumatoid arthritis: A population-based study. Arthritis Rheum. 2005, 52, 722–732. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.A.; Creager, M.A.; Libby, P. Diabetes and atherosclerosis: Epidemiology, pathophysiology, and management. Jama 2002, 287, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- van der Velde, M.; Matsushita, K.; Coresh, J.; Astor, B.C.; Woodward, M.; Levey, A.; de Jong, P.; Gansevoort, R.T.; van der Velde, M.; Matsushita, K.; et al. Lower estimated glomerular filtration rate and higher albuminuria are associated with all-cause and cardiovascular mortality. A collaborative meta-analysis of high-risk population cohorts. Kidney Int. 2011, 79, 1341–1352. [Google Scholar] [CrossRef] [Green Version]
- Ljunggren, S.; Bengtsson, T.; Karlsson, H.; Starkhammar Johansson, C.; Palm, E.; Nayeri, F.; Ghafouri, B.; Davies, J.; Svensäter, G.; Lönn, J. Modified lipoproteins in periodontitis: A link to cardiovascular disease? Biosci. Rep. 2019, 39, BSR20181665. [Google Scholar] [CrossRef] [Green Version]
- Kasper, P.; Martin, A.; Lang, S.; Kütting, F.; Goeser, T.; Demir, M.; Steffen, H.M. NAFLD and cardiovascular diseases: A clinical review. Clin. Res. Cardiol. 2021, 110, 921–937. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Chavakis, T. Local and systemic mechanisms linking periodontal disease and inflammatory comorbidities. Nat. Rev. Immunol. 2021, 21, 426–440. [Google Scholar] [CrossRef]
- Pašková, Ľ. Lipid profile and risks of cardiovascular diseases in conditions of rheumatoid arthritis. Ceska Slov. Farm. 2019, 68, 219–228. [Google Scholar]
- Watanabe, J.; Charles-Schoeman, C.; Miao, Y.; Elashoff, D.; Lee, Y.Y.; Katselis, G.; Lee, T.D.; Reddy, S.T. Proteomic profiling following immunoaffinity capture of high-density lipoprotein: Association of acute-phase proteins and complement factors with proinflammatory high-density lipoprotein in rheumatoid arthritis. Arthritis Rheum. 2012, 64, 1828–1837. [Google Scholar] [CrossRef] [Green Version]
- Di Angelantonio, E.; Sarwar, N.; Perry, P.; Kaptoge, S.; Ray, K.K.; Thompson, A.; Wood, A.M.; Lewington, S.; Sattar, N.; Packard, C.J.; et al. Major lipids, apolipoproteins, and risk of vascular disease. Jama 2009, 302, 1993–2000. [Google Scholar] [CrossRef] [Green Version]
- Charles-Schoeman, C.; Watanabe, J.; Lee, Y.Y.; Furst, D.E.; Amjadi, S.; Elashoff, D.; Park, G.; McMahon, M.; Paulus, H.E.; Fogelman, A.M.; et al. Abnormal function of high-density lipoprotein is associated with poor disease control and an altered protein cargo in rheumatoid arthritis. Arthritis Rheum. 2009, 60, 2870–2879. [Google Scholar] [CrossRef] [PubMed]
- Khovidhunkit, W.; Kim, M.S.; Memon, R.A.; Shigenaga, J.K.; Moser, A.H.; Feingold, K.R.; Grunfeld, C. Effects of infection and inflammation on lipid and lipoprotein metabolism: Mechanisms and consequences to the host. J. Lipid Res. 2004, 45, 1169–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navab, M.; Van Lenten, B.J.; Reddy, S.T.; Fogelman, A.M. High-density lipoprotein and the dynamics of atherosclerotic lesions. Circulation 2001, 104, 2386–2387. [Google Scholar] [CrossRef] [Green Version]
- Rosenson, R.S.; Brewer, H.B., Jr.; Ansell, B.J.; Barter, P.; Chapman, M.J.; Heinecke, J.W.; Kontush, A.; Tall, A.R.; Webb, N.R. Dysfunctional HDL and atherosclerotic cardiovascular disease. Nat. Rev. Cardiol. 2016, 13, 48–60. [Google Scholar] [CrossRef]
- Navab, M.; Berliner, J.A.; Subbanagounder, G.; Hama, S.; Lusis, A.J.; Castellani, L.W.; Reddy, S.; Shih, D.; Shi, W.; Watson, A.D.; et al. HDL and the inflammatory response induced by LDL-derived oxidized phospholipids. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 481–488. [Google Scholar] [CrossRef] [Green Version]
- Speer, T.; Zewinger, S. High-density lipoprotein (HDL) and infections: A versatile culprit. Eur. Heart J. 2018, 39, 1191–1193. [Google Scholar] [CrossRef] [PubMed]
- Hima, B.G.; Rao, V.S.; Kakkar, V.V. Friend Turns Foe: Transformation of Anti-Inflammatory HDL to Proinflammatory HDL during Acute-Phase Response. Cholesterol 2011, 2011, 274629. [Google Scholar] [CrossRef] [Green Version]
- Tall, A.R.; Yvan-Charvet, L. Cholesterol, inflammation and innate immunity. Nat. Rev. Immunol. 2015, 15, 104–116. [Google Scholar] [CrossRef] [Green Version]
- Yu, B.L.; Wang, S.H.; Peng, D.Q.; Zhao, S.P. HDL and immunomodulation: An emerging role of HDL against atherosclerosis. Immunol. Cell Biol. 2010, 88, 285–290. [Google Scholar] [CrossRef]
- Tölle, M.; Huang, T.; Schuchardt, M.; Jankowski, V.; Prüfer, N.; Jankowski, J.; Tietge, U.J.; Zidek, W.; van der Giet, M. High-density lipoprotein loses its anti-inflammatory capacity by accumulation of pro-inflammatory-serum amyloid A. Cardiovasc. Res. 2012, 94, 154–162. [Google Scholar] [CrossRef] [Green Version]
- Säemann, M.D.; Poglitsch, M.; Kopecky, C.; Haidinger, M.; Hörl, W.H.; Weichhart, T. The versatility of HDL: A crucial anti-inflammatory regulator. Eur. J. Clin. Investig. 2010, 40, 1131–1143. [Google Scholar] [CrossRef] [PubMed]
- Van Lenten, B.J.; Hama, S.Y.; de Beer, F.C.; Stafforini, D.M.; McIntyre, T.M.; Prescott, S.M.; La Du, B.N.; Fogelman, A.M.; Navab, M. Anti-inflammatory HDL becomes pro-inflammatory during the acute phase response. Loss of protective effect of HDL against LDL oxidation in aortic wall cell cocultures. J. Clin. Investig. 1995, 96, 2758–2767. [Google Scholar] [CrossRef] [PubMed]
- Holzer, M.; Zangger, K.; El-Gamal, D.; Binder, V.; Curcic, S.; Konya, V.; Schuligoi, R.; Heinemann, A.; Marsche, G. Myeloperoxidase-derived chlorinating species induce protein carbamylation through decomposition of thiocyanate and urea: Novel pathways generating dysfunctional high-density lipoprotein. Antioxid. Redox Signal 2012, 17, 1043–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, W.; Abe-Dohmae, S.; Tsujita, M.; Iwamoto, N.; Ogikubo, O.; Otsuka, T.; Kumon, Y.; Yokoyama, S. Biogenesis of HDL by SAA is dependent on ABCA1 in the liver in vivo. J. Lipid Res. 2008, 49, 386–393. [Google Scholar] [CrossRef] [Green Version]
- Marsche, G.; Heine, G.H.; Stadler, J.T.; Holzer, M. Current Understanding of the Relationship of HDL Composition, Structure and Function to Their Cardioprotective Properties in Chronic Kidney Disease. Biomolecules 2020, 10, 1348. [Google Scholar] [CrossRef]
- Shen, Y.; Ding, F.H.; Sun, J.T.; Pu, L.J.; Zhang, R.Y.; Zhang, Q.; Chen, Q.J.; Shen, W.F.; Lu, L. Association of elevated apoA-I glycation and reduced HDL-associated paraoxonase1, 3 activity, and their interaction with angiographic severity of coronary artery disease in patients with type 2 diabetes mellitus. Cardiovasc. Diabetol. 2015, 14, 52. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Fan, J.; Su, Q.; Yang, Z. Cytokines and Abnormal Glucose and Lipid Metabolism. Front. Endocrinol. 2019, 10, 703. [Google Scholar] [CrossRef] [Green Version]
- Kleemann, R.; Verschuren, L.; van Erk, M.J.; Nikolsky, Y.; Cnubben, N.H.; Verheij, E.R.; Smilde, A.K.; Hendriks, H.F.; Zadelaar, S.; Smith, G.J.; et al. Atherosclerosis and liver inflammation induced by increased dietary cholesterol intake: A combined transcriptomics and metabolomics analysis. Genome Biol. 2007, 8, R200. [Google Scholar] [CrossRef] [Green Version]
- Tripathy, D.; Mohanty, P.; Dhindsa, S.; Syed, T.; Ghanim, H.; Aljada, A.; Dandona, P. Elevation of free fatty acids induces inflammation and impairs vascular reactivity in healthy subjects. Diabetes 2003, 52, 2882–2887. [Google Scholar] [CrossRef] [Green Version]
- Poznyak, A.; Grechko, A.V.; Poggio, P.; Myasoedova, V.A.; Alfieri, V.; Orekhov, A.N. The Diabetes Mellitus-Atherosclerosis Connection: The Role of Lipid and Glucose Metabolism and Chronic Inflammation. Int. J. Mol. Sci. 2020, 21, 1835. [Google Scholar] [CrossRef] [Green Version]
- Marie, D.; Ateba, G.; Felicite, K.; Fernando, K.; Chia, M.; Henry, L. Oxidative Stress in Patients with Chronic Inflammatory Diseases in a Tertiary Health Care Setting in Africa. J. Autoimmun. Disord. 2017, 3, 47. [Google Scholar]
- Smallwood, M.J.; Nissim, A.; Knight, A.R.; Whiteman, M.; Haigh, R.; Winyard, P.G. Oxidative stress in autoimmune rheumatic diseases. Free Radic. Biol. Med. 2018, 125, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z. The metabolic syndrome. Lancet 2005, 365, 1415–1428. [Google Scholar] [CrossRef]
- Kahn, B.B.; Flier, J.S. Obesity and insulin resistance. J. Clin. Investig. 2000, 106, 473–481. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Nasr, A.; Tfayli, H.; Bacha, F.; Michaliszyn, S.F.; Arslanian, S. Increased Lipolysis, Diminished Adipose Tissue Insulin Sensitivity, and Impaired β-Cell Function Relative to Adipose Tissue Insulin Sensitivity in Obese Youth with Impaired Glucose Tolerance. Diabetes 2017, 66, 3085–3090. [Google Scholar] [CrossRef] [Green Version]
- Xia, M.F.; Bian, H.; Gao, X. NAFLD and Diabetes: Two Sides of the Same Coin? Rationale for Gene-Based Personalized NAFLD Treatment. Front. Pharmacol. 2019, 10, 877. [Google Scholar] [CrossRef] [Green Version]
- Fabbrini, E.; Sullivan, S.; Klein, S. Obesity and nonalcoholic fatty liver disease: Biochemical, metabolic, and clinical implications. Hepatology 2010, 51, 679–689. [Google Scholar] [CrossRef] [Green Version]
- Gaggini, M.; Morelli, M.; Buzzigoli, E.; DeFronzo, R.A.; Bugianesi, E.; Gastaldelli, A. Non-alcoholic fatty liver disease (NAFLD) and its connection with insulin resistance, dyslipidemia, atherosclerosis and coronary heart disease. Nutrients 2013, 5, 1544–1560. [Google Scholar] [CrossRef]
- Tomah, S.; Alkhouri, N.; Hamdy, O. Nonalcoholic fatty liver disease and type 2 diabetes: Where do Diabetologists stand? Clin. Diabetes Endocrinol. 2020, 6, 9. [Google Scholar] [CrossRef]
- Suryawanshi, N.P.; Bhutey, A.K.; Nagdeote, A.N.; Jadhav, A.A.; Manoorkar, G.S. Study of lipid peroxide and lipid profile in diabetes mellitus. Indian J. Clin. Biochem. 2006, 21, 126–130. [Google Scholar] [CrossRef] [Green Version]
- Deprince, A.; Haas, J.T.; Staels, B. Dysregulated lipid metabolism links NAFLD to cardiovascular disease. Mol. Metab. 2020, 42, 101092. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.G.; Robson, S.C.; Yao, Z. Lipoprotein metabolism in nonalcoholic fatty liver disease. J. Biomed. Res 2013, 27, 1–13. [Google Scholar] [CrossRef] [PubMed]
- DeFilippis, A.P.; Blaha, M.J.; Martin, S.S.; Reed, R.M.; Jones, S.R.; Nasir, K.; Blumenthal, R.S.; Budoff, M.J. Nonalcoholic fatty liver disease and serum lipoproteins: The Multi-Ethnic Study of Atherosclerosis. Atherosclerosis 2013, 227, 429–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, L.; Shao, X.; Qiu, C.; Shao, X.; Wang, X.; Niu, R.; Wang, Y. Hepatic steatosis is associated with abnormal hepatic enzymes, visceral adiposity, altered myocardial glucose uptake measured by (18)F-FDG PET/CT. BMC Endocr. Disord. 2020, 20, 75. [Google Scholar] [CrossRef] [PubMed]
- Chao, H.W.; Chao, S.W.; Lin, H.; Ku, H.C.; Cheng, C.F. Homeostasis of Glucose and Lipid in Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2019, 20, 298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Zheng, J.; Zhang, S.; Wang, B.; Wu, C.; Guo, X. Advances in the Involvement of Gut Microbiota in Pathophysiology of NAFLD. Front. Med. 2020, 7, 361. [Google Scholar] [CrossRef]
- Luci, C.; Bourinet, M.; Leclère, P.S.; Anty, R.; Gual, P. Chronic Inflammation in Non-Alcoholic Steatohepatitis: Molecular Mechanisms and Therapeutic Strategies. Front. Endocrinol. 2020, 11, 597648. [Google Scholar] [CrossRef]
- Papa, A.; Danese, S.; Urgesi, R.; Grillo, A.; Guglielmo, S.; Roberto, I.; Bonizzi, M.; Guidi, L.; De Vitis, I.; Santoliquido, A.; et al. Early atherosclerosis in patients with inflammatory bowel disease. Eur. Rev. Med. Pharmacol. Sci. 2006, 10, 7–11. [Google Scholar]
- Pan, X.; Kaminga, A.C.; Liu, A.; Wen, S.W.; Luo, M.; Luo, J. Gut Microbiota, Glucose, Lipid, and Water-Electrolyte Metabolism in Children with Nonalcoholic Fatty Liver Disease. Front. Cell Infect. Microbiol. 2021, 11, 683743. [Google Scholar] [CrossRef]
- Kessoku, T.; Kobayashi, T.; Imajo, K.; Tanaka, K.; Yamamoto, A.; Takahashi, K.; Kasai, Y.; Ozaki, A.; Iwaki, M.; Nogami, A.; et al. Endotoxins and Non-Alcoholic Fatty Liver Disease. Front. Endocrinol. 2021, 12, 770986. [Google Scholar] [CrossRef]
- Miura, K.; Ohnishi, H. Role of gut microbiota and Toll-like receptors in nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 7381–7391. [Google Scholar] [CrossRef] [PubMed]
- Nesse, W.; Dijkstra, P.U.; Abbas, F.; Spijkervet, F.K.; Stijger, A.; Tromp, J.A.; van Dijk, J.L.; Vissink, A. Increased prevalence of cardiovascular and autoimmune diseases in periodontitis patients: A cross-sectional study. J. Periodontol. 2010, 81, 1622–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plutzky, J.; Liao, K.P. Lipids in RA: Is Less Not Necessarily More? Curr. Rheumatol. Rep. 2018, 20, 8. [Google Scholar] [CrossRef] [PubMed]
- González-Gay, M.A.; González-Juanatey, C. Inflammation and lipid profile in rheumatoid arthritis: Bridging an apparent paradox. Ann. Rheum. Dis. 2014, 73, 1281–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mok, C.C.; Lau, C.S. Pathogenesis of systemic lupus erythematosus. J. Clin. Pathol. 2003, 56, 481–490. [Google Scholar] [CrossRef] [Green Version]
- Asanuma, Y.; Chung, C.P.; Oeser, A.; Shintani, A.; Stanley, E.; Raggi, P.; Stein, C.M. Increased concentration of proatherogenic inflammatory cytokines in systemic lupus erythematosus: Relationship to cardiovascular risk factors. J. Rheumatol. 2006, 33, 539–545. [Google Scholar]
- Szabó, M.Z.; Szodoray, P.; Kiss, E. Dyslipidemia in systemic lupus erythematosus. Immunol. Res. 2017, 65, 543–550. [Google Scholar] [CrossRef]
- Ward, M.M. Premature morbidity from cardiovascular and cerebrovascular diseases in women with systemic lupus erythematosus. Arthritis Rheum. 1999, 42, 338–346. [Google Scholar] [CrossRef]
- Asanuma, Y.; Oeser, A.; Shintani, A.K.; Turner, E.; Olsen, N.; Fazio, S.; Linton, M.F.; Raggi, P.; Stein, C.M. Premature coronary-artery atherosclerosis in systemic lupus erythematosus. N. Engl. J. Med. 2003, 349, 2407–2415. [Google Scholar] [CrossRef]
- Coates, L.C.; FitzGerald, O.; Helliwell, P.S.; Paul, C. Psoriasis, psoriatic arthritis, and rheumatoid arthritis: Is all inflammation the same? Semin. Arthritis Rheum. 2016, 46, 291–304. [Google Scholar] [CrossRef] [Green Version]
- Akkara Veetil, B.M.; Matteson, E.L.; Maradit-Kremers, H.; McEvoy, M.T.; Crowson, C.S. Trends in lipid profiles in patients with psoriasis: A population-based analysis. BMC Dermatol. 2012, 12, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhyani, M.; Ehsani, A.H.; Robati, R.M.; Robati, A.M. The lipid profile in psoriasis: A controlled study. J. Eur. Acad. Dermatol. Venereol. 2007, 21, 1330–1332. [Google Scholar] [CrossRef] [PubMed]
- Miller, I.M.; Ellervik, C.; Yazdanyar, S.; Jemec, G.B. Meta-analysis of psoriasis, cardiovascular disease, and associated risk factors. J. Am. Acad. Dermatol. 2013, 69, 1014–1024. [Google Scholar] [CrossRef] [PubMed]
- Masson, W.; Lobo, M.; Molinero, G. Psoriasis and Cardiovascular Risk: A Comprehensive Review. Adv. Ther. 2020, 37, 2017–2033. [Google Scholar] [CrossRef]
- Matsushita, K.; van der Velde, M.; Astor, B.C.; Woodward, M.; Levey, A.S.; de Jong, P.E.; Coresh, J.; Gansevoort, R.T. Association of estimated glomerular filtration rate and albuminuria with all-cause and cardiovascular mortality in general population cohorts: A collaborative meta-analysis. Lancet 2010, 375, 2073–2081. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Bogle, R.; Banerjee, D. Why do young people with chronic kidney disease die early? World J. Nephrol. 2014, 3, 143–155. [Google Scholar] [CrossRef]
- Valdivielso, J.M.; Rodríguez-Puyol, D.; Pascual, J.; Barrios, C.; Bermúdez-López, M.; Sánchez-Niño, M.D.; Pérez-Fernández, M.; Ortiz, A. Atherosclerosis in Chronic Kidney Disease: More, Less, or Just Different? Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1938–1966. [Google Scholar] [CrossRef]
- Gansevoort, R.T.; Correa-Rotter, R.; Hemmelgarn, B.R.; Jafar, T.H.; Heerspink, H.J.; Mann, J.F.; Matsushita, K.; Wen, C.P. Chronic kidney disease and cardiovascular risk: Epidemiology, mechanisms, and prevention. Lancet 2013, 382, 339–352. [Google Scholar] [CrossRef]
- Linton, M.F.; Yancey, P.G.; Davies, S.S.; Jerome, W.G.; Linton, E.F.; Song, W.L.; Doran, A.C.; Vickers, K.C. The Role of Lipids and Lipoproteins in Atherosclerosis. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Hershman, J.M., Hofland, J., Kalra, S., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Gisterå, A.; Hansson, G.K. The immunology of atherosclerosis. Nat. Rev. Nephrol. 2017, 13, 368–380. [Google Scholar] [CrossRef]
- Bergheanu, S.C.; Bodde, M.C.; Jukema, J.W. Pathophysiology and treatment of atherosclerosis: Current view and future perspective on lipoprotein modification treatment. Neth. Heart J. 2017, 25, 231–242. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Li, Y.; Li, Y.; Ren, X.; Zhang, X.; Hu, D.; Gao, Y.; Xing, Y.; Shang, H. Oxidative Stress-Mediated Atherosclerosis: Mechanisms and Therapies. Front. Physiol. 2017, 8, 600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cyrus, T.; Witztum, J.L.; Rader, D.J.; Tangirala, R.; Fazio, S.; Linton, M.F.; Funk, C.D. Disruption of the 12/15-lipoxygenase gene diminishes atherosclerosis in apo E-deficient mice. J. Clin. Investig. 1999, 103, 1597–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robbins, C.S.; Hilgendorf, I.; Weber, G.F.; Theurl, I.; Iwamoto, Y.; Figueiredo, J.L.; Gorbatov, R.; Sukhova, G.K.; Gerhardt, L.M.; Smyth, D.; et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat. Med. 2013, 19, 1166–1172. [Google Scholar] [CrossRef] [PubMed]
- Riwanto, M.; Rohrer, L.; Roschitzki, B.; Besler, C.; Mocharla, P.; Mueller, M.; Perisa, D.; Heinrich, K.; Altwegg, L.; von Eckardstein, A.; et al. Altered activation of endothelial anti- and proapoptotic pathways by high-density lipoprotein from patients with coronary artery disease: Role of high-density lipoprotein-proteome remodeling. Circulation 2013, 127, 891–904. [Google Scholar] [CrossRef]
- Arida, A.; Protogerou, A.D.; Kitas, G.D.; Sfikakis, P.P. Systemic Inflammatory Response and Atherosclerosis: The Paradigm of Chronic Inflammatory Rheumatic Diseases. Int. J. Mol. Sci. 2018, 19, 1890. [Google Scholar] [CrossRef] [Green Version]
- Greenow, K.; Pearce, N.J.; Ramji, D.P. The key role of apolipoprotein E in atherosclerosis. J. Mol. Med. 2005, 83, 329–342. [Google Scholar] [CrossRef]
- Mineo, C.; Shaul, P.W. Regulation of signal transduction by HDL. J. Lipid Res. 2013, 54, 2315–2324. [Google Scholar] [CrossRef] [Green Version]
- Mineo, C.; Shaul, P.W. Novel biological functions of high-density lipoprotein cholesterol. Circ. Res. 2012, 111, 1079–1090. [Google Scholar] [CrossRef] [Green Version]
- Marsche, G.; Frank, S.; Raynes, J.G.; Kozarsky, K.F.; Sattler, W.; Malle, E. The lipidation status of acute-phase protein serum amyloid A determines cholesterol mobilization via scavenger receptor class B, type I. Biochem. J. 2007, 402, 117–124. [Google Scholar] [CrossRef] [Green Version]
- Yvan-Charvet, L.; Welch, C.; Pagler, T.A.; Ranalletta, M.; Lamkanfi, M.; Han, S.; Ishibashi, M.; Li, R.; Wang, N.; Tall, A.R. Increased inflammatory gene expression in ABC transporter-deficient macrophages: Free cholesterol accumulation, increased signaling via toll-like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation 2008, 118, 1837–1847. [Google Scholar] [CrossRef] [Green Version]
- Van Lenten, B.J.; Navab, M.; Shih, D.; Fogelman, A.M.; Lusis, A.J. The role of high-density lipoproteins in oxidation and inflammation. Trends Cardiovasc. Med. 2001, 11, 155–161. [Google Scholar] [CrossRef]
- Litvinov, D.; Mahini, H.; Garelnabi, M. Antioxidant and anti-inflammatory role of paraoxonase 1: Implication in arteriosclerosis diseases. N. Am. J. Med. Sci. 2012, 4, 523–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackness, M.I.; Arrol, S.; Abbott, C.; Durrington, P.N. Protection of low-density lipoprotein against oxidative modification by high-density lipoprotein associated paraoxonase. Atherosclerosis 1993, 104, 129–135. [Google Scholar] [CrossRef]
- van den Oever, I.A.; Raterman, H.G.; Nurmohamed, M.T.; Simsek, S. Endothelial dysfunction, inflammation, and apoptosis in diabetes mellitus. Mediat. Inflamm. 2010, 2010, 792393. [Google Scholar] [CrossRef] [PubMed]
- Yuhanna, I.S.; Zhu, Y.; Cox, B.E.; Hahner, L.D.; Osborne-Lawrence, S.; Lu, P.; Marcel, Y.L.; Anderson, R.G.; Mendelsohn, M.E.; Hobbs, H.H.; et al. High-density lipoprotein binding to scavenger receptor-BI activates endothelial nitric oxide synthase. Nat. Med. 2001, 7, 853–857. [Google Scholar] [CrossRef]
- Mineo, C.; Shaul, P.W. Role of high-density lipoprotein and scavenger receptor B type I in the promotion of endothelial repair. Trends Cardiovasc. Med. 2007, 17, 156–161. [Google Scholar] [CrossRef]
- Li, X.A.; Guo, L.; Dressman, J.L.; Asmis, R.; Smart, E.J. A novel ligand-independent apoptotic pathway induced by scavenger receptor class B, type I and suppressed by endothelial nitric-oxide synthase and high density lipoprotein. J. Biol. Chem. 2005, 280, 19087–19096. [Google Scholar] [CrossRef] [Green Version]
- Seetharam, D.; Mineo, C.; Gormley, A.K.; Gibson, L.L.; Vongpatanasin, W.; Chambliss, K.L.; Hahner, L.D.; Cummings, M.L.; Kitchens, R.L.; Marcel, Y.L.; et al. High-density lipoprotein promotes endothelial cell migration and reendothelialization via scavenger receptor-B type I. Circ. Res. 2006, 98, 63–72. [Google Scholar] [CrossRef] [Green Version]
- Terasaka, N.; Wang, N.; Yvan-Charvet, L.; Tall, A.R. High-density lipoprotein protects macrophages from oxidized low-density lipoprotein-induced apoptosis by promoting efflux of 7-ketocholesterol via ABCG1. Proc. Natl. Acad. Sci. USA 2007, 104, 15093–15098. [Google Scholar] [CrossRef] [Green Version]
- van der Stoep, M.; Korporaal, S.J.; Van Eck, M. High-density lipoprotein as a modulator of platelet and coagulation responses. Cardiovasc. Res. 2014, 103, 362–371. [Google Scholar] [CrossRef] [Green Version]
- Kontush, A.; Lindahl, M.; Lhomme, M.; Calabresi, L.; Chapman, M.J.; Davidson, W.S. Structure of HDL: Particle subclasses and molecular components. Handb. Exp. Pharmacol. 2015, 224, 3–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, S. HDL Proteome Watch. Available online: https://homepages.uc.edu/~davidswm/HDLproteome.html (accessed on 1 July 2022).
- Davidson, S. LDL Proteome Watch. Available online: https://homepages.uc.edu/~davidswm/LDLproteome.html (accessed on 1 July 2022).
- Davidson, W.S.; Shah, A.S.; Sexmith, H.; Gordon, S.M. The HDL Proteome Watch: Compilation of studies leads to new insights on HDL function. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2022, 1867, 159072. [Google Scholar] [CrossRef] [PubMed]
- Jorge, I.; Burillo, E.; Mesa, R.; Baila-Rueda, L.; Moreno, M.; Trevisan-Herraz, M.; Silla-Castro, J.C.; Camafeita, E.; Ortega-Muñoz, M.; Bonzon-Kulichenko, E.; et al. The human HDL proteome displays high inter-individual variability and is altered dynamically in response to angioplasty-induced atheroma plaque rupture. J. Proteom. 2014, 106, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Vaisar, T.; Pennathur, S.; Green, P.S.; Gharib, S.A.; Hoofnagle, A.N.; Cheung, M.C.; Byun, J.; Vuletic, S.; Kassim, S.; Singh, P.; et al. Shotgun proteomics implicates protease inhibition and complement activation in the antiinflammatory properties of HDL. J. Clin. Investig. 2007, 117, 746–756. [Google Scholar] [CrossRef]
- Ludovico, I.D.; Gisonno, R.A.; Gonzalez, M.C.; Garda, H.A.; Ramella, N.A.; Tricerri, M.A. Understanding the role of apolipoproteinA-I in atherosclerosis. Post-translational modifications synergize dysfunction? Biochim. Biophys. Acta-Gen. Subj. 2021, 1865, 129732. [Google Scholar] [CrossRef]
- Mei, X.; Atkinson, D. Lipid-free Apolipoprotein A-I Structure: Insights into HDL Formation and Atherosclerosis Development. Arch. Med. Res. 2015, 46, 351–360. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.M.; Handa, P.; Tateya, S.; Schwartz, J.; Tang, C.; Mitra, P.; Oram, J.F.; Chait, A.; Kim, F. Apolipoprotein A-I attenuates palmitate-mediated NF-κB activation by reducing Toll-like receptor-4 recruitment into lipid rafts. PLoS ONE 2012, 7, e33917. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.; Di Bartolo, B.A.; Nakhla, S.; Heather, A.K.; Mitchell, T.W.; Jessup, W.; Celermajer, D.S.; Barter, P.J.; Rye, K.A. Anti-inflammatory effects of apolipoprotein A-I in the rabbit. Atherosclerosis 2010, 212, 392–397. [Google Scholar] [CrossRef]
- Murphy, A.J.; Woollard, K.J.; Suhartoyo, A.; Stirzaker, R.A.; Shaw, J.; Sviridov, D.; Chin-Dusting, J.P. Neutrophil activation is attenuated by high-density lipoprotein and apolipoprotein A-I in in vitro and in vivo models of inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1333–1341. [Google Scholar] [CrossRef] [Green Version]
- Mineo, C.; Shaul, P.W. HDL stimulation of endothelial nitric oxide synthase: A novel mechanism of HDL action. Trends Cardiovasc. Med. 2003, 13, 226–231. [Google Scholar] [CrossRef]
- Li, Y.; Dong, J.B.; Wu, M.P. Human ApoA-I overexpression diminishes LPS-induced systemic inflammation and multiple organ damage in mice. Eur. J. Pharmacol. 2008, 590, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Juonala, M.; Viikari, J.S.; Kähönen, M.; Solakivi, T.; Helenius, H.; Jula, A.; Marniemi, J.; Taittonen, L.; Laitinen, T.; Nikkari, T.; et al. Childhood levels of serum apolipoproteins B and A-I predict carotid intima-media thickness and brachial endothelial function in adulthood: The cardiovascular risk in young Finns study. J. Am. Coll. Cardiol. 2008, 52, 293–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charles-Schoeman, C.; Fleischmann, R.; Davignon, J.; Schwartz, H.; Turner, S.M.; Beysen, C.; Milad, M.; Hellerstein, M.K.; Luo, Z.; Kaplan, I.V.; et al. Potential mechanisms leading to the abnormal lipid profile in patients with rheumatoid arthritis versus healthy volunteers and reversal by tofacitinib. Arthritis Rheumatol. 2015, 67, 616–625. [Google Scholar] [CrossRef] [PubMed]
- Gkolfinopoulou, C.; Soukou, F.; Dafnis, I.; Kellici, T.F.; Sanoudou, D.; Mavromoustakos, T.; Stratikos, E.; Chroni, A. Structure-function analysis of naturally occurring apolipoprotein A-I L144R, A164S and L178P mutants provides insight on their role on HDL levels and cardiovascular risk. Cell Mol. Life Sci. 2021, 78, 1523–1544. [Google Scholar] [CrossRef] [PubMed]
- Haase, C.L.; Frikke-Schmidt, R.; Nordestgaard, B.G.; Kateifides, A.K.; Kardassis, D.; Nielsen, L.B.; Andersen, C.B.; Køber, L.; Johnsen, A.H.; Grande, P.; et al. Mutation in APOA1 predicts increased risk of ischaemic heart disease and total mortality without low HDL cholesterol levels. J. Intern. Med. 2011, 270, 136–146. [Google Scholar] [CrossRef]
- Yang, M.; Liu, Y.; Dai, J.; Li, L.; Ding, X.; Xu, Z.; Mori, M.; Miyahara, H.; Sawashita, J.; Higuchi, K. Apolipoprotein A-II induces acute-phase response associated AA amyloidosis in mice through conformational changes of plasma lipoprotein structure. Sci. Rep. 2018, 8, 5620. [Google Scholar] [CrossRef]
- Hedrick, C.C.; Castellani, L.W.; Wong, H.; Lusis, A.J. In vivo interactions of apoA-II, apoA-I, and hepatic lipase contributing to HDL structure and antiatherogenic functions. J. Lipid Res. 2001, 42, 563–570. [Google Scholar] [CrossRef]
- Tailleux, A.; Duriez, P.; Fruchart, J.C.; Clavey, V. Apolipoprotein A-II, HDL metabolism and atherosclerosis. Atherosclerosis 2002, 164, 1–13. [Google Scholar] [CrossRef]
- Koike, T.; Koike, Y.; Yang, D.; Guo, Y.; Rom, O.; Song, J.; Xu, J.; Chen, Y.; Wang, Y.; Zhu, T.; et al. Human apolipoprotein A-II reduces atherosclerosis in knock-in rabbits. Atherosclerosis 2021, 316, 32–40. [Google Scholar] [CrossRef]
- Zhong, S.; Goldberg, I.J.; Bruce, C.; Rubin, E.; Breslow, J.L.; Tall, A. Human ApoA-II inhibits the hydrolysis of HDL triglyceride and the decrease of HDL size induced by hypertriglyceridemia and cholesteryl ester transfer protein in transgenic mice. J. Clin. Investig. 1994, 94, 2457–2467. [Google Scholar] [CrossRef]
- Castellani, L.W.; Navab, M.; Van Lenten, B.J.; Hedrick, C.C.; Hama, S.Y.; Goto, A.M.; Fogelman, A.M.; Lusis, A.J. Overexpression of apolipoprotein AII in transgenic mice converts high density lipoproteins to proinflammatory particles. J. Clin. Investig. 1997, 100, 464–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carnuta, M.G.; Stancu, C.S.; Toma, L.; Sanda, G.M.; Niculescu, L.S.; Deleanu, M.; Popescu, A.C.; Popescu, M.R.; Vlad, A.; Dimulescu, D.R.; et al. Dysfunctional high-density lipoproteins have distinct composition, diminished anti-inflammatory potential and discriminate acute coronary syndrome from stable coronary artery disease patients. Sci. Rep. 2017, 7, 7295. [Google Scholar] [CrossRef] [Green Version]
- Perelas, A.; Safarika, V.; Vlachos, I.S.; Tzanetakou, I.; Korou, L.M.; Konstantopoulos, P.; Doulamis, I.; Ioannidis, I.; Kornezos, I.; Gargas, D.; et al. Correlation between mesenteric fat thickness and serum apolipoproteins in patients with peripheral arterial occlusive disease. Lipids Health Dis. 2012, 11, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, J.; Ko, C.W.; Tso, P.; Bhargava, A. Apolipoprotein A-IV: A Multifunctional Protein Involved in Protection against Atherosclerosis and Diabetes. Cells 2019, 8, 319. [Google Scholar] [CrossRef] [PubMed]
- Kohan, A.B.; Wang, F.; Lo, C.M.; Liu, M.; Tso, P. ApoA-IV: Current and emerging roles in intestinal lipid metabolism, glucose homeostasis, and satiety. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G472–G481. [Google Scholar] [CrossRef] [Green Version]
- Simon, T.; Cook, V.R.; Rao, A.; Weinberg, R.B. Impact of murine intestinal apolipoprotein A-IV expression on regional lipid absorption, gene expression, and growth. J. Lipid Res. 2011, 52, 1984–1994. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Wang, L.; Zhang, Z.; Feng, L.; Song, X.; Wu, J. Apolipoprotein A-IV involves in glucose and lipid metabolism of rat. Nutr. Metab. 2019, 16, 41. [Google Scholar] [CrossRef]
- Vowinkel, T.; Mori, M.; Krieglstein, C.F.; Russell, J.; Saijo, F.; Bharwani, S.; Turnage, R.H.; Davidson, W.S.; Tso, P.; Granger, D.N.; et al. Apolipoprotein A-IV inhibits experimental colitis. J. Clin. Investig. 2004, 114, 260–269. [Google Scholar] [CrossRef] [Green Version]
- Shimabukuro Okuda, L.; Tallada Iborra, R.; Ramos Pinto, P.; Fabres Machado, U.; Corrêa-Giannella, M.L.; Pickford, R.; Woods, T.; Brimble, M.A.; Rye, K.A.; Passarelli, M. Advanced Glycated apoA-IV Loses Its Ability to Prevent the LPS-Induced Reduction in Cholesterol Efflux-Related Gene Expression in Macrophages. Mediat. Inflamm. 2020, 2020, 6515401. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.R.; Wang, Y.; Adili, R.; Ju, L.; Spring, C.M.; Jin, J.W.; Yang, H.; Neves, M.A.D.; Chen, P.; Yang, Y.; et al. Apolipoprotein A-IV binds αIIbβ3 integrin and inhibits thrombosis. Nat. Commun. 2018, 9, 3608. [Google Scholar] [CrossRef] [Green Version]
- Remaley, A.T.; Stonik, J.A.; Demosky, S.J.; Neufeld, E.B.; Bocharov, A.V.; Vishnyakova, T.G.; Eggerman, T.L.; Patterson, A.P.; Duverger, N.J.; Santamarina-Fojo, S.; et al. Apolipoprotein specificity for lipid efflux by the human ABCAI transporter. Biochem. Biophys. Res. Commun. 2001, 280, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Steinmetz, A.; Utermann, G. Activation of lecithin: Cholesterol acyltransferase by human apolipoprotein A-IV. J. Biol. Chem. 1985, 260, 2258–2264. [Google Scholar] [CrossRef]
- Wong, W.M.; Gerry, A.B.; Putt, W.; Roberts, J.L.; Weinberg, R.B.; Humphries, S.E.; Leake, D.S.; Talmud, P.J. Common variants of apolipoprotein A-IV differ in their ability to inhibit low density lipoprotein oxidation. Atherosclerosis 2007, 192, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Recalde, D.; Ostos, M.A.; Badell, E.; Garcia-Otin, A.L.; Pidoux, J.; Castro, G.; Zakin, M.M.; Scott-Algara, D. Human apolipoprotein A-IV reduces secretion of proinflammatory cytokines and atherosclerotic effects of a chronic infection mimicked by lipopolysaccharide. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 756–761. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Shen, Y.; Li, Q.R.; Ding, F.H.; Wang, X.Q.; Liu, H.J.; Yan, X.X.; Wang, L.J.; Yang, K.; Wang, H.B.; et al. Glycated Apolipoprotein A-IV Induces Atherogenesis in Patients with CAD in Type 2 Diabetes. J. Am. Coll. Cardiol. 2017, 70, 2006–2019. [Google Scholar] [CrossRef]
- Getz, G.S.; Reardon, C.A. Apoprotein E as a lipid transport and signaling protein in the blood, liver, and artery wall. J. Lipid Res. 2009, 50, S156–S161. [Google Scholar] [CrossRef] [Green Version]
- Jofre-Monseny, L.; Minihane, A.M.; Rimbach, G. Impact of apoE genotype on oxidative stress, inflammation and disease risk. Mol. Nutr. Food Res. 2008, 52, 131–145. [Google Scholar] [CrossRef]
- Baitsch, D.; Bock, H.H.; Engel, T.; Telgmann, R.; Müller-Tidow, C.; Varga, G.; Bot, M.; Herz, J.; Robenek, H.; von Eckardstein, A.; et al. Apolipoprotein E induces antiinflammatory phenotype in macrophages. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1160–1168. [Google Scholar] [CrossRef] [Green Version]
- Vedhachalam, C.; Narayanaswami, V.; Neto, N.; Forte, T.M.; Phillips, M.C.; Lund-Katz, S.; Bielicki, J.K. The C-terminal lipid-binding domain of apolipoprotein E is a highly efficient mediator of ABCA1-dependent cholesterol efflux that promotes the assembly of high-density lipoproteins. Biochemistry 2007, 46, 2583–2593. [Google Scholar] [CrossRef]
- Tudorache, I.F.; Trusca, V.G.; Gafencu, A.V. Apolipoprotein E—A Multifunctional Protein with Implications in Various Pathologies as a Result of Its Structural Features. Comput. Struct. Biotechnol. J. 2017, 15, 359–365. [Google Scholar] [CrossRef]
- Nolte, R.T.; Atkinson, D. Conformational analysis of apolipoprotein A-I and E-3 based on primary sequence and circular dichroism. Biophys. J. 1992, 63, 1221–1239. [Google Scholar] [CrossRef] [Green Version]
- Filou, S.; Lhomme, M.; Karavia, E.A.; Kalogeropoulou, C.; Theodoropoulos, V.; Zvintzou, E.; Sakellaropoulos, G.C.; Petropoulou, P.I.; Constantinou, C.; Kontush, A.; et al. Distinct Roles of Apolipoproteins A1 and E in the Modulation of High-Density Lipoprotein Composition and Function. Biochemistry 2016, 55, 3752–3762. [Google Scholar] [CrossRef] [PubMed]
- Kypreos, K.E.; Zannis, V.I. Pathway of biogenesis of apolipoprotein E-containing HDL in vivo with the participation of ABCA1 and LCAT. Biochem. J. 2007, 403, 359–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hafiane, A.; Bielicki, J.K.; Johansson, J.O.; Genest, J. Apolipoprotein E derived HDL mimetic peptide ATI-5261 promotes nascent HDL formation and reverse cholesterol transport in vitro. Biochim. Biophys. Acta 2014, 1842, 1498–1512. [Google Scholar] [CrossRef] [PubMed]
- Mahley, R.W.; Huang, Y.; Weisgraber, K.H. Putting cholesterol in its place: apoE and reverse cholesterol transport. J. Clin. Investig. 2006, 116, 1226–1229. [Google Scholar] [CrossRef]
- Yancey, P.G.; Yu, H.; Linton, M.F.; Fazio, S. A pathway-dependent on apoE, ApoAI, and ABCA1 determines formation of buoyant high-density lipoprotein by macrophage foam cells. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1123–1131. [Google Scholar] [CrossRef] [Green Version]
- Rigotti, A.; Miettinen, H.E.; Krieger, M. The role of the high-density lipoprotein receptor SR-BI in the lipid metabolism of endocrine and other tissues. Endocr. Rev. 2003, 24, 357–387. [Google Scholar] [CrossRef] [Green Version]
- Morton, A.M.; Koch, M.; Mendivil, C.O.; Furtado, J.D.; Tjønneland, A.; Overvad, K.; Wang, L.; Jensen, M.K.; Sacks, F.M. Apolipoproteins E and CIII interact to regulate HDL metabolism and coronary heart disease risk. JCI Insight 2018, 3, e98045. [Google Scholar] [CrossRef] [Green Version]
- Arai, T.; Rinninger, F.; Varban, L.; Fairchild-Huntress, V.; Liang, C.P.; Chen, W.; Seo, T.; Deckelbaum, R.; Huszar, D.; Tall, A.R. Decreased selective uptake of high density lipoprotein cholesteryl esters in apolipoprotein E knock-out mice. Proc. Natl. Acad. Sci. USA 1999, 96, 12050–12055. [Google Scholar] [CrossRef] [Green Version]
- Valanti, E.K.; Dalakoura-Karagkouni, K.; Sanoudou, D. Current and Emerging Reconstituted HDL-apoA-I and HDL-apoE Approaches to Treat Atherosclerosis. J. Pers. Med. 2018, 8, 34. [Google Scholar] [CrossRef] [Green Version]
- Gaidukov, L.; Viji, R.I.; Yacobson, S.; Rosenblat, M.; Aviram, M.; Tawfik, D.S. ApoE induces serum paraoxonase PON1 activity and stability similar to ApoA-I. Biochemistry 2010, 49, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Fan, P.; Liu, H.; Wang, Y.; Zhang, F.; Bai, H. Apolipoprotein E-containing HDL-associated platelet-activating factor acetylhydrolase activities and malondialdehyde concentrations in patients with PCOS. Reprod. Biomed. Online 2012, 24, 197–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, K.; Bruckdorfer, K.R.; Hutton, R.A.; Owen, J.S. Binding of apoE-rich high density lipoprotein particles by saturable sites on human blood platelets inhibits agonist-induced platelet aggregation. J. Lipid Res. 1989, 30, 831–840. [Google Scholar] [CrossRef]
- Riddell, D.R.; Graham, A.; Owen, J.S. Apolipoprotein E inhibits platelet aggregation through the L-arginine:nitric oxide pathway. Implications for vascular disease. J. Biol. Chem. 1997, 272, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Kothapalli, D.; Fuki, I.; Ali, K.; Stewart, S.A.; Zhao, L.; Yahil, R.; Kwiatkowski, D.; Hawthorne, E.A.; FitzGerald, G.A.; Phillips, M.C.; et al. Antimitogenic effects of HDL and APOE mediated by Cox-2-dependent IP activation. J. Clin. Investig. 2004, 113, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Kothapalli, D.; Liu, S.L.; Bae, Y.H.; Monslow, J.; Xu, T.; Hawthorne, E.A.; Byfield, F.J.; Castagnino, P.; Rao, S.; Rader, D.J.; et al. Cardiovascular protection by ApoE and ApoE-HDL linked to suppression of ECM gene expression and arterial stiffening. Cell Rep. 2012, 2, 1259–1271. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, P.J.; Alborn, W.E.; Sloan, J.H.; Ulmer, M.; Boodhoo, A.; Knierman, M.D.; Schultze, A.E.; Konrad, R.J. The novel apolipoprotein A5 is present in human serum, is associated with VLDL, HDL, and chylomicrons, and circulates at very low concentrations compared with other apolipoproteins. Clin. Chem. 2005, 51, 351–359. [Google Scholar] [CrossRef] [Green Version]
- Priore Oliva, C.; Carubbi, F.; Schaap, F.G.; Bertolini, S.; Calandra, S. Hypertriglyceridaemia and low plasma HDL in a patient with apolipoprotein A-V deficiency due to a novel mutation in the APOA5 gene. J. Intern. Med. 2008, 263, 450–458. [Google Scholar] [CrossRef]
- Shu, X.; Nelbach, L.; Weinstein, M.M.; Burgess, B.L.; Beckstead, J.A.; Young, S.G.; Ryan, R.O.; Forte, T.M. Intravenous injection of apolipoprotein A-V reconstituted high-density lipoprotein decreases hypertriglyceridemia in apoav−/− mice and requires glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2504–2509. [Google Scholar] [CrossRef] [Green Version]
- Qu, S.; Perdomo, G.; Su, D.; D’Souza, F.M.; Shachter, N.S.; Dong, H.H. Effects of apoA-V on HDL and VLDL metabolism in APOC3 transgenic mice. J. Lipid Res. 2007, 48, 1476–1487. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Kim, D.H.; Slusher, S.; Fan, Y.; Forte, T.; Dong, H. Abstract 16: ApoA5 Reduces Atherosclerosis in LDL Receptor-Deficient Mice. Arterioscler. Thromb. Vasc. Biol. 2012, 32, A16. [Google Scholar] [CrossRef]
- Khovidhunkit, W.; Duchateau, P.N.; Medzihradszky, K.F.; Moser, A.H.; Naya-Vigne, J.; Shigenaga, J.K.; Kane, J.P.; Grunfeld, C.; Feingold, K.R. Apolipoproteins A-IV and A-V are acute-phase proteins in mouse HDL. Atherosclerosis 2004, 176, 37–44. [Google Scholar] [CrossRef]
- Becker, S.; Schomburg, L.; Renko, K.; Tölle, M.; van der Giet, M.; Tietge, U.J. Altered apolipoprotein A-V expression during the acute phase response is independent of plasma triglyceride levels in mice and humans. Biochem. Biophys. Res. Commun. 2006, 339, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Genoux, A.; Gervois, P.; Vu-Dac, N.; Pennacchio, L.; Rubin, E.; Fruchart-Najib, J.; Fruchart, J.C. 3P-0816∗ Apolipoprotein A5 is an inflammatory responsive gene down-regulated by tumor necrosis factorα and interleukin-1. Atheroscler. Suppl. 2003, 4, 241. [Google Scholar] [CrossRef]
- Yamazaki, A.; Ohkawa, R.; Yamagata, Y.; Horiuchi, Y.; Lai, S.J.; Kameda, T.; Ichimura, N.; Tohda, S.; Tozuka, M. Apolipoprotein C-II and C-III preferably transfer to both high-density lipoprotein (HDL)2 and the larger HDL3 from very low-density lipoprotein (VLDL). Biol. Chem. 2021, 402, 439–449. [Google Scholar] [CrossRef]
- Cohn, J.S.; Tremblay, M.; Batal, R.; Jacques, H.; Veilleux, L.; Rodriguez, C.; Bernier, L.; Mamer, O.; Davignon, J. Plasma kinetics of VLDL and HDL apoC-I in normolipidemic and hypertriglyceridemic subjects. J. Lipid Res. 2002, 43, 1680–1687. [Google Scholar] [CrossRef] [Green Version]
- Wolska, A.; Dunbar, R.L.; Freeman, L.A.; Ueda, M.; Amar, M.J.; Sviridov, D.O.; Remaley, A.T. Apolipoprotein C-II: New findings related to genetics, biochemistry, and role in triglyceride metabolism. Atherosclerosis 2017, 267, 49–60. [Google Scholar] [CrossRef]
- Kotite, L.; Zhang, L.H.; Yu, Z.; Burlingame, A.L.; Havel, R.J. Human apoC-IV: Isolation, characterization, and immunochemical quantification in plasma and plasma lipoproteins. J. Lipid Res. 2003, 44, 1387–1394. [Google Scholar] [CrossRef] [Green Version]
- Curry, M.D.; McConathy, W.J.; Fesmire, J.D.; Alaupovic, P. Quantitative determination of apolipoproteins C-I and C-II in human plasma by separate electroimmunoassays. Clin. Chem. 1981, 27, 543–548. [Google Scholar] [CrossRef]
- Gautier, T.; Masson, D.; de Barros, J.P.; Athias, A.; Gambert, P.; Aunis, D.; Metz-Boutigue, M.H.; Lagrost, L. Human apolipoprotein C-I accounts for the ability of plasma high density lipoproteins to inhibit the cholesteryl ester transfer protein activity. J. Biol. Chem. 2000, 275, 37504–37509. [Google Scholar] [CrossRef] [Green Version]
- Berbée, J.F.; van der Hoogt, C.C.; Sundararaman, D.; Havekes, L.M.; Rensen, P.C. Severe hypertriglyceridemia in human APOC1 transgenic mice is caused by apoC-I-induced inhibition of LPL. J. Lipid Res. 2005, 46, 297–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westerterp, M.; Berbée, J.F.; Delsing, D.J.; Jong, M.C.; Gijbels, M.J.; Dahlmans, V.E.; Offerman, E.H.; Romijn, J.A.; Havekes, L.M.; Rensen, P.C. Apolipoprotein C-I binds free fatty acids and reduces their intracellular esterification. J. Lipid Res. 2007, 48, 1353–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berbée, J.F.; van der Hoogt, C.C.; Kleemann, R.; Schippers, E.F.; Kitchens, R.L.; van Dissel, J.T.; Bakker-Woudenberg, I.A.; Havekes, L.M.; Rensen, P.C. Apolipoprotein CI stimulates the response to lipopolysaccharide and reduces mortality in gram-negative sepsis. FASEB J. 2006, 20, 2162–2164. [Google Scholar] [CrossRef] [PubMed]
- Sacks, F.M. The crucial roles of apolipoproteins E and C-III in apoB lipoprotein metabolism in normolipidemia and hypertriglyceridemia. Curr. Opin. Lipidol. 2015, 26, 56–63. [Google Scholar] [CrossRef]
- Zewinger, S.; Reiser, J.; Jankowski, V.; Alansary, D.; Hahm, E.; Triem, S.; Klug, M.; Schunk, S.J.; Schmit, D.; Kramann, R.; et al. Apolipoprotein C3 induces inflammation and organ damage by alternative inflammasome activation. Nat. Immunol. 2020, 21, 30–41. [Google Scholar] [CrossRef]
- Goldberg, I.J.; Scheraldi, C.A.; Yacoub, L.K.; Saxena, U.; Bisgaier, C.L. Lipoprotein ApoC-II activation of lipoprotein lipase. Modulation by apolipoprotein A-IV. J. Biol. Chem. 1990, 265, 4266–4272. [Google Scholar] [CrossRef]
- Tian, L.; Xu, Y.; Fu, M.; Jia, L.; Yang, Y. Influence of apolipoproteinCII concentrations on HDL subclass distribution. J. Atheroscler. Thromb. 2009, 16, 611–620. [Google Scholar] [CrossRef] [Green Version]
- Kei, A.A.; Filippatos, T.D.; Tsimihodimos, V.; Elisaf, M.S. A review of the role of apolipoprotein C-II in lipoprotein metabolism and cardiovascular disease. Metabolism 2012, 61, 906–921. [Google Scholar] [CrossRef]
- Jong, M.C.; Hofker, M.H.; Havekes, L.M. Role of ApoCs in lipoprotein metabolism: Functional differences between ApoC1, ApoC2, and ApoC3. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 472–484. [Google Scholar] [CrossRef] [Green Version]
- Roselli della Rovere, G.; Lapolla, A.; Sartore, G.; Rossetti, C.; Zambon, S.; Minicuci, N.; Crepaldi, G.; Fedele, D.; Manzato, E. Plasma lipoproteins, apoproteins and cardiovascular disease in type 2 diabetic patients. A nine-year follow-up study. Nutr. Metab. Cardiovasc. Dis. 2003, 13, 46–51. [Google Scholar] [CrossRef]
- Gerber, Y.; Goldbourt, U.; Cohen, H.; Harats, D. Association between serum apolipoprotein C(II) concentration and coronary heart disease. Prev. Med. 2002, 35, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Li, K.; Lieu, C.; Tong, S.; Kawai, S.; Fukutomi, T.; Zhou, Y.; Wands, J.; Li, J. Expression of apolipoprotein C-IV is regulated by Ku antigen/peroxisome proliferator-activated receptor gamma complex and correlates with liver steatosis. J. Hepatol. 2008, 49, 787–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Cheng, J.; Li, N.H.; Chen, Y.N.; Cai, M.Y.; Tang, S.S.; Huang, H.; Zhang, B.; Cen, J.M.; Yang, X.L.; et al. The association of APOC4 polymorphisms with premature coronary artery disease in a Chinese Han population. Lipids Health Dis. 2015, 14, 63. [Google Scholar] [CrossRef] [Green Version]
- Rull, A.; Ordóñez-Llanos, J.; Sánchez-Quesada, J.L. The role of LDL-bound apoJ in the development of atherosclerosis. Clin. Lipidol. 2015, 10, 321–328. [Google Scholar] [CrossRef]
- Navab, M.; Hama-Levy, S.; Van Lenten, B.J.; Fonarow, G.C.; Cardinez, C.J.; Castellani, L.W.; Brennan, M.L.; Lusis, A.J.; Fogelman, A.M.; La Du, B.N. Mildly oxidized LDL induces an increased apolipoprotein J/paraoxonase ratio. J. Clin. Investig. 1997, 99, 2005–2019. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Yoo, E.K.; Kim, J.Y.; Choi, Y.K.; Lee, H.J.; Kim, J.K.; Jeoung, N.H.; Lee, K.U.; Park, I.S.; Min, B.H.; et al. Protective role of clusterin/apolipoprotein J against neointimal hyperplasia via antiproliferative effect on vascular smooth muscle cells and cytoprotective effect on endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1558–1564. [Google Scholar] [CrossRef]
- Hardardóttir, I.; Kunitake, S.T.; Moser, A.H.; Doerrler, W.T.; Rapp, J.H.; Grünfeld, C.; Feingold, K.R. Endotoxin and cytokines increase hepatic messenger RNA levels and serum concentrations of apolipoprotein J (clusterin) in Syrian hamsters. J. Clin. Investig. 1994, 94, 1304–1309. [Google Scholar] [CrossRef]
- Yanni, A.E.; Agrogiannis, G.; Gkekas, C.; Perrea, D. Clusterin/Apolipoprotein J immunolocalization on carotid artery is affected by TNF-alpha, cigarette smoking and anti-platelet treatment. Lipids Health Dis. 2014, 13, 70. [Google Scholar] [CrossRef] [Green Version]
- Won, J.C.; Park, C.Y.; Oh, S.W.; Lee, E.S.; Youn, B.S.; Kim, M.S. Plasma clusterin (ApoJ) levels are associated with adiposity and systemic inflammation. PLoS ONE 2014, 9, e103351. [Google Scholar] [CrossRef] [Green Version]
- Fernández-de-Retana, S.; Cano-Sarabia, M.; Marazuela, P.; Sánchez-Quesada, J.L.; Garcia-Leon, A.; Montañola, A.; Montaner, J.; Maspoch, D.; Hernández-Guillamon, M. Characterization of ApoJ-reconstituted high-density lipoprotein (rHDL) nanodisc for the potential treatment of cerebral β-amyloidosis. Sci. Rep. 2017, 7, 14637. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.P.; Wei, S.L.; Chiang, S.C.; Lee-Chen, G.J. Association of apolipoprotein J polymorphism 1598delT with coronary artery disease and lipoprotein levels. Cardiology 2011, 118, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Trougakos, I.P.; Poulakou, M.; Stathatos, M.; Chalikia, A.; Melidonis, A.; Gonos, E.S. Serum levels of the senescence biomarker clusterin/apolipoprotein J increase significantly in diabetes type II and during development of coronary heart disease or at myocardial infarction. Exp. Gerontol. 2002, 37, 1175–1187. [Google Scholar] [CrossRef]
- Koudinov, A.R.; Berezov, T.T.; Kumar, A.; Koudinova, N.V. Alzheimer’s amyloid beta interaction with normal human plasma high density lipoprotein: Association with apolipoprotein and lipids. Clin. Chim. Acta 1998, 270, 75–84. [Google Scholar] [CrossRef]
- Mackness, B.; Hunt, R.; Durrington, P.N.; Mackness, M.I. Increased immunolocalization of paraoxonase, clusterin, and apolipoprotein A-I in the human artery wall with the progression of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 1233–1238. [Google Scholar] [CrossRef]
- Christoffersen, C.; Nielsen, L.B.; Axler, O.; Andersson, A.; Johnsen, A.H.; Dahlbäck, B. Isolation and characterization of human apolipoprotein M-containing lipoproteins. J. Lipid Res. 2006, 47, 1833–1843. [Google Scholar] [CrossRef] [Green Version]
- Christoffersen, C.; Jauhiainen, M.; Moser, M.; Porse, B.; Ehnholm, C.; Boesl, M.; Dahlbäck, B.; Nielsen, L.B. Effect of apolipoprotein M on high density lipoprotein metabolism and atherosclerosis in low density lipoprotein receptor knock-out mice. J. Biol. Chem. 2008, 283, 1839–1847. [Google Scholar] [CrossRef] [Green Version]
- Yao, S.; Luo, G.; Liu, H.; Zhang, J.; Zhan, Y.; Xu, N.; Zhang, X.; Zheng, L. Apolipoprotein M promotes the anti-inflammatory effect of high-density lipoprotein by binding to scavenger receptor BI. Ann. Transl. Med. 2020, 8, 1676. [Google Scholar] [CrossRef] [PubMed]
- Hajny, S.; Borup, A.; Elsøe, S.; Christoffersen, C. Increased plasma apoM levels impair triglyceride turnover in mice. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 158969. [Google Scholar] [CrossRef]
- Huang, Y.; Liu, Y.; Jiang, L.; Sun, R.; Zhang, H.; Liu, R.; Xu, N. Apolipoprotein m (APOM) levels and APOM rs805297 G/T polymorphism are associated with increased risk of rheumatoid arthritis. Jt. Bone Spine 2014, 81, 32–36. [Google Scholar] [CrossRef]
- Perdomo, G.; Henry Dong, H. Apolipoprotein D in lipid metabolism and its functional implication in atherosclerosis and aging. Aging 2009, 1, 17–27. [Google Scholar] [CrossRef]
- Lagor, W.R.; Fields, D.W.; Khetarpal, S.A.; Kumaravel, A.; Lin, W.; Weintraub, N.; Wu, K.; Hamm-Alvarez, S.F.; Drazul-Schrader, D.; de la Llera-Moya, M.; et al. The effects of apolipoprotein F deficiency on high density lipoprotein cholesterol metabolism in mice. PLoS ONE 2012, 7, e31616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Main, L.A.; Ohnishi, T.; Yokoyama, S. Activation of human plasma cholesteryl ester transfer protein by human apolipoprotein A-IV. Biochim. Biophys. Acta 1996, 1300, 17–24. [Google Scholar] [CrossRef]
- de Grooth, G.J.; Klerkx, A.H.; Stroes, E.S.; Stalenhoef, A.F.; Kastelein, J.J.; Kuivenhoven, J.A. A review of CETP and its relation to atherosclerosis. J. Lipid Res. 2004, 45, 1967–1974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inazu, A.; Brown, M.L.; Hesler, C.B.; Agellon, L.B.; Koizumi, J.; Takata, K.; Maruhama, Y.; Mabuchi, H.; Tall, A.R. Increased high-density lipoprotein levels caused by a common cholesteryl-ester transfer protein gene mutation. N. Engl. J. Med. 1990, 323, 1234–1238. [Google Scholar] [CrossRef] [PubMed]
- Nordestgaard, L.T.; Christoffersen, M.; Lauridsen, B.K.; Afzal, S.; Nordestgaard, B.G.; Frikke-Schmidt, R.; Tybjærg-Hansen, A. Long-term Benefits and Harms Associated with Genetic Cholesteryl Ester Transfer Protein Deficiency in the General Population. JAMA Cardiol. 2022, 7, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Calabresi, L.; Nilsson, P.; Pinotti, E.; Gomaraschi, M.; Favari, E.; Adorni, M.P.; Bernini, F.; Sirtori, C.R.; Calandra, S.; Franceschini, G.; et al. A novel homozygous mutation in CETP gene as a cause of CETP deficiency in a Caucasian kindred. Atherosclerosis 2009, 205, 506–511. [Google Scholar] [CrossRef]
- Zhong, S.; Sharp, D.S.; Grove, J.S.; Bruce, C.; Yano, K.; Curb, J.D.; Tall, A.R. Increased coronary heart disease in Japanese-American men with mutation in the cholesteryl ester transfer protein gene despite increased HDL levels. J. Clin. Investig. 1996, 97, 2917–2923. [Google Scholar] [CrossRef]
- Bruce, C.; Sharp, D.S.; Tall, A.R. Relationship of HDL and coronary heart disease to a common amino acid polymorphism in the cholesteryl ester transfer protein in men with and without hypertriglyceridemia. J. Lipid Res. 1998, 39, 1071–1078. [Google Scholar] [CrossRef]
- Deguchi, H.; Banerjee, Y.; Elias, D.J.; Griffin, J.H. Elevated CETP Lipid Transfer Activity is Associated with the Risk of Venous Thromboembolism. J. Atheroscler. Thromb. 2016, 23, 1159–1167. [Google Scholar] [CrossRef] [Green Version]
- McIntyre, T.M.; Prescott, S.M.; Stafforini, D.M. The emerging roles of PAF acetylhydrolase. J. Lipid Res. 2009, 50, S255–S259. [Google Scholar] [CrossRef] [Green Version]
- Tselepis, A.D.; John Chapman, M. Inflammation, bioactive lipids and atherosclerosis: Potential roles of a lipoprotein-associated phospholipase A2, platelet activating factor-acetylhydrolase. Atheroscler. Suppl. 2002, 3, 57–68. [Google Scholar] [CrossRef]
- Carpenter, K.L.; Dennis, I.F.; Challis, I.R.; Osborn, D.P.; Macphee, C.H.; Leake, D.S.; Arends, M.J.; Mitchinson, M.J. Inhibition of lipoprotein-associated phospholipase A2 diminishes the death-inducing effects of oxidised LDL on human monocyte-macrophages. FEBS Lett. 2001, 505, 357–363. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Zhang, P.; Zhang, L.; Osman, H.; Mohler, E.R., 3rd; Macphee, C.; Zalewski, A.; Postle, A.; Wilensky, R.L. Role of lipoprotein-associated phospholipase A2 in leukocyte activation and inflammatory responses. Atherosclerosis 2007, 191, 54–62. [Google Scholar] [CrossRef]
- Tellis, C.C.; Tselepis, A.D. The role of lipoprotein-associated phospholipase A2 in atherosclerosis may depend on its lipoprotein carrier in plasma. Biochim. Biophys. Acta 2009, 1791, 327–338. [Google Scholar] [CrossRef]
- Thompson, A.; Gao, P.; Orfei, L.; Watson, S.; Di Angelantonio, E.; Kaptoge, S.; Ballantyne, C.; Cannon, C.P.; Criqui, M.; Cushman, M.; et al. Lipoprotein-associated phospholipase A(2) and risk of coronary disease, stroke, and mortality: Collaborative analysis of 32 prospective studies. Lancet 2010, 375, 1536–1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pussinen, P.J.; Jauhiainen, M.; Metso, J.; Pyle, L.E.; Marcel, Y.L.; Fidge, N.H.; Ehnholm, C. Binding of phospholipid transfer protein (PLTP) to apolipoproteins A-I and A-II: Location of a PLTP binding domain in the amino terminal region of apoA-I. J. Lipid Res. 1998, 39, 152–161. [Google Scholar] [CrossRef]
- Cheung, M.C.; Albers, J.J. Active plasma phospholipid transfer protein is associated with apoA-I- but not apoE-containing lipoproteins. J. Lipid Res. 2006, 47, 1315–1321. [Google Scholar] [CrossRef] [Green Version]
- Francis, G.A. Chapter 15—High-Density Lipoproteins: Metabolism and Protective Roles Against Atherosclerosis. In Biochemistry of Lipids, Lipoproteins and Membranes, 6th ed.; Ridgway, N.D., McLeod, R.S., Eds.; Elsevier: Boston, MA, USA, 2016; pp. 437–457. [Google Scholar]
- Oram, J.F.; Wolfbauer, G.; Tang, C.; Davidson, W.S.; Albers, J.J. An amphipathic helical region of the N-terminal barrel of phospholipid transfer protein is critical for ABCA1-dependent cholesterol efflux. J. Biol. Chem. 2008, 283, 11541–11549. [Google Scholar] [CrossRef] [Green Version]
- Oram, J.F.; Wolfbauer, G.; Vaughan, A.M.; Tang, C.; Albers, J.J. Phospholipid transfer protein interacts with and stabilizes ATP-binding cassette transporter A1 and enhances cholesterol efflux from cells. J. Biol. Chem. 2003, 278, 52379–52385. [Google Scholar] [CrossRef] [Green Version]
- Chowaniec, Z.; Skoczyńska, A. Plasma lipid transfer proteins: The role of PLTP and CETP in atherogenesis. Adv. Clin. Exp. Med. 2018, 27, 429–436. [Google Scholar] [CrossRef]
- van Haperen, R.; van Tol, A.; van Gent, T.; Scheek, L.; Visser, P.; van der Kamp, A.; Grosveld, F.; de Crom, R. Increased risk of atherosclerosis by elevated plasma levels of phospholipid transfer protein. J. Biol. Chem. 2002, 277, 48938–48943. [Google Scholar] [CrossRef] [Green Version]
- van Haperen, R.; van Gent, T.; van Tol, A.; de Crom, R. Elevated expression of PLTP is atherogenic in apolipoprotein E deficient mice. Atherosclerosis 2013, 227, 37–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.P.; Yan, D.; Qiao, C.; Liu, R.J.; Chen, J.G.; Li, J.; Schneider, M.; Lagrost, L.; Xiao, X.; Jiang, X.C. Increased atherosclerotic lesions in apoE mice with plasma phospholipid transfer protein overexpression. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1601–1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.M.; Wang, Y.; Yu, Y.; Jiang, H.; Babinska, A.; Chen, X.Y.; He, K.G.; Min, X.D.; Han, J.J.; Yang, C.X.; et al. Plasma Phospholipid Transfer Protein Promotes Platelet Aggregation. Thromb. Haemost. 2018, 118, 2086–2097. [Google Scholar] [CrossRef] [PubMed]
- Audo, R.; Deckert, V.; Daien, C.I.; Che, H.; Elhmioui, J.; Lemaire, S.; Pais de Barros, J.P.; Desrumaux, C.; Combe, B.; Hahne, M.; et al. PhosphoLipid transfer protein (PLTP) exerts a direct pro-inflammatory effect on rheumatoid arthritis (RA) fibroblasts-like-synoviocytes (FLS) independently of its lipid transfer activity. PLoS ONE 2018, 13, e0193815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlitt, A.; Liu, J.; Yan, D.; Mondragon-Escorpizo, M.; Norin, A.J.; Jiang, X.C. Anti-inflammatory effects of phospholipid transfer protein (PLTP) deficiency in mice. Biochim. Biophys. Acta 2005, 1733, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Shelly, L.; Royer, L.; Sand, T.; Jensen, H.; Luo, Y. Phospholipid transfer protein deficiency ameliorates diet-induced hypercholesterolemia and inflammation in mice. J. Lipid Res. 2008, 49, 773–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desrumaux, C.; Lemaire-Ewing, S.; Ogier, N.; Yessoufou, A.; Hammann, A.; Sequeira-Le Grand, A.; Deckert, V.; Pais de Barros, J.P.; Le Guern, N.; Guy, J.; et al. Plasma phospholipid transfer protein (PLTP) modulates adaptive immune functions through alternation of T helper cell polarization. Cell Mol. Immunol. 2016, 13, 795–804. [Google Scholar] [CrossRef] [Green Version]
- Gautier, T.; Klein, A.; Deckert, V.; Desrumaux, C.; Ogier, N.; Sberna, A.L.; Paul, C.; Le Guern, N.; Athias, A.; Montange, T.; et al. Effect of plasma phospholipid transfer protein deficiency on lethal endotoxemia in mice. J. Biol. Chem. 2008, 283, 18702–18710. [Google Scholar] [CrossRef] [Green Version]
- Brehm, A.; Geraghty, P.; Campos, M.; Garcia-Arcos, I.; Dabo, A.J.; Gaffney, A.; Eden, E.; Jiang, X.C.; D’Armiento, J.; Foronjy, R. Cathepsin G degradation of phospholipid transfer protein (PLTP) augments pulmonary inflammation. FASEB J. 2014, 28, 2318–2331. [Google Scholar] [CrossRef] [Green Version]
- Vuletic, S.; Dong, W.; Wolfbauer, G.; Tang, C.; Albers, J.J. PLTP regulates STAT3 and NFκB in differentiated THP1 cells and human monocyte-derived macrophages. Biochim. Biophys. Acta 2011, 1813, 1917–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deckert, V.; Lemaire, S.; Ripoll, P.J.; de Barros, J.P.; Labbé, J.; Borgne, C.C.; Turquois, V.; Maquart, G.; Larose, D.; Desroche, N.; et al. Recombinant human plasma phospholipid transfer protein (PLTP) to prevent bacterial growth and to treat sepsis. Sci. Rep. 2017, 7, 3053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautier, T.; Lagrost, L. Plasma PLTP (phospholipid-transfer protein): An emerging role in ‘reverse lipopolysaccharide transport’ and innate immunity. Biochem. Soc. Trans. 2011, 39, 984–988. [Google Scholar] [CrossRef] [Green Version]
- Yan, D.; Navab, M.; Bruce, C.; Fogelman, A.M.; Jiang, X.C. PLTP deficiency improves the anti-inflammatory properties of HDL and reduces the ability of LDL to induce monocyte chemotactic activity. J. Lipid Res. 2004, 45, 1852–1858. [Google Scholar] [CrossRef] [PubMed]
- Robins, S.J.; Lyass, A.; Brocia, R.W.; Massaro, J.M.; Vasan, R.S. Plasma lipid transfer proteins and cardiovascular disease. The Framingham Heart Study. Atherosclerosis 2013, 228, 230–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akira, S. Toll-like Receptors and Innate Immunity. In Advances in Immunology; Dixon, F.J., Ed.; Academic Press: San Diego, CA, USA, 2001; Volume 78, pp. 1–56. [Google Scholar]
- Levels, J.H.; Marquart, J.A.; Abraham, P.R.; van den Ende, A.E.; Molhuizen, H.O.; van Deventer, S.J.; Meijers, J.C. Lipopolysaccharide is transferred from high-density to low-density lipoproteins by lipopolysaccharide-binding protein and phospholipid transfer protein. Infect. Immun. 2005, 73, 2321–2326. [Google Scholar] [CrossRef] [Green Version]
- Asada, M.; Oishi, E.; Sakata, S.; Hata, J.; Yoshida, D.; Honda, T.; Furuta, Y.; Shibata, M.; Suzuki, K.; Watanabe, H.; et al. Serum Lipopolysaccharide-Binding Protein Levels and the Incidence of Cardiovascular Disease in a General Japanese Population: The Hisayama Study. J. Am. Heart Assoc. 2019, 8, e013628. [Google Scholar] [CrossRef]
- Serrano, M.; Moreno-Navarrete, J.M.; Puig, J.; Moreno, M.; Guerra, E.; Ortega, F.; Xifra, G.; Ricart, W.; Fernández-Real, J.M. Serum lipopolysaccharide-binding protein as a marker of atherosclerosis. Atherosclerosis 2013, 230, 223–227. [Google Scholar] [CrossRef]
- Lepper, P.M.; Kleber, M.E.; Grammer, T.B.; Hoffmann, K.; Dietz, S.; Winkelmann, B.R.; Boehm, B.O.; März, W. Lipopolysaccharide-binding protein (LBP) is associated with total and cardiovascular mortality in individuals with or without stable coronary artery disease—Results from the Ludwigshafen Risk and Cardiovascular Health Study (LURIC). Atherosclerosis 2011, 219, 291–297. [Google Scholar] [CrossRef]
- Cooke, A.L.; Morris, J.; Melchior, J.T.; Street, S.E.; Jerome, W.G.; Huang, R.; Herr, A.B.; Smith, L.E.; Segrest, J.P.; Remaley, A.T.; et al. A thumbwheel mechanism for APOA1 activation of LCAT activity in HDL. J. Lipid Res. 2018, 59, 1244–1255. [Google Scholar] [CrossRef] [Green Version]
- Lambert, G.; Sakai, N.; Vaisman, B.L.; Neufeld, E.B.; Marteyn, B.; Chan, C.C.; Paigen, B.; Lupia, E.; Thomas, A.; Striker, L.J.; et al. Analysis of glomerulosclerosis and atherosclerosis in lecithin cholesterol acyltransferase-deficient mice. J. Biol. Chem. 2001, 276, 15090–15098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuivenhoven, J.A.; Pritchard, H.; Hill, J.; Frohlich, J.; Assmann, G.; Kastelein, J. The molecular pathology of lecithin:cholesterol acyltransferase (LCAT) deficiency syndromes. J. Lipid Res. 1997, 38, 191–205. [Google Scholar] [CrossRef]
- Rader, D.J. Lecithin: Cholesterol acyltransferase and atherosclerosis: Another high-density lipoprotein story that doesn’t quite follow the script. Circulation 2009, 120, 549–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanigawa, H.; Billheimer, J.T.; Tohyama, J.; Fuki, I.V.; Ng, D.S.; Rothblat, G.H.; Rader, D.J. Lecithin: Cholesterol acyltransferase expression has minimal effects on macrophage reverse cholesterol transport in vivo. Circulation 2009, 120, 160–169. [Google Scholar] [CrossRef]
- Brousseau, M.E.; Santamarina-Fojo, S.; Vaisman, B.L.; Applebaum-Bowden, D.; Bérard, A.M.; Talley, G.D.; Brewer, H.B., Jr.; Hoeg, J.M. Overexpression of human lecithin:cholesterol acyltransferase in cholesterol-fed rabbits: LDL metabolism and HDL metabolism are affected in a gene dose-dependent manner. J. Lipid Res. 1997, 38, 2537–2547. [Google Scholar] [CrossRef]
- Brites, F.; Martin, M.; Guillas, I.; Kontush, A. Antioxidative activity of high-density lipoprotein (HDL): Mechanistic insights into potential clinical benefit. BBA Clin. 2017, 8, 66–77. [Google Scholar] [CrossRef]
- Chen, N.; Liu, Y.; Greiner, C.D.; Holtzman, J.L. Physiologic concentrations of homocysteine inhibit the human plasma GSH peroxidase that reduces organic hydroperoxides. J. Lab. Clin. Med. 2000, 136, 58–65. [Google Scholar] [CrossRef]
- Tabet, F.; Touyz, R.M. Chapter 30—Reactive Oxygen Species, Oxidative Stress, and Vascular Biology in Hypertension. In Comprehensive Hypertension; Lip, G.Y.H., Hall, J.E., Eds.; Mosby: Philadelphia, PA, USA, 2007; pp. 337–347. [Google Scholar]
- Kornhauser, C.; Garcia-Ramirez, J.R.; Wrobel, K.; Pérez-Luque, E.L.; Garay-Sevilla, M.E.; Wrobel, K. Serum selenium and glutathione peroxidase concentrations in type 2 diabetes mellitus patients. Prim. Care Diabetes 2008, 2, 81–85. [Google Scholar] [CrossRef]
- Chung, S.S.; Kim, M.; Youn, B.S.; Lee, N.S.; Park, J.W.; Lee, I.K.; Lee, Y.S.; Kim, J.B.; Cho, Y.M.; Lee, H.K.; et al. Glutathione peroxidase 3 mediates the antioxidant effect of peroxisome proliferator-activated receptor gamma in human skeletal muscle cells. Mol. Cell Biol. 2009, 29, 20–30. [Google Scholar] [CrossRef] [Green Version]
- Jin, R.C.; Mahoney, C.E.; Coleman Anderson, L.; Ottaviano, F.; Croce, K.; Leopold, J.A.; Zhang, Y.Y.; Tang, S.S.; Handy, D.E.; Loscalzo, J. Glutathione peroxidase-3 deficiency promotes platelet-dependent thrombosis in vivo. Circulation 2011, 123, 1963–1973. [Google Scholar] [CrossRef]
- Buijsse, B.; Lee, D.H.; Steffen, L.; Erickson, R.R.; Luepker, R.V.; Jacobs, D.R., Jr.; Holtzman, J.L. Low serum glutathione peroxidase activity is associated with increased cardiovascular mortality in individuals with low HDLc’s. PLoS ONE 2012, 7, e38901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aviram, M.; Rosenblat, M.; Bisgaier, C.L.; Newton, R.S.; Primo-Parmo, S.L.; La Du, B.N. Paraoxonase inhibits high-density lipoprotein oxidation and preserves its functions. A possible peroxidative role for paraoxonase. J. Clin. Investig. 1998, 101, 1581–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aviram, M.; Hardak, E.; Vaya, J.; Mahmood, S.; Milo, S.; Hoffman, A.; Billicke, S.; Draganov, D.; Rosenblat, M. Human serum paraoxonases (PON1) Q and R selectively decrease lipid peroxides in human coronary and carotid atherosclerotic lesions: PON1 esterase and peroxidase-like activities. Circulation 2000, 101, 2510–2517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berrougui, H.; Loued, S.; Khalil, A. Purified human paraoxonase-1 interacts with plasma membrane lipid rafts and mediates cholesterol efflux from macrophages. Free Radic. Biol. Med. 2012, 52, 1372–1381. [Google Scholar] [CrossRef]
- Rodrigo, L.; Mackness, B.; Durrington, P.N.; Hernandez, A.; Mackness, M.I. Hydrolysis of platelet-activating factor by human serum paraoxonase. Biochem. J. 2001, 354, 1–7. [Google Scholar] [CrossRef]
- Ohmori, T.; Yano, Y.; Sakata, A.; Ikemoto, T.; Shimpo, M.; Madoiwa, S.; Katsuki, T.; Mimuro, J.; Shimada, K.; Kario, K.; et al. Lack of association between serum paraoxonase-1 activity and residual platelet aggregation during dual anti-platelet therapy. Thromb. Res. 2012, 129, e36–e40. [Google Scholar] [CrossRef]
- Besler, C.; Heinrich, K.; Rohrer, L.; Doerries, C.; Riwanto, M.; Shih, D.M.; Chroni, A.; Yonekawa, K.; Stein, S.; Schaefer, N.; et al. Mechanisms underlying adverse effects of HDL on eNOS-activating pathways in patients with coronary artery disease. J. Clin. Investig. 2011, 121, 2693–2708. [Google Scholar] [CrossRef] [Green Version]
- Ogun, A.S.; Adeyinka, A. Biochemistry, Transferrin. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2022. [Google Scholar]
- Nitin, R.; Bowman, A.B. Chapter Three—Connections Between Manganese Neurotoxicity and Neurological Disease. In Advances in Neurotoxicology; Aschner, M., Costa, L.G., Eds.; Academic Press: Cambridge, MA, USA, 2018; Volume 2, pp. 87–113. [Google Scholar]
- Matusiewicz, M.; Neubauer, K.; Lewandowska, P.; Gamian, A.; Krzystek-Korpacka, M. Reduced Transferrin Levels in Active Inflammatory Bowel Disease. BioMed Res. Int. 2017, 2017, 9541370. [Google Scholar] [CrossRef] [Green Version]
- Nemeth, E.; Ganz, T. Anemia of inflammation. Hematol. Oncol. Clin. N. Am. 2014, 28, 671–681. [Google Scholar] [CrossRef] [Green Version]
- van Campenhout, A.; van Campenhout, C.M.; Lagrou, A.R.; Manuel-y-Keenoy, B. Transferrin modifications and lipid peroxidation: Implications in diabetes mellitus. Free Radic. Res. 2003, 37, 1069–1077. [Google Scholar] [CrossRef]
- Sack, G.H., Jr. Serum amyloid A—A review. Mol. Med. 2018, 24, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Zhou, H.; Cheng, N.; Qian, F.; Ye, R.D. Serum amyloid A1 isoforms display different efficacy at Toll-like receptor 2 and formyl peptide receptor 2. Immunobiology 2014, 219, 916–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zewinger, S.; Drechsler, C.; Kleber, M.E.; Dressel, A.; Riffel, J.; Triem, S.; Lehmann, M.; Kopecky, C.; Säemann, M.D.; Lepper, P.M.; et al. Serum amyloid A: High-density lipoproteins interaction and cardiovascular risk. Eur. Heart J. 2015, 36, 3007–3016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, N.R. High-Density Lipoproteins and Serum Amyloid A (SAA). Curr. Atheroscler. Rep. 2021, 23, 7. [Google Scholar] [CrossRef]
- Kosuge, M.; Ebina, T.; Ishikawa, T.; Hibi, K.; Tsukahara, K.; Okuda, J.; Iwahashi, N.; Ozaki, H.; Yano, H.; Kusama, I.; et al. Serum amyloid A is a better predictor of clinical outcomes than C-reactive protein in non-ST-segment elevation acute coronary syndromes. Circ. J. 2007, 71, 186–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, V.L.; Thompson, J.; Tannock, L.R. Serum amyloid A in atherosclerosis. Curr. Opin. Lipidol. 2011, 22, 302–307. [Google Scholar] [CrossRef]
- Thompson, J.C.; Wilson, P.G.; Shridas, P.; Ji, A.; de Beer, M.; de Beer, F.C.; Webb, N.R.; Tannock, L.R. Serum amyloid A3 is pro-atherogenic. Atherosclerosis 2018, 268, 32–35. [Google Scholar] [CrossRef]
- Sato, M.; Ohkawa, R.; Yoshimoto, A.; Yano, K.; Ichimura, N.; Nishimori, M.; Okubo, S.; Yatomi, Y.; Tozuka, M. Effects of serum amyloid A on the structure and antioxidant ability of high-density lipoprotein. Biosci. Rep. 2016, 36, e00369. [Google Scholar] [CrossRef] [Green Version]
- Márquez, A.B.; Nazir, S.; van der Vorst, E.P.C. High-Density Lipoprotein Modifications: A Pathological Consequence or Cause of Disease Progression? Biomedicines 2020, 8, 549. [Google Scholar] [CrossRef]
- Linke, R.P.; Bock, V.; Valet, G.; Rothe, G. Inhibition of the oxidative burst response of N-formyl peptide-stimulated neutrophils by serum amyloid-A protein. Biochem. Biophys. Res. Commun. 1991, 176, 1100–1105. [Google Scholar] [CrossRef]
- Schuchardt, M.; Prüfer, N.; Tu, Y.; Herrmann, J.; Hu, X.P.; Chebli, S.; Dahlke, K.; Zidek, W.; van der Giet, M.; Tölle, M. Dysfunctional high-density lipoprotein activates toll-like receptors via serum amyloid A in vascular smooth muscle cells. Sci. Rep. 2019, 9, 3421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shridas, P.; De Beer, M.C.; Webb, N.R. High-density lipoprotein inhibits serum amyloid A-mediated reactive oxygen species generation and NLRP3 inflammasome activation. J. Biol. Chem. 2018, 293, 13257–13269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.H.; Park, J.Y.; Kim, J.Y.; Choi, C.S.; Kim, Y.I.; Chung, Y.E.; Lee, M.S.; Hong, S.K.; Lee, K.U. Elevated serum ceruloplasmin levels in subjects with metabolic syndrome: A population-based study. Metabolism 2002, 51, 838–842. [Google Scholar] [CrossRef] [PubMed]
- Shukla, N.; Maher, J.; Masters, J.; Angelini, G.D.; Jeremy, J.Y. Does oxidative stress change ceruloplasmin from a protective to a vasculopathic factor? Atherosclerosis 2006, 187, 238–250. [Google Scholar] [CrossRef]
- Weisel, J.W. Fibrinogen and fibrin. Adv. Protein Chem. 2005, 70, 247–299. [Google Scholar] [CrossRef]
- Davalos, D.; Akassoglou, K. Fibrinogen as a key regulator of inflammation in disease. Semin. Immunopathol. 2012, 34, 43–62. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhu, C.G.; Guo, Y.L.; Xu, R.X.; Li, S.; Dong, Q.; Li, J.J. Higher fibrinogen level is independently linked with the presence and severity of new-onset coronary atherosclerosis among Han Chinese population. PLoS ONE 2014, 9, e113460. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhu, C.G.; Guo, Y.L.; Li, S.; Xu, R.X.; Dong, Q.; Li, J.J. Fibrinogen and the Severity of Coronary Atherosclerosis among Adults with and without Statin Treatment: Lipid as a mediator. Heart Lung Circ. 2016, 25, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Sabeti, S.; Exner, M.; Mlekusch, W.; Amighi, J.; Quehenberger, P.; Rumpold, H.; Maurer, G.; Minar, E.; Wagner, O.; Schillinger, M. Prognostic impact of fibrinogen in carotid atherosclerosis: Nonspecific indicator of inflammation or independent predictor of disease progression? Stroke 2005, 36, 1400–1404. [Google Scholar] [CrossRef]
- Green, D.; Foiles, N.; Chan, C.; Schreiner, P.J.; Liu, K. Elevated fibrinogen levels and subsequent subclinical atherosclerosis: The CARDIA Study. Atherosclerosis 2009, 202, 623–631. [Google Scholar] [CrossRef] [Green Version]
- Fowkes, F.G.; Connor, J.M.; Smith, F.B.; Wood, J.; Donnan, P.T.; Lowe, G.D. Fibrinogen genotype and risk of peripheral atherosclerosis. Lancet 1992, 339, 693–696. [Google Scholar] [CrossRef]
- Raijmakers, R.; van Beers, J.J.; El-Azzouny, M.; Visser, N.F.; Božič, B.; Pruijn, G.J.; Heck, A.J. Elevated levels of fibrinogen-derived endogenous citrullinated peptides in synovial fluid of rheumatoid arthritis patients. Arthritis Res. Ther. 2012, 14, R114. [Google Scholar] [CrossRef] [Green Version]
- Lind, P.; Hedblad, B.; Stavenow, L.; Janzon, L.; Eriksson, K.F.; Lindgärde, F. Influence of plasma fibrinogen levels on the incidence of myocardial infarction and death is modified by other inflammation-sensitive proteins: A long-term cohort study. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 452–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.; McCulloh, R.J. Hemopexin and haptoglobin: Allies against heme toxicity from hemoglobin not contenders. Front. Physiol. 2015, 6, 187. [Google Scholar] [CrossRef]
- Watanabe, J.; Grijalva, V.; Hama, S.; Barbour, K.; Berger, F.G.; Navab, M.; Fogelman, A.M.; Reddy, S.T. Hemoglobin and its scavenger protein haptoglobin associate with apoA-1-containing particles and influence the inflammatory properties and function of high density lipoprotein. J. Biol. Chem. 2009, 284, 18292–18301. [Google Scholar] [CrossRef] [Green Version]
- Goldenstein, H.; Levy, N.S.; Ward, J.; Costacou, T.; Levy, A.P. Haptoglobin Genotype Is a Determinant of Hemoglobin Adducts and Vitamin E Content in HDL. J. Diabetes Res. 2018, 2018, 6125420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, B.; Oda, M.N.; Vaisar, T.; Oram, J.F.; Heinecke, J.W. Pathways for oxidation of high-density lipoprotein in human cardiovascular disease. Curr. Opin. Mol. Ther. 2006, 8, 198–205. [Google Scholar]
- Asleh, R.; Levy, A.P.; Levy, N.S.; Asleh, A.; Goldenstein, H.; Segol, I.; Gulati, R.; Lerman, L.O.; Lerman, A. Haptoglobin Phenotype Is Associated with High-Density Lipoprotein-Bound Hemoglobin Content and Coronary Endothelial Dysfunction in Patients with Mild Nonobstructive Coronary Artery Disease. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 774–786. [Google Scholar] [CrossRef] [Green Version]
- Janciauskiene, S.; Welte, T. Well-Known and Less Well-Known Functions of Alpha-1 Antitrypsin. Its Role in Chronic Obstructive Pulmonary Disease and Other Disease Developments. Ann. Am. Thorac. Soc. 2016, 13 (Suppl. S4), S280–S288. [Google Scholar] [CrossRef]
- Gordon, S.M.; Sviridov, D.; Sakurai, T.; Freeman, L.; Remaley, A.T. Abstract 29: Alpha-1-antitrypsin Protects High Density Lipoprotein from Functional Inactivation by Elastase. Arterioscler. Thromb. Vasc. Biol. 2016, 36, A29. [Google Scholar] [CrossRef]
- Moreno, J.A.; Ortega-Gomez, A.; Rubio-Navarro, A.; Louedec, L.; Ho-Tin-Noé, B.; Caligiuri, G.; Nicoletti, A.; Levoye, A.; Plantier, L.; Meilhac, O. High-density lipoproteins potentiate α1-antitrypsin therapy in elastase-induced pulmonary emphysema. Am. J. Respir. Cell Mol. Biol. 2014, 51, 536–549. [Google Scholar] [CrossRef] [PubMed]
- Mahta, A.; Yaghi, S.; Reznik, M.E.; Thompson, B.B.; Wendell, L.C.; Rao, S.; Potter, N.S.; Dakay, K.B.; Cutting, S.; Mac Grory, B.; et al. Serum alpha-1 antitrypsin in acute ischemic stroke: A prospective pilot study. J. Clin. Neurosci. 2020, 76, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Talmud, P.J.; Martin, S.; Steiner, G.; Flavell, D.M.; Whitehouse, D.B.; Nagl, S.; Jackson, R.; Taskinen, M.R.; Frick, M.H.; Nieminen, M.S.; et al. Progression of atherosclerosis is associated with variation in the alpha1-antitrypsin gene. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 644–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Z.; Lei, H.; Sun, Y.; Liu, X.; Su, D.F. Orosomucoid, an acute response protein with multiple modulating activities. J. Physiol. Biochem. 2015, 71, 329–340. [Google Scholar] [CrossRef]
- Berntsson, J.; Östling, G.; Persson, M.; Smith, J.G.; Hedblad, B.; Engström, G. Orosomucoid, Carotid Plaque, and Incidence of Stroke. Stroke 2016, 47, 1858–1863. [Google Scholar] [CrossRef]
- El-Beblawy, N.M.; Andrawes, N.G.; Ismail, E.A.; Enany, B.E.; El-Seoud, H.S.; Erfan, M.A. Serum and Urinary Orosomucoid in Young Patients with Type 1 Diabetes: A Link Between Inflammation, Microvascular Complications, and Subclinical Atherosclerosis. Clin. Appl. Thromb. Hemost. 2016, 22, 718–726. [Google Scholar] [CrossRef]
- de Boer, J.P.; Creasey, A.A.; Chang, A.; Abbink, J.J.; Roem, D.; Eerenberg, A.J.; Hack, C.E.; Taylor, F.B., Jr. Alpha-2-macroglobulin functions as an inhibitor of fibrinolytic, clotting, and neutrophilic proteinases in sepsis: Studies using a baboon model. Infect. Immun. 1993, 61, 5035–5043. [Google Scholar] [CrossRef] [Green Version]
- Yoshino, S.; Fujimoto, K.; Takada, T.; Kawamura, S.; Ogawa, J.; Kamata, Y.; Kodera, Y.; Shichiri, M. Molecular form and concentration of serum α(2)-macroglobulin in diabetes. Sci. Rep. 2019, 9, 12927. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Liu, X.; Xiang, Y.; Ding, X.; Wang, T.; Liu, Y.; Yin, M.; Tan, C.; Deng, F.; Chen, L. Alpha-2-macroglobulin and heparin cofactor II and the vulnerability of carotid atherosclerotic plaques: An iTRAQ-based analysis. Biochem. Biophys. Res. Commun. 2017, 483, 964–971. [Google Scholar] [CrossRef]
- Sacks, F.M.; Liang, L.; Furtado, J.D.; Cai, T.; Davidson, W.S.; He, Z.; McClelland, R.L.; Rimm, E.B.; Jensen, M.K. Protein-Defined Subspecies of HDLs (High-Density Lipoproteins) and Differential Risk of Coronary Heart Disease in 4 Prospective Studies. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2714–2727. [Google Scholar] [CrossRef]
- Li, L.; Dong, M.; Wang, X.G. The Implication and Significance of Beta 2 Microglobulin: A Conservative Multifunctional Regulator. Chin. Med. J. 2016, 129, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Yılmaz, B.; Köklü, S.; Yüksel, O.; Arslan, S. Serum beta 2-microglobulin as a biomarker in inflammatory bowel disease. World J. Gastroenterol. 2014, 20, 10916–10920. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.A.; Jeon, J.Y.; Yoon, J.M.; Suh, C.H. Beta 2-microglobulin can be a disease activity marker in systemic lupus erythematosus. Am. J. Med. Sci. 2010, 339, 337–340. [Google Scholar] [CrossRef] [PubMed]
- Topçiu-Shufta, V.; Miftari, R.; Haxhibeqiri, V.; Haxhibeqiri, S. Association of Beta-2 Microglobulin with Inflammation and Dislipidemia in High-Flux Membrane Hemodialysis Patients. Med. Arch. 2016, 70, 348–350. [Google Scholar] [CrossRef] [Green Version]
- Sjöblom, K.G.; Saxne, T.; Wollheim, F.A. Plasma levels of beta 2-microglobulin in rheumatoid arthritis. Ann. Rheum. Dis. 1980, 39, 333–339. [Google Scholar] [CrossRef]
- Amighi, J.; Hoke, M.; Mlekusch, W.; Schlager, O.; Exner, M.; Haumer, M.; Pernicka, E.; Koppensteiner, R.; Minar, E.; Rumpold, H.; et al. Beta 2 microglobulin and the risk for cardiovascular events in patients with asymptomatic carotid atherosclerosis. Stroke 2011, 42, 1826–1833. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.M.; Kimura, E.; Harada, R.K.; Nair, N.; Narasimhan, B.; Meng, X.Y.; Zhang, F.; Beck, K.R.; Olin, J.W.; Fung, E.T.; et al. Beta2-microglobulin as a biomarker in peripheral arterial disease: Proteomic profiling and clinical studies. Circulation 2007, 116, 1396–1403. [Google Scholar] [CrossRef] [Green Version]
- You, L.; Xie, R.; Hu, H.; Gu, G.; Zheng, H.; Zhang, J.; Yang, X.; He, X.; Cui, W. High levels of serum β2-microglobulin predict severity of coronary artery disease. BMC Cardiovasc. Disord. 2017, 17, 71. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.M.; Kim, S.S.; Kim, H.; Koo, T.; Im, E.Y.; Kim, S.B. Higher serum beta2-microglobulin levels are associated with better survival in chronic hemodialysis patients: A reverse epidemiology. Clin. Nephrol. 2011, 75, 458–465. [Google Scholar] [CrossRef]
- Kim, R.R.; Chen, Z.; Mann, T.J.; Bastard, K.; Scott, K.F.; Church, W.B. Structural and Functional Aspects of Targeting the Secreted Human Group IIA Phospholipase A(2). Molecules 2020, 25, 4459. [Google Scholar] [CrossRef]
- Boilard, E.; Lai, Y.; Larabee, K.; Balestrieri, B.; Ghomashchi, F.; Fujioka, D.; Gobezie, R.; Coblyn, J.S.; Weinblatt, M.E.; Massarotti, E.M.; et al. A novel anti-inflammatory role for secretory phospholipase A2 in immune complex-mediated arthritis. EMBO Mol. Med. 2010, 2, 172–187. [Google Scholar] [CrossRef] [PubMed]
- de Beer, F.C.; Connell, P.M.; Yu, J.; de Beer, M.C.; Webb, N.R.; van der Westhuyzen, D.R. HDL modification by secretory phospholipase A(2) promotes scavenger receptor class B type I interaction and accelerates HDL catabolism. J. Lipid Res. 2000, 41, 1849–1857. [Google Scholar] [CrossRef]
- Tietge, U.J.; Maugeais, C.; Lund-Katz, S.; Grass, D.; deBeer, F.C.; Rader, D.J. Human secretory phospholipase A2 mediates decreased plasma levels of HDL cholesterol and apoA-I in response to inflammation in human apoA-I transgenic mice. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1213–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura-Matsumoto, M.; Ishikawa, Y.; Komiyama, K.; Tsuruta, T.; Murakami, M.; Masuda, S.; Akasaka, Y.; Ito, K.; Ishiguro, S.; Morita, H.; et al. Expression of secretory phospholipase A2s in human atherosclerosis development. Atherosclerosis 2008, 196, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Niessen, H.W.; Krijnen, P.A.; Visser, C.A.; Meijer, C.J.; Erik Hack, C. Type II secretory phospholipase A2 in cardiovascular disease: A mediator in atherosclerosis and ischemic damage to cardiomyocytes? Cardiovasc. Res. 2003, 60, 68–77. [Google Scholar] [CrossRef]
- Sun, C.Q.; Zhong, C.Y.; Sun, W.W.; Xiao, H.; Zhu, P.; Lin, Y.Z.; Zhang, C.L.; Gao, H.; Song, Z.Y. Elevated Type II Secretory Phospholipase A2 Increases the Risk of Early Atherosclerosis in Patients with Newly Diagnosed Metabolic Syndrome. Sci. Rep. 2016, 6, 34929. [Google Scholar] [CrossRef] [Green Version]
- Janssen, B.J.; Huizinga, E.G.; Raaijmakers, H.C.; Roos, A.; Daha, M.R.; Nilsson-Ekdahl, K.; Nilsson, B.; Gros, P. Structures of complement component C3 provide insights into the function and evolution of immunity. Nature 2005, 437, 505–511. [Google Scholar] [CrossRef] [Green Version]
- Fehérvári, M.; Krepuska, M.; Széplaki, G.; Apor, A.; Sótonyi, P.; Prohászka, Z.; Acsády, G.; Szeberin, Z. The level of complement C3 is associated with the severity of atherosclerosis but not with arterial calcification in peripheral artery disease. Int. Angiol. 2014, 33, 35–41. [Google Scholar]
- Széplaki, G.; Prohászka, Z.; Duba, J.; Rugonfalvi-Kiss, S.; Karádi, I.; Kókai, M.; Kramer, J.; Füst, G.; Kleiber, M.; Romics, L.; et al. Association of high serum concentration of the third component of complement (C3) with pre-existing severe coronary artery disease and new vascular events in women. Atherosclerosis 2004, 177, 383–389. [Google Scholar] [CrossRef]
- Feingold, K.R.; Grunfeld, C. Effect of inflammation on HDL structure and function. Curr. Opin. Lipidol. 2016, 27, 521–530. [Google Scholar] [CrossRef]
- Ungurianu, A.; Margină, D.; Grădinaru, D.; Băcanu, C.; Ilie, M.; Tsitsimpikou, C.; Tsarouhas, K.; Spandidos, D.A.; Tsatsakis, A.M. Lipoprotein redox status evaluation as a marker of cardiovascular disease risk in patients with inflammatory disease. Mol. Med. Rep. 2017, 15, 256–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bigagli, E.; Lodovici, M. Circulating Oxidative Stress Biomarkers in Clinical Studies on Type 2 Diabetes and Its Complications. Oxid. Med. Cell Longev. 2019, 2019, 5953685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardner, M.; Yalcinkaya, M.; Goetze, S.; Luca, E.; Balaz, M.; Hunjadi, M.; Hartung, J.; Shemet, A.; Kränkel, N.; Radosavljevic, S.; et al. Structure-function relationships of HDL in diabetes and coronary heart disease. JCI Insight 2020, 5, e131491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feingold, K.R.; Grunfeld, C. The Effect of Inflammation and Infection on Lipids and Lipoproteins. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Hershman, J.M., Hofland, J., Kalra, S., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- McMahon, M.; Grossman, J.; FitzGerald, J.; Dahlin-Lee, E.; Wallace, D.J.; Thong, B.Y.; Badsha, H.; Kalunian, K.; Charles, C.; Navab, M.; et al. Proinflammatory high-density lipoprotein as a biomarker for atherosclerosis in patients with systemic lupus erythematosus and rheumatoid arthritis. Arthritis Rheum. 2006, 54, 2541–2549. [Google Scholar] [CrossRef] [PubMed]
- Skaggs, B.J.; Hahn, B.H.; Sahakian, L.; Grossman, J.; McMahon, M. Dysfunctional, pro-inflammatory HDL directly upregulates monocyte PDGFRβ, chemotaxis and TNFα production. Clin. Immunol. 2010, 137, 147–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, S.M.; Chung, J.H.; Playford, M.P.; Dey, A.K.; Sviridov, D.; Seifuddin, F.; Chen, Y.C.; Pirooznia, M.; Chen, M.Y.; Mehta, N.N.; et al. High density lipoprotein proteome is associated with cardiovascular risk factors and atherosclerosis burden as evaluated by coronary CT angiography. Atherosclerosis 2018, 278, 278–285. [Google Scholar] [CrossRef]
- Van Lenten, B.J.; Wagner, A.C.; Nayak, D.P.; Hama, S.; Navab, M.; Fogelman, A.M. High-density lipoprotein loses its anti-inflammatory properties during acute influenza a infection. Circulation 2001, 103, 2283–2288. [Google Scholar] [CrossRef] [Green Version]
- Vachatova, S.; Andrys, C.; Krejsek, J.; Salavec, M.; Ettler, K.; Rehacek, V.; Cermakova, E.; Malkova, A.; Fiala, Z.; Borska, L. Metabolic Syndrome and Selective Inflammatory Markers in Psoriatic Patients. J. Immunol. Res. 2016, 2016, 5380792. [Google Scholar] [CrossRef] [Green Version]
- Cederholm, A.; Svenungsson, E.; Stengel, D.; Fei, G.Z.; Pockley, A.G.; Ninio, E.; Frostegård, J. Platelet-activating factor-acetylhydrolase and other novel risk and protective factors for cardiovascular disease in systemic lupus erythematosus. Arthritis. Rheum. 2004, 50, 2869–2876. [Google Scholar] [CrossRef] [Green Version]
- Holzer, M.; Wolf, P.; Inzinger, M.; Trieb, M.; Curcic, S.; Pasterk, L.; Weger, W.; Heinemann, A.; Marsche, G. Anti-psoriatic therapy recovers high-density lipoprotein composition and function. J. Investig. Dermatol. 2014, 134, 635–642. [Google Scholar] [CrossRef] [Green Version]
- Holzer, M.; Wolf, P.; Curcic, S.; Birner-Gruenberger, R.; Weger, W.; Inzinger, M.; El-Gamal, D.; Wadsack, C.; Heinemann, A.; Marsche, G. Psoriasis alters HDL composition and cholesterol efflux capacity. J. Lipid Res. 2012, 53, 1618–1624. [Google Scholar] [CrossRef] [Green Version]
- Tselepis, A.D.; Elisaf, M.; Besis, S.; Karabina, S.A.; Chapman, M.J.; Siamopoulou, A. Association of the inflammatory state in active juvenile rheumatoid arthritis with hypo-high-density lipoproteinemia and reduced lipoprotein-associated platelet-activating factor acetylhydrolase activity. Arthritis Rheum. 1999, 42, 373–383. [Google Scholar] [CrossRef]
- Dulioust, A.; Hilliquin, P.; Menkes, C.J.; Benveniste, J.; Arnoux, B. Paf-acether acetylhydrolase activity is increased in patients with rheumatic diseases. Scand. J. Rheumatol. 1992, 21, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Parra, S.; Heras, M.; Herrero, P.; Amigó, N.; Garcés, E.; Girona, J.; Correig, X.; Canela, N.; Castro, A. Gelsolin: A new biomarker of disease activity in SLE patients associated with HDL-c. Rheumatology 2020, 59, 650–661. [Google Scholar] [CrossRef]
- Shao, B.; de Boer, I.; Tang, C.; Mayer, P.S.; Zelnick, L.; Afkarian, M.; Heinecke, J.W.; Himmelfarb, J. A Cluster of Proteins Implicated in Kidney Disease Is Increased in High-Density Lipoprotein Isolated from Hemodialysis Subjects. J. Proteome Res. 2015, 14, 2792–2806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holzer, M.; Birner-Gruenberger, R.; Stojakovic, T.; El-Gamal, D.; Binder, V.; Wadsack, C.; Heinemann, A.; Marsche, G. Uremia alters HDL composition and function. J. Am. Soc. Nephrol. 2011, 22, 1631–1641. [Google Scholar] [CrossRef]
- Mangé, A.; Goux, A.; Badiou, S.; Patrier, L.; Canaud, B.; Maudelonde, T.; Cristol, J.P.; Solassol, J. HDL proteome in hemodialysis patients: A quantitative nanoflow liquid chromatography-tandem mass spectrometry approach. PLoS ONE 2012, 7, e34107. [Google Scholar] [CrossRef] [Green Version]
- Kasumov, T. Abstract 454: High Density Lipoprotein Proteome Dynamics is Altered in Non-alcoholic Fatty Liver Disease. Arterioscler. Thromb. Vasc. Biol. 2016, 36, A454. [Google Scholar] [CrossRef]
- Vaisar, T.; Tang, C.; Babenko, I.; Hutchins, P.; Wimberger, J.; Suffredini, A.F.; Heinecke, J.W. Inflammatory remodeling of the HDL proteome impairs cholesterol efflux capacity. J. Lipid Res. 2015, 56, 1519–1530. [Google Scholar] [CrossRef] [Green Version]
- Juárez-Rojas, J.; Medina-Urrutia, A.; Posadas-Sánchez, R.; Jorge-Galarza, E.; Mendoza-Pérez, E.; Caracas-Portilla, N.; Cardoso-Saldaña, G.; Muñoz-Gallegos, G.; Posadas-Romero, C. High-density lipoproteins are abnormal in young women with uncomplicated systemic lupus erythematosus. Lupus 2008, 17, 981–987. [Google Scholar] [CrossRef]
- Reisinger, A.C.; Schuller, M.; Sourij, H.; Stadler, J.T.; Hackl, G.; Eller, P.; Marsche, G. Impact of Sepsis on High-Density Lipoprotein Metabolism. Front. Cell Dev. Biol. 2021, 9, 795460. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Lee, E.Y.; Park, J.K.; Song, Y.W.; Kim, J.R.; Cho, K.H. Patients with Rheumatoid Arthritis Show Altered Lipoprotein Profiles with Dysfunctional High-Density Lipoproteins that Can Exacerbate Inflammatory and Atherogenic Process. PLoS ONE 2016, 11, e0164564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.K.; Kim, J.Y.; Moon, J.Y.; Ahn, E.Y.; Lee, E.Y.; Lee, E.B.; Cho, K.H.; Song, Y.W. Altered lipoproteins in patients with systemic lupus erythematosus are associated with augmented oxidative stress: A potential role in atherosclerosis. Arthritis Res. Ther. 2016, 18, 306. [Google Scholar] [CrossRef] [Green Version]
- Torkhovskaia, T.I.; Fortinskaia, E.S.; Ivanova, L.I.; Nikitina, N.A.; Zakharova, T.S.; Kochetova, M.M.; Kliuchnikova Zh, I.; Sharapova, G.; Khalilov, E.M. Characteristics of the lipid transport system in psoriasis. Vopr. Med. Khim. 2002, 48, 297–303. [Google Scholar] [PubMed]
- Guérin, M.; Le Goff, W.; Lassel, T.S.; Van Tol, A.; Steiner, G.; Chapman, M.J. Atherogenic role of elevated CE transfer from HDL to VLDL(1) and dense LDL in type 2 diabetes: Impact of the degree of triglyceridemia. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 282–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riemens, S.C.; van Tol, A.; Sluiter, W.J.; Dullaart, R.P. Plasma phospholipid transfer protein activity is lowered by 24-h insulin and acipimox administration: Blunted response to insulin in type 2 diabetic patients. Diabetes 1999, 48, 1631–1637. [Google Scholar] [CrossRef] [PubMed]
- Cacciagiú, L.D.; González, A.I.; Gomez Rosso, L.; Meroño, T.; De Marziani, G.; Elbert, A.; Berg, G.; Brites, F.; Schreier, L. HDL-associated enzymes and proteins in hemodialysis patients. Clin. Biochem. 2012, 45, 243–248. [Google Scholar] [CrossRef] [PubMed]
- McCullough, A.; Previs, S.F.; Dasarathy, J.; Lee, K.; Osme, A.; Kim, C.; Ilchenko, S.; Lorkowski, S.W.; Smith, J.D.; Dasarathy, S.; et al. HDL flux is higher in patients with nonalcoholic fatty liver disease. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E852–E862. [Google Scholar] [CrossRef]
- Fadaei, R.; Poustchi, H.; Meshkani, R.; Moradi, N.; Golmohammadi, T.; Merat, S. Impaired HDL cholesterol efflux capacity in patients with non-alcoholic fatty liver disease is associated with subclinical atherosclerosis. Sci. Rep. 2018, 8, 11691. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, S.S.; Na, S.P.; Yu, C.Y.; Ji, Y.; Zhao, S.L.; Xie, R.J.; Bao, Y.S. Characterization of Lipoprotein-associated Phospholipase A2 in Serum in Patients with Stage 3-5 Chronic Kidney Disease. Am. J. Med. Sci. 2016, 352, 348–353. [Google Scholar] [CrossRef]
- Khovidhunkit, W.; Shigenaga, J.K.; Moser, A.H.; Feingold, K.R.; Grunfeld, C. Cholesterol efflux by acute-phase high density lipoprotein: Role of lecithin: Cholesterol acyltransferase. J. Lipid Res. 2001, 42, 967–975. [Google Scholar] [CrossRef]
- de la Llera Moya, M.; McGillicuddy, F.C.; Hinkle, C.C.; Byrne, M.; Joshi, M.R.; Nguyen, V.; Tabita-Martinez, J.; Wolfe, M.L.; Badellino, K.; Pruscino, L.; et al. Inflammation modulates human HDL composition and function in vivo. Atherosclerosis 2012, 222, 390–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanimoto, N.; Kumon, Y.; Suehiro, T.; Ohkubo, S.; Ikeda, Y.; Nishiya, K.; Hashimoto, K. Serum paraoxonase activity decreases in rheumatoid arthritis. Life Sci. 2003, 72, 2877–2885. [Google Scholar] [CrossRef]
- Shakoei, S.; Nakhjavani, M.; Mirmiranpoor, H.; Motlagh, M.A.; Azizpour, A.; Abedini, R. The Serum Level of Oxidative Stress and Antioxidant Markers in Patients with Psoriasis: A Cross-sectional Study. J. Clin. Aesthet. Dermatol. 2021, 14, 38–41. [Google Scholar] [PubMed]
- Trakaki, A.; Wolf, P.; Weger, W.; Eichmann, T.O.; Scharnagl, H.; Stadler, J.T.; Salmhofer, W.; Knuplez, E.; Holzer, M.; Marsche, G. Biological anti-psoriatic therapy profoundly affects high-density lipoprotein function. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 158943. [Google Scholar] [CrossRef] [PubMed]
- Riemens, S.; van Tol, A.; Sluiter, W.; Dullaart, R. Elevated plasma cholesteryl ester transfer in NIDDM: Relationships with apolipoprotein B-containing lipoproteins and phospholipid transfer protein. Atherosclerosis 1998, 140, 71–79. [Google Scholar] [CrossRef]
- Nakhjavani, M.; Morteza, A.; Karimi, R.; Banihashmi, Z.; Esteghamati, A. Diabetes induces gender gap on LCAT levels and activity. Life Sci. 2013, 92, 51–54. [Google Scholar] [CrossRef]
- Calabresi, L.; Simonelli, S.; Conca, P.; Busnach, G.; Cabibbe, M.; Gesualdo, L.; Gigante, M.; Penco, S.; Veglia, F.; Franceschini, G. Acquired lecithin:cholesterol acyltransferase deficiency as a major factor in lowering plasma HDL levels in chronic kidney disease. J. Intern. Med. 2015, 277, 552–561. [Google Scholar] [CrossRef]
- Janac, J.; Zeljkovic, A.; Jelic-Ivanovic, Z.; Dimitrijevic-Sreckovic, V.; Miljkovic, M.; Stefanovic, A.; Munjas, J.; Vekic, J.; Kotur-Stevuljevic, J.; Spasojević-Kalimanovska, V. The association between lecithin-cholesterol acyltransferase activity and fatty liver index. Ann. Clin. Biochem. 2019, 56, 583–592. [Google Scholar] [CrossRef]
- García-González, A.; Gaxiola-Robles, R.; Zenteno-Savín, T. Oxidative stress in patients with rheumatoid arthritis. Rev. Investig. Clin. 2015, 67, 46–53. [Google Scholar]
- Hassan, M.Q.; Hadi, R.A.; Al-Rawi, Z.S.; Padron, V.A.; Stohs, S.J. The glutathione defense system in the pathogenesis of rheumatoid arthritis. J. Appl. Toxicol. 2001, 21, 69–73. [Google Scholar] [CrossRef] [PubMed]
- ZZborovsky, A.; Martemyanov, V.; Stazharov, M.; Bedina, S.; Chernykh, T.; Mozgovaya, E. AB0024 Activities of purine metabolism enzymes and antioxidant system in rheumatoid arthritis patients. Anna. Rheum. Dis. 2001, 60, A367. [Google Scholar] [CrossRef]
- Turgay, M.; Durak, I.; Erten, S.; Ertugrul, E.; Devrim, E.; Avci, A.; Turgay, F. Oxidative stress and antioxidant parameters in a Turkish group of patients with active and inactive systemic lupus erythematosus. APLAR J. Rheumatol. 2007, 10, 101–106. [Google Scholar] [CrossRef]
- Shah, D.; Wanchu, A.; Bhatnagar, A. Interaction between oxidative stress and chemokines: Possible pathogenic role in systemic lupus erythematosus and rheumatoid arthritis. Immunobiology 2011, 216, 1010–1017. [Google Scholar] [CrossRef] [PubMed]
- Taysi, S.; Gul, M.; Sari, R.A.; Akcay, F.; Bakan, N. Serum oxidant/antioxidant status of patients with systemic lupus erythematosus. Clin. Chem. Lab. Med. 2002, 40, 684–688. [Google Scholar] [CrossRef]
- Kökçam, I.; Naziroğlu, M. Antioxidants and lipid peroxidation status in the blood of patients with psoriasis. Clin. Chim. Acta 1999, 289, 23–31. [Google Scholar] [CrossRef]
- Panjamurthy, K.; Manoharan, S.; Ramachandran, C.R. Lipid peroxidation and antioxidant status in patients with periodontitis. Cell Mol. Biol. Lett. 2005, 10, 255–264. [Google Scholar]
- Sreeram, M.; Suryakar, A.N.; Dani, N.H. Is gamma-glutamyl transpeptidase a biomarker for oxidative stress in periodontitis? J. Indian Soc. Periodontol. 2015, 19, 150–154. [Google Scholar] [CrossRef]
- Dhotre, P.S.; Suryakar, A.N.; Bhogade, R.B. Oxidative stress in periodontitis. Eur. J. Gen. Med. 2012, 9, 81–84. [Google Scholar] [CrossRef]
- Kaji, H.; Kurasaki, M.; Ito, K.; Saito, T.; Saito, K.; Niioka, T.; Kojima, Y.; Ohsaki, Y.; Ide, H.; Tsuji, M.; et al. Increased lipoperoxide value and glutathione peroxidase activity in blood plasma of type 2 (non-insulin-dependent) diabetic women. Klin. Wochenschr. 1985, 63, 765–768. [Google Scholar] [CrossRef]
- Ozdemir, G.; Ozden, M.; Maral, H.; Kuskay, S.; Cetinalp, P.; Tarkun, I. Malondialdehyde, glutathione, glutathione peroxidase and homocysteine levels in type 2 diabetic patients with and without microalbuminuria. Ann. Clin. Biochem. 2005, 42, 99–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zachara, B.A.; Salak, A.; Koterska, D.; Manitius, J.; Wasowicz, W. Selenium and glutathione peroxidases in blood of patients with different stages of chronic renal failure. J. Trace Elem. Med. Biol. 2004, 17, 291–299. [Google Scholar] [CrossRef]
- Roxborough, H.E.; Mercer, C.; McMaster, D.; Maxwell, A.P.; Young, I.S. Plasma glutathione peroxidase activity is reduced in haemodialysis patients. Nephron 1999, 81, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Świderska, M.; Maciejczyk, M.; Zalewska, A.; Pogorzelska, J.; Flisiak, R.; Chabowski, A. Oxidative stress biomarkers in the serum and plasma of patients with non-alcoholic fatty liver disease (NAFLD). Can plasma AGE be a marker of NAFLD? Oxidative stress biomarkers in NAFLD patients. Free Radic Res. 2019, 53, 841–850. [Google Scholar] [CrossRef]
- O’Neill, F.; Riwanto, M.; Charakida, M.; Colin, S.; Manz, J.; McLoughlin, E.; Khan, T.; Klein, N.; Kay, C.W.; Patel, K.; et al. Structural and functional changes in HDL with low grade and chronic inflammation. Int. J. Cardiol. 2015, 188, 111–116. [Google Scholar] [CrossRef]
- Rosenblat, M.; Karry, R.; Aviram, M. Paraoxonase 1 (PON1) is a more potent antioxidant and stimulant of macrophage cholesterol efflux, when present in HDL than in lipoprotein-deficient serum: Relevance to diabetes. Atherosclerosis 2006, 187, 74–81. [Google Scholar] [CrossRef]
- Abbott, C.A.; Mackness, M.I.; Kumar, S.; Boulton, A.J.; Durrington, P.N. Serum paraoxonase activity, concentration, and phenotype distribution in diabetes mellitus and its relationship to serum lipids and lipoproteins. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 1812–1818. [Google Scholar] [CrossRef]
- Ikeda, Y.; Suehiro, T.; Inoue, M.; Nakauchi, Y.; Morita, T.; Arii, K.; Ito, H.; Kumon, Y.; Hashimoto, K. Serum paraoxonase activity and its relationship to diabetic complications in patients with non-insulin-dependent diabetes mellitus. Metabolism 1998, 47, 598–602. [Google Scholar] [CrossRef]
- Samouilidou, E.; Kostopoulos, V.; Liaouri, A.; Kioussi, E.; Vassiliou, K.; Bountou, E.; Grapsa, E. Association of lipid profile with serum PON1 concentration in patients with chronic kidney disease. Ren. Fail 2016, 38, 1601–1606. [Google Scholar] [CrossRef] [Green Version]
- Sellam, J.; Proulle, V.; Jüngel, A.; Ittah, M.; Miceli Richard, C.; Gottenberg, J.E.; Toti, F.; Benessiano, J.; Gay, S.; Freyssinet, J.M.; et al. Increased levels of circulating microparticles in primary Sjögren’s syndrome, systemic lupus erythematosus and rheumatoid arthritis and relation with disease activity. Arthritis Res. Ther. 2009, 11, R156. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.K.; Farewell, V.; Vadas, P.; Bookman, A.A.; Keystone, E.C.; Pruzanski, W. Secretory phospholipase A2 as an index of disease activity in rheumatoid arthritis. Prospective double blind study of 212 patients. J. Rheumatol. 1996, 23, 1162–1166. [Google Scholar] [PubMed]
- Tejera-Segura, B.; Macía-Díaz, M.; Machado, J.D.; de Vera-González, A.; García-Dopico, J.A.; Olmos, J.M.; Hernández, J.L.; Díaz-González, F.; González-Gay, M.A.; Ferraz-Amaro, I. HDL cholesterol efflux capacity in rheumatoid arthritis patients: Contributing factors and relationship with subclinical atherosclerosis. Arthritis Res. Ther. 2017, 19, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Mills, D.; Bala, M. Psoriasis: Cardiovascular risk factors and other disease comorbidities. J. Drugs Dermatol. 2008, 7, 373–377. [Google Scholar] [PubMed]
- Persson, G.R.; Persson, R.E. Cardiovascular disease and periodontitis: An update on the associations and risk. J. Clin. Periodontol. 2008, 35, 362–379. [Google Scholar] [CrossRef]
- Monteiro, A.M.; Jardini, M.A.; Alves, S.; Giampaoli, V.; Aubin, E.C.; Figueiredo Neto, A.M.; Gidlund, M. Cardiovascular disease parameters in periodontitis. J. Periodontol. 2009, 80, 378–388. [Google Scholar] [CrossRef]
- Taleghani, F.; Shamaei, M.; Shamaei, M. Association between chronic periodontitis and serum lipid levels. Acta Med. Iran 2010, 48, 47–50. [Google Scholar]
- Iacopino, A.M.; Cutler, C.W. Pathophysiological relationships between periodontitis and systemic disease: Recent concepts involving serum lipids. J. Periodontol. 2000, 71, 1375–1384. [Google Scholar] [CrossRef]
- Riemens, S.C.; van Tol, A.; Sluiter, W.J.; Dullaart, R.P. Plasma phospholipid transfer protein activity is related to insulin resistance: Impaired acute lowering by insulin in obese Type II diabetic patients. Diabetologia 1998, 41, 929–934. [Google Scholar] [CrossRef] [Green Version]
- Arii, K.; Suehiro, T.; Yamamoto, M.; Ito, H.; Hashimoto, K. Suppression of plasma cholesteryl ester transfer protein activity in acute hyperinsulinemia and effect of plasma nonesterified fatty acid. Metabolism 1997, 46, 1166–1170. [Google Scholar] [CrossRef]
- Apro, J.; Tietge, U.J.; Dikkers, A.; Parini, P.; Angelin, B.; Rudling, M. Impaired Cholesterol Efflux Capacity of High-Density Lipoprotein Isolated From Interstitial Fluid in Type 2 Diabetes Mellitus-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 787–791. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Ronsein, G.E.; Tang, C.; Jarvik, G.P.; Davidson, W.S.; Kothari, V.; Song, H.D.; Segrest, J.P.; Bornfeldt, K.E.; Heinecke, J.W. Diabetes Impairs Cellular Cholesterol Efflux From ABCA1 to Small HDL Particles. Circ Res. 2020, 127, 1198–1210. [Google Scholar] [CrossRef] [PubMed]
- Gall, J.; Frisdal, E.; Bittar, R.; Le Goff, W.; Bruckert, E.; Lesnik, P.; Guerin, M.; Giral, P. Association of Cholesterol Efflux Capacity with Clinical Features of Metabolic Syndrome: Relevance to Atherosclerosis. J. Am. Heart Assoc. 2016, 5, e004808. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Shiu, S.W.; Wong, Y.; Tan, K.C. Impaired serum capacity to induce cholesterol efflux is associated with endothelial dysfunction in type 2 diabetes mellitus. Diab. Vasc. Dis. Res. 2009, 6, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Rysz, J.; Gluba-Brzózka, A.; Rysz-Górzyńska, M.; Franczyk, B. The Role and Function of HDL in Patients with Chronic Kidney Disease and the Risk of Cardiovascular Disease. Int. J. Mol. Sci. 2020, 21, 601. [Google Scholar] [CrossRef] [Green Version]
- Florens, N.; Calzada, C.; Delolme, F.; Page, A.; Guebre Egziabher, F.; Juillard, L.; Soulage, A.C.O. Proteomic Characterization of High-Density Lipoprotein Particles from Non-Diabetic Hemodialysis Patients. Toxins 2019, 11, 671. [Google Scholar] [CrossRef] [Green Version]
- Pasterk, L.; Lemesch, S.; Leber, B.; Trieb, M.; Curcic, S.; Stadlbauer, V.; Schuligoi, R.; Schicho, R.; Heinemann, A.; Marsche, G. Oxidized plasma albumin promotes platelet-endothelial crosstalk and endothelial tissue factor expression. Sci. Rep. 2016, 6, 22104. [Google Scholar] [CrossRef]
- Chatterjee, S. Chapter Two—Oxidative Stress, Inflammation, and Disease. In Oxidative Stress and Biomaterials; Dziubla, T., Butterfield, D.A., Eds.; Academic Press: Cambridge, MA, USA, 2016; pp. 35–58. [Google Scholar]
- Huang, Y.; DiDonato, J.A.; Levison, B.S.; Schmitt, D.; Li, L.; Wu, Y.; Buffa, J.; Kim, T.; Gerstenecker, G.S.; Gu, X.; et al. An abundant dysfunctional apolipoprotein A1 in human atheroma. Nat. Med. 2014, 20, 193–203. [Google Scholar] [CrossRef] [Green Version]
- Gao, D.; Ashraf, M.Z.; Zhang, L.; Kar, N.; Byzova, T.V.; Podrez, E.A. Cross-linking modifications of HDL apoproteins by oxidized phospholipids: Structural characterization, in vivo detection, and functional implications. J. Biol. Chem. 2020, 295, 1973–1984. [Google Scholar] [CrossRef]
- Luo, S.; Lei, H.; Qin, H.; Xia, Y. Molecular mechanisms of endothelial NO synthase uncoupling. Curr. Pharm. Des. 2014, 20, 3548–3553. [Google Scholar] [CrossRef]
- Zhang, R.; Brennan, M.L.; Fu, X.; Aviles, R.J.; Pearce, G.L.; Penn, M.S.; Topol, E.J.; Sprecher, D.L.; Hazen, S.L. Association between myeloperoxidase levels and risk of coronary artery disease. Jama 2001, 286, 2136–2142. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Nukuna, B.; Brennan, M.L.; Sun, M.; Goormastic, M.; Settle, M.; Schmitt, D.; Fu, X.; Thomson, L.; Fox, P.L.; et al. Apolipoprotein A-I is a selective target for myeloperoxidase-catalyzed oxidation and functional impairment in subjects with cardiovascular disease. J. Clin. Investig. 2004, 114, 529–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podrez, E.A.; Abu-Soud, H.M.; Hazen, S.L. Myeloperoxidase-generated oxidants and atherosclerosis. Free Radic. Biol. Med. 2000, 28, 1717–1725. [Google Scholar] [CrossRef]
- Undurti, A.; Huang, Y.; Lupica, J.A.; Smith, J.D.; DiDonato, J.A.; Hazen, S.L. Modification of high density lipoprotein by myeloperoxidase generates a pro-inflammatory particle. J. Biol. Chem. 2009, 284, 30825–30835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Nicholls, S.J.; Rodriguez, E.R.; Kummu, O.; Hörkkö, S.; Barnard, J.; Reynolds, W.F.; Topol, E.J.; DiDonato, J.A.; Hazen, S.L. Protein carbamylation links inflammation, smoking, uremia and atherogenesis. Nat. Med. 2007, 13, 1176–1184. [Google Scholar] [CrossRef]
- Verbrugge, F.H.; Tang, W.H.; Hazen, S.L. Protein carbamylation and cardiovascular disease. Kidney Int. 2015, 88, 474–478. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.P.; Bali, A.; Singh, N.; Jaggi, A.S. Advanced glycation end products and diabetic complications. Korean J. Physiol. Pharmacol. 2014, 18, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Kasumov, T.; Golizeh, M.; Kashyap, S. Glycation and deamidation result in HDL dysfunction in patients with type 2 diabetes. Diabetes 2018, 67 (Suppl. S1), 330-OR. [Google Scholar] [CrossRef]
- Kashyap, S.R.; Osme, A.; Ilchenko, S.; Golizeh, M.; Lee, K.; Wang, S.; Bena, J.; Previs, S.F.; Smith, J.D.; Kasumov, T. Glycation Reduces the Stability of ApoAI and Increases HDL Dysfunction in Diet-Controlled Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2018, 103, 388–396. [Google Scholar] [CrossRef] [Green Version]
- Hoang, A.; Murphy, A.J.; Coughlan, M.T.; Thomas, M.C.; Forbes, J.M.; O’Brien, R.; Cooper, M.E.; Chin-Dusting, J.P.; Sviridov, D. Advanced glycation of apolipoprotein A-I impairs its anti-atherogenic properties. Diabetologia 2007, 50, 1770–1779. [Google Scholar] [CrossRef]
- Hedrick, C.C.; Thorpe, S.R.; Fu, M.X.; Harper, C.M.; Yoo, J.; Kim, S.M.; Wong, H.; Peters, A.L. Glycation impairs high-density lipoprotein function. Diabetologia 2000, 43, 312–320. [Google Scholar] [CrossRef] [Green Version]
- Nagata, H.; Lyu, J.; Imachi, H.; Fukunaga, K.; Sato, S.; Kobayashi, T.; Saheki, T.; Seo, K.; Salimah, J.B.; Iwama, H.; et al. AGEs inhibit scavenger receptor class B type I gene expression via Smad1 in HUVECs. J. Mol. Endocrinol. 2021, 66, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Matsuki, K.; Tamasawa, N.; Yamashita, M.; Tanabe, J.; Murakami, H.; Matsui, J.; Imaizumi, T.; Satoh, K.; Suda, T. Metformin restores impaired HDL-mediated cholesterol efflux due to glycation. Atherosclerosis 2009, 206, 434–438. [Google Scholar] [CrossRef] [PubMed]
- de Leeuw, K.; Graaff, R.; de Vries, R.; Dullaart, R.P.; Smit, A.J.; Kallenberg, C.G.; Bijl, M. Accumulation of advanced glycation endproducts in patients with systemic lupus erythematosus. Rheumatology 2007, 46, 1551–1556. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Sato, E.; Fujiwara, N.; Kawagoe, Y.; Ueda, Y.; Suzuki, T.; Yamada, S.; Takeuchi, M.; Fukami, K.; Ueda, S.; et al. Positive association of serum levels of advanced glycation end products and high mobility group box-1 with asymmetric dimethylarginine in nondiabetic chronic kidney disease patients. Metabolism 2009, 58, 1624–1628. [Google Scholar] [CrossRef]
- Bijnen, M.; van Greevenbroek, M.M.J.; van der Kallen, C.J.H.; Scheijen, J.L.; van de Waarenburg, M.P.H.; Stehouwer, C.D.A.; Wouters, K.; Schalkwijk, C.G. Hepatic Fat Content and Liver Enzymes Are Associated with Circulating Free and Protein-Bound Advanced Glycation End Products, Which Are Associated with Low-Grade Inflammation: The CODAM Study. J. Diabetes Res. 2019, 2019, 6289831. [Google Scholar] [CrossRef] [Green Version]
- Duell, P.B.; Oram, J.F.; Bierman, E.L. Nonenzymatic glycosylation of HDL and impaired HDL-receptor-mediated cholesterol efflux. Diabetes 1991, 40, 377–384. [Google Scholar] [CrossRef]
- Liu, D.; Ji, L.; Zhang, D.; Tong, X.; Pan, B.; Liu, P.; Zhang, Y.; Huang, Y.; Su, J.; Willard, B.; et al. Nonenzymatic glycation of high-density lipoprotein impairs its anti-inflammatory effects in innate immunity. Diabetes Metab. Res. Rev. 2012, 28, 186–195. [Google Scholar] [CrossRef]
- Bäck, M.; Hansson, G.K. Anti-inflammatory therapies for atherosclerosis. Nat. Rev. Cardiol. 2015, 12, 199–211. [Google Scholar] [CrossRef]
- Kashyap, S.; Kheniser, K.; Li, L.; Bena, J.; Kasumov, T. The therapeutic efficacy of intensive medical therapy in ameliorating high-density lipoprotein dysfunction in subjects with type two diabetes. Lipids Health Dis. 2016, 15, 141. [Google Scholar] [CrossRef]
- Westlake, S.L.; Colebatch, A.N.; Baird, J.; Kiely, P.; Quinn, M.; Choy, E.; Ostor, A.J.; Edwards, C.J. The effect of methotrexate on cardiovascular disease in patients with rheumatoid arthritis: A systematic literature review. Rheumatology 2010, 49, 295–307. [Google Scholar] [CrossRef] [Green Version]
- Charles-Schoeman, C.; Gugiu, G.B.; Ge, H.; Shahbazian, A.; Lee, Y.Y.; Wang, X.; Furst, D.E.; Ranganath, V.K.; Maldonado, M.; Lee, T.; et al. Remodeling of the HDL proteome with treatment response to abatacept or adalimumab in the AMPLE trial of patients with rheumatoid arthritis. Atherosclerosis 2018, 275, 107–114. [Google Scholar] [CrossRef]
- Jafri, K.; Taylor, L.; Nezamzadeh, M.; Baker, J.F.; Mehta, N.N.; Bartels, C.; Williams, C.T.; Ogdie, A. Management of hyperlipidemia among patients with rheumatoid arthritis in the primary care setting. BMC Musculoskelet. Disord. 2015, 16, 237. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.H.; Kim, S.G. Fibrates Revisited: Potential Role in Cardiovascular Risk Reduction. Diabetes Metab. J. 2020, 44, 213–221. [Google Scholar] [CrossRef]
- Yamashita, S.; Masuda, D.; Matsuzawa, Y. Pemafibrate, a New Selective PPARα Modulator: Drug Concept and Its Clinical Applications for Dyslipidemia and Metabolic Diseases. Curr. Atheroscler. Rep. 2020, 22, 5. [Google Scholar] [CrossRef] [Green Version]
- Marx, N.; Sukhova, G.K.; Collins, T.; Libby, P.; Plutzky, J. PPARalpha activators inhibit cytokine-induced vascular cell adhesion molecule-1 expression in human endothelial cells. Circulation 1999, 99, 3125–3131. [Google Scholar] [CrossRef] [Green Version]
- Meade, T.; Zuhrie, R.; Cook, C.; Cooper, J. Bezafibrate in men with lower extremity arterial disease: Randomised controlled trial. Bmj 2002, 325, 1139. [Google Scholar] [CrossRef] [Green Version]
- Keech, A.; Simes, R.J.; Barter, P.; Best, J.; Scott, R.; Taskinen, M.R.; Forder, P.; Pillai, A.; Davis, T.; Glasziou, P.; et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): Randomised controlled trial. Lancet 2005, 366, 1849–1861. [Google Scholar] [CrossRef]
- Bezafibrate Infarction Prevention (BIP) Study. Secondary prevention by raising HDL cholesterol and reducing triglycerides in patients with coronary artery disease. Circulation 2000, 102, 21–27. [CrossRef]
- Kim, N.H.; Han, K.H.; Choi, J.; Lee, J.; Kim, S.G. Use of fenofibrate on cardiovascular outcomes in statin users with metabolic syndrome: Propensity matched cohort study. Bmj 2019, 366, l5125. [Google Scholar] [CrossRef]
- Rubins, H.B.; Robins, S.J.; Collins, D.; Fye, C.L.; Anderson, J.W.; Elam, M.B.; Faas, F.H.; Linares, E.; Schaefer, E.J.; Schectman, G.; et al. Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial Study Group. N. Engl. J. Med. 1999, 341, 410–418. [Google Scholar] [CrossRef]
- Zhu, L.; Hayen, A.; Bell, K.J.L. Legacy effect of fibrate add-on therapy in diabetic patients with dyslipidemia: A secondary analysis of the ACCORDION study. Cardiovasc. Diabetol. 2020, 19, 28. [Google Scholar] [CrossRef] [Green Version]
- Shirinsky, I.; Polovnikova, O.; Kalinovskaya, N.; Shirinsky, V. The effects of fenofibrate on inflammation and cardiovascular markers in patients with active rheumatoid arthritis: A pilot study. Rheumatol. Int. 2013, 33, 3045–3048. [Google Scholar] [CrossRef]
- Jun, M.; Foote, C.; Lv, J.; Neal, B.; Patel, A.; Nicholls, S.J.; Grobbee, D.E.; Cass, A.; Chalmers, J.; Perkovic, V. Effects of fibrates on cardiovascular outcomes: A systematic review and meta-analysis. Lancet 2010, 375, 1875–1884. [Google Scholar] [CrossRef]
- Lee, M.; Saver, J.L.; Towfighi, A.; Chow, J.; Ovbiagele, B. Efficacy of fibrates for cardiovascular risk reduction in persons with atherogenic dyslipidemia: A meta-analysis. Atherosclerosis 2011, 217, 492–498. [Google Scholar] [CrossRef]
- Bruckert, E.; Labreuche, J.; Deplanque, D.; Touboul, P.J.; Amarenco, P. Fibrates effect on cardiovascular risk is greater in patients with high triglyceride levels or atherogenic dyslipidemia profile: A systematic review and meta-analysis. J. Cardiovasc. Pharmacol. 2011, 57, 267–272. [Google Scholar] [CrossRef]
- Jakob, T.; Nordmann, A.J.; Schandelmaier, S.; Ferreira-González, I.; Briel, M. Fibrates for primary prevention of cardiovascular disease events. Cochrane Database Syst. Rev. 2016, 11, Cd009753. [Google Scholar] [CrossRef]
- Sgro, C.; Escousse, A. Side effects of fibrates (except liver and muscle). Therapie 1991, 46, 351–354. [Google Scholar]
- Yokote, K.; Yamashita, S.; Arai, H.; Araki, E.; Suganami, H.; Ishibashi, S. A pooled analysis of pemafibrate phase II/III clinical trials indicated significant improvement in glycemic and liver function-related parameters. Atheroscler. Suppl. 2018, 32, 154–155. [Google Scholar] [CrossRef]
- Yokote, K.; Yamashita, S.; Arai, H.; Araki, E.; Suganami, H.; Ishibashi, S.; on Behalf of the K-877 Study Group. Long-Term Efficacy and Safety of Pemafibrate, a Novel Selective Peroxisome Proliferator-Activated Receptor-α Modulator (SPPARMα), in Dyslipidemic Patients with Renal Impairment. Int. J. Mol. Sci. 2019, 20, 706. [Google Scholar] [CrossRef] [Green Version]
- Ida, S.; Kaneko, R.; Murata, K. Efficacy and safety of pemafibrate administration in patients with dyslipidemia: A systematic review and meta-analysis. Cardiovasc. Diabetol. 2019, 18, 38. [Google Scholar] [CrossRef]
- Blair, H.A. Pemafibrate: First Global Approval. Drugs 2017, 77, 1805–1810. [Google Scholar] [CrossRef]
- Kowa Research Institute. Kowa to Discontinue K-877 (Pemafibrate) “prominent” Cardiovascular Outcomes Study. Available online: https://www.prnewswire.com/news-releases/kowa-to-discontinue-k-877-pemafibrate-prominent-cardiovascular-outcomes-study-301520956.html (accessed on 2 July 2022).
- Remaley, A.T.; Norata, G.D.; Catapano, A.L. Novel concepts in HDL pharmacology. Cardiovasc. Res. 2014, 103, 423–428. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.H.; Kamanna, V.S.; Zhang, M.C.; Kashyap, M.L. Niacin inhibits surface expression of ATP synthase beta chain in HepG2 cells: Implications for raising HDL. J. Lipid Res. 2008, 49, 1195–1201. [Google Scholar] [CrossRef] [Green Version]
- Boden, W.E.; Probstfield, J.L.; Anderson, T.; Chaitman, B.R.; Desvignes-Nickens, P.; Koprowicz, K.; McBride, R.; Teo, K.; Weintraub, W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N. Engl. J. Med. 2011, 365, 2255–2267. [Google Scholar] [CrossRef] [Green Version]
- Michos, E.D.; Sibley, C.T.; Baer, J.T.; Blaha, M.J.; Blumenthal, R.S. Niacin and statin combination therapy for atherosclerosis regression and prevention of cardiovascular disease events: Reconciling the AIM-HIGH (Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglycerides: Impact on Global Health Outcomes) trial with previous surrogate endpoint trials. J. Am. Coll. Cardiol. 2012, 59, 2058–2064. [Google Scholar] [CrossRef] [Green Version]
- Schandelmaier, S.; Briel, M.; Saccilotto, R.; Olu, K.K.; Arpagaus, A.; Hemkens, L.G.; Nordmann, A.J. Niacin for primary and secondary prevention of cardiovascular events. Cochrane Database Syst. Rev. 2017, 6, Cd009744. [Google Scholar] [CrossRef]
- Li, H.; Xu, X.; Lu, L.; Sun, R.; Guo, Q.; Chen, Q.; Wang, J.; He, Z.; Zhang, Y. The comparative impact among different intensive statins and combination therapies with niacin/ezetimibe on carotid intima-media thickness: A systematic review, traditional meta-analysis, and network meta-analysis of randomized controlled trials. Eur. J. Clin. Pharmacol. 2021, 77, 1133–1145. [Google Scholar] [CrossRef]
- Maron, D.J.; Fazio, S.; Linton, M.F. Current perspectives on statins. Circulation 2000, 101, 207–213. [Google Scholar] [CrossRef] [Green Version]
- Jacob, B.G.; Möhrle, W.; Richter, W.O.; Schwandt, P. Short- and long-term effects of lovastatin and pravastatin alone and in combination with cholestyramine on serum lipids, lipoproteins and apolipoproteins in primary hypercholesterolaemia. Eur. J. Clin. Pharmacol. 1992, 42, 353–358. [Google Scholar] [CrossRef]
- Cuchel, M.; Schaefer, E.J.; Millar, J.S.; Jones, P.J.; Dolnikowski, G.G.; Vergani, C.; Lichtenstein, A.H. Lovastatin decreases de novo cholesterol synthesis and LDL Apo B-100 production rates in combined-hyperlipidemic males. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 1910–1917. [Google Scholar] [CrossRef]
- McTaggart, F.; Jones, P. Effects of statins on high-density lipoproteins: A potential contribution to cardiovascular benefit. Cardiovasc. Drugs Ther. 2008, 22, 321–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, G.; Duez, H.; Blanquart, C.; Berezowski, V.; Poulain, P.; Fruchart, J.C.; Najib-Fruchart, J.; Glineur, C.; Staels, B. Statin-induced inhibition of the Rho-signaling pathway activates PPARalpha and induces HDL apoA-I. J. Clin. Investig. 2001, 107, 1423–1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.W.; Kang, H.J.; Jhon, M.; Kim, J.W.; Lee, J.Y.; Walker, A.J.; Agustini, B.; Kim, J.M.; Berk, M. Statins and Inflammation: New Therapeutic Opportunities in Psychiatry. Front. Psychiatry 2019, 10, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahalwar, R.; Khanna, D. Pleiotropic antioxidant potential of rosuvastatin in preventing cardiovascular disorders. Eur. J. Pharmacol. 2013, 711, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Hilgendorff, A.; Muth, H.; Parviz, B.; Staubitz, A.; Haberbosch, W.; Tillmanns, H.; Hölschermann, H. Statins differ in their ability to block NF-kappaB activation in human blood monocytes. Int. J. Clin. Pharmacol. Ther 2003, 41, 397–401. [Google Scholar] [CrossRef]
- Geluk, C.A.; Asselbergs, F.W.; Hillege, H.L.; Bakker, S.J.; de Jong, P.E.; Zijlstra, F.; van Gilst, W.H. Impact of statins in microalbuminuric subjects with the metabolic syndrome: A substudy of the PREVEND Intervention Trial. Eur. Heart J. 2005, 26, 1314–1320. [Google Scholar] [CrossRef]
- Chen, Y.H.; Feng, B.; Chen, Z.W. Statins for primary prevention of cardiovascular and cerebrovascular events in diabetic patients without established cardiovascular diseases: A meta-analysis. Exp. Clin. Endocrinol. Diabetes 2012, 120, 116–120. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.H.; Chen, P.C.; Yang, Y.H.; Wang, L.C.; Lee, J.H.; Lin, Y.T.; Chiang, B.L. Statin reduces mortality and morbidity in systemic lupus erythematosus patients with hyperlipidemia: A nationwide population-based cohort study. Atherosclerosis 2015, 243, 11–18. [Google Scholar] [CrossRef]
- McCarey, D.W.; McInnes, I.B.; Madhok, R.; Hampson, R.; Scherbakov, O.; Ford, I.; Capell, H.A.; Sattar, N. Trial of Atorvastatin in Rheumatoid Arthritis (TARA): Double-blind, randomised placebo-controlled trial. Lancet 2004, 363, 2015–2021. [Google Scholar] [CrossRef]
- Kitas, G.D.; Nightingale, P.; Armitage, J.; Sattar, N.; Belch, J.J.F.; Symmons, D.P.M. A Multicenter, Randomized, Placebo-Controlled Trial of Atorvastatin for the Primary Prevention of Cardiovascular Events in Patients with Rheumatoid Arthritis. Arthritis Rheumatol. 2019, 71, 1437–1449. [Google Scholar] [CrossRef]
- Cholesterol Treatment Trialists’ Collaboration. Efficacy and safety of statin therapy in older people: A meta-analysis of individual participant data from 28 randomised controlled trials. Lancet 2019, 393, 407–415. [Google Scholar] [CrossRef] [Green Version]
- Taylor, F.; Huffman, M.D.; Macedo, A.F.; Moore, T.H.; Burke, M.; Davey Smith, G.; Ward, K.; Ebrahim, S. Statins for the primary prevention of cardiovascular disease. Cochrane Database Syst. Rev. 2013, 2013, Cd004816. [Google Scholar] [CrossRef] [PubMed]
- Sirtori, C.R. The pharmacology of statins. Pharmacol. Res. 2014, 88, 3–11. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Waylivra. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/waylivra (accessed on 2 July 2022).
- Sacks Editorial Board, F.M.; Zheng, C.; Cohn Editorial Board, J.S. Complexities of plasma apolipoprotein C-III metabolism. J. Lipid Res. 2011, 52, 1067–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witztum, J.L.; Gaudet, D.; Freedman, S.D.; Alexander, V.J.; Digenio, A.; Williams, K.R.; Yang, Q.; Hughes, S.G.; Geary, R.S.; Arca, M.; et al. Volanesorsen and Triglyceride Levels in Familial Chylomicronemia Syndrome. N. Engl. J. Med. 2019, 381, 531–542. [Google Scholar] [CrossRef]
- Fogacci, F.; Norata, G.D.; Toth, P.P.; Arca, M.; Cicero, A.F.G. Efficacy and Safety of Volanesorsen (ISIS 304801): The Evidence from Phase 2 and 3 Clinical Trials. Curr. Atheroscler. Rep. 2020, 22, 18. [Google Scholar] [CrossRef]
- Gouni-Berthold, I.; Alexander, V.J.; Yang, Q.; Hurh, E.; Steinhagen-Thiessen, E.; Moriarty, P.M.; Hughes, S.G.; Gaudet, D.; Hegele, R.A.; O’Dea, L.S.L.; et al. Efficacy and safety of volanesorsen in patients with multifactorial chylomicronaemia (COMPASS): A multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Diabetes Endocrinol. 2021, 9, 264–275. [Google Scholar] [CrossRef]
- Digenio, A.; Dunbar, R.L.; Alexander, V.J.; Hompesch, M.; Morrow, L.; Lee, R.G.; Graham, M.J.; Hughes, S.G.; Yu, R.; Singleton, W.; et al. Antisense-Mediated Lowering of Plasma Apolipoprotein C-III by Volanesorsen Improves Dyslipidemia and Insulin Sensitivity in Type 2 Diabetes. Diabetes Care 2016, 39, 1408–1415. [Google Scholar] [CrossRef] [Green Version]
- Tall, A.R.; Rader, D.J. Trials and Tribulations of CETP Inhibitors. Circ Res. 2018, 122, 106–112. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Olsson, A.G.; Abt, M.; Ballantyne, C.M.; Barter, P.J.; Brumm, J.; Chaitman, B.R.; Holme, I.M.; Kallend, D.; Leiter, L.A.; et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N. Engl. J. Med. 2012, 367, 2089–2099. [Google Scholar] [CrossRef]
- Lincoff, A.M.; Nicholls, S.J.; Riesmeyer, J.S.; Barter, P.J.; Brewer, H.B.; Fox, K.A.A.; Gibson, C.M.; Granger, C.; Menon, V.; Montalescot, G.; et al. Evacetrapib and Cardiovascular Outcomes in High-Risk Vascular Disease. N. Engl. J. Med. 2017, 376, 1933–1942. [Google Scholar] [CrossRef] [PubMed]
- Barter, P.J.; Caulfield, M.; Eriksson, M.; Grundy, S.M.; Kastelein, J.J.; Komajda, M.; Lopez-Sendon, J.; Mosca, L.; Tardif, J.C.; Waters, D.D.; et al. Effects of torcetrapib in patients at high risk for coronary events. N. Engl. J. Med. 2007, 357, 2109–2122. [Google Scholar] [CrossRef] [Green Version]
- Furtado, J.D.; Ruotolo, G.; Nicholls, S.J.; Dullea, R.; Carvajal-Gonzalez, S.; Sacks, F.M. Pharmacological Inhibition of CETP (Cholesteryl Ester Transfer Protein) Increases HDL (High-Density Lipoprotein) That Contains ApoC3 and Other HDL Subspecies Associated with Higher Risk of Coronary Heart Disease. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Luu, T.; Emfinger, C.H.; Parks, B.A.; Shi, J.; Trefts, E.; Zeng, F.; Kuklenyik, Z.; Harris, R.C.; Wasserman, D.H.; et al. CETP Inhibition Improves HDL Function but Leads to Fatty Liver and Insulin Resistance in CETP-Expressing Transgenic Mice on a High-Fat Diet. Diabetes 2018, 67, 2494–2506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, L.; Hopewell, J.C.; Chen, F.; Wallendszus, K.; Stevens, W.; Collins, R.; Wiviott, S.D.; Cannon, C.P.; Braunwald, E.; Sammons, E.; et al. Effects of Anacetrapib in Patients with Atherosclerotic Vascular Disease. N. Engl. J. Med. 2017, 377, 1217–1227. [Google Scholar] [CrossRef]
- Merck & Co., Inc., Kenilworth, NJ, USA. Merck Provides Update on Anacetrapib Development Program. Available online: https://www.merck.com/news/merck-provides-update-on-anacetrapib-development-program/ (accessed on 2 July 2022).
- Sammons, E.; Hopewell, J.C.; Chen, F.; Stevens, W.; Wallendszus, K.; Valdes-Marquez, E.; Dayanandan, R.; Knott, C.; Murphy, K.; Wincott, E.; et al. Long-term safety and efficacy of anacetrapib in patients with atherosclerotic vascular disease. Eur. Heart J. 2022, 43, 1416–1424. [Google Scholar] [CrossRef]
- Whitlock, M.E.; Swenson, T.L.; Ramakrishnan, R.; Leonard, M.T.; Marcel, Y.L.; Milne, R.W.; Tall, A.R. Monoclonal antibody inhibition of cholesteryl ester transfer protein activity in the rabbit. Effects on lipoprotein composition and high density lipoprotein cholesteryl ester metabolism. J. Clin. Investig. 1989, 84, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Komori, T. CETi-1. AVANT. Curr. Opin. Investig. Drugs 2004, 5, 334–338. [Google Scholar]
- Davidson, M.H.; Maki, K.; Umporowicz, D.; Wheeler, A.; Rittershaus, C.; Ryan, U. The safety and immunogenicity of a CETP vaccine in healthy adults. Atherosclerosis 2003, 169, 113–120. [Google Scholar] [CrossRef]
- Kramer, W. Novel drug approaches in development for the treatment of lipid disorders. Exp. Clin. Endocrinol. Diabetes 2013, 121, 567–580. [Google Scholar] [CrossRef]
- Carballo-Jane, E.; Chen, Z.; O’Neill, E.; Wang, J.; Burton, C.; Chang, C.H.; Chen, X.; Eveland, S.; Frantz-Wattley, B.; Gagen, K.; et al. ApoA-I mimetic peptides promote pre-β HDL formation in vivo causing remodeling of HDL and triglyceride accumulation at higher dose. Bioorg. Med. Chem. 2010, 18, 8669–8678. [Google Scholar] [CrossRef] [PubMed]
- Van Lenten, B.J.; Wagner, A.C.; Anantharamaiah, G.M.; Navab, M.; Reddy, S.T.; Buga, G.M.; Fogelman, A.M. Apolipoprotein A-I mimetic peptides. Curr. Atheroscler. Rep. 2009, 11, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Navab, M.; Anantharamaiah, G.M.; Reddy, S.T.; Hama, S.; Hough, G.; Grijalva, V.R.; Yu, N.; Ansell, B.J.; Datta, G.; Garber, D.W.; et al. Apolipoprotein A-I mimetic peptides. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1325–1331. [Google Scholar] [CrossRef] [Green Version]
- Dunbar, R.L.; Movva, R.; Bloedon, L.T.; Duffy, D.; Norris, R.B.; Navab, M.; Fogelman, A.M.; Rader, D.J. Oral Apolipoprotein A-I Mimetic D-4F Lowers HDL-Inflammatory Index in High-Risk Patients: A First-in-Human Multiple-Dose, Randomized Controlled Trial. Clin. Transl. Sci. 2017, 10, 455–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, C.E.; Weissbach, N.; Kjems, L.; Ayalasomayajula, S.; Zhang, Y.; Chang, I.; Navab, M.; Hama, S.; Hough, G.; Reddy, S.T.; et al. Treatment of patients with cardiovascular disease with L-4F, an apo-A1 mimetic, did not improve select biomarkers of HDL function. J. Lipid Res. 2011, 52, 361–373. [Google Scholar] [CrossRef] [Green Version]
- Wolska, A.; Reimund, M.; Sviridov, D.O.; Amar, M.J.; Remaley, A.T. Apolipoprotein Mimetic Peptides: Potential New Therapies for Cardiovascular Diseases. Cells 2021, 10, 597. [Google Scholar] [CrossRef] [PubMed]
- Abudukeremu, A.; Li, H.; Sun, R.; Liu, X.; Wu, X.; Xie, X.; Huang, J.; Zhang, J.; Bao, J.; Zhang, Y. Efficacy and safety of HDL/apoA-1 mimetics on human and mice with atherosclerosis: A systematic review and meta-analysis. Eur. Heart J. 2022, 43, ehab849.075. [Google Scholar] [CrossRef]
- Huang, J.; Wang, D.; Huang, L.H.; Huang, H. Roles of Reconstituted High-Density Lipoprotein Nanoparticles in Cardiovascular Disease: A New Paradigm for Drug Discovery. Int. J. Mol. Sci. 2020, 21, 739. [Google Scholar] [CrossRef] [Green Version]
- Nissen, S.E.; Tsunoda, T.; Tuzcu, E.M.; Schoenhagen, P.; Cooper, C.J.; Yasin, M.; Eaton, G.M.; Lauer, M.A.; Sheldon, W.S.; Grines, C.L.; et al. Effect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: A randomized controlled trial. Jama 2003, 290, 2292–2300. [Google Scholar] [CrossRef]
- Reijers, J.A.A.; Kallend, D.G.; Malone, K.E.; Jukema, J.W.; Wijngaard, P.L.J.; Burggraaf, J.; Moerland, M. MDCO-216 Does Not Induce Adverse Immunostimulation, in Contrast to Its Predecessor ETC-216. Cardiovasc. Drugs Ther. 2017, 31, 381–389. [Google Scholar] [CrossRef] [Green Version]
- Nicholls, S.J.; Puri, R.; Ballantyne, C.M.; Jukema, J.W.; Kastelein, J.J.P.; Koenig, W.; Wright, R.S.; Kallend, D.; Wijngaard, P.; Borgman, M.; et al. Effect of Infusion of High-Density Lipoprotein Mimetic Containing Recombinant Apolipoprotein A-I Milano on Coronary Disease in Patients with an Acute Coronary Syndrome in the MILANO-PILOT Trial: A Randomized Clinical Trial. JAMA Cardiol. 2018, 3, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, S.J.; Andrews, J.; Kastelein, J.J.P.; Merkely, B.; Nissen, S.E.; Ray, K.K.; Schwartz, G.G.; Worthley, S.G.; Keyserling, C.; Dasseux, J.L.; et al. Effect of Serial Infusions of CER-001, a Pre-β High-Density Lipoprotein Mimetic, on Coronary Atherosclerosis in Patients Following Acute Coronary Syndromes in the CER-001 Atherosclerosis Regression Acute Coronary Syndrome Trial: A Randomized Clinical Trial. JAMA Cardiol. 2018, 3, 815–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tardif, J.C.; Grégoire, J.; L’Allier, P.L.; Ibrahim, R.; Lespérance, J.; Heinonen, T.M.; Kouz, S.; Berry, C.; Basser, R.; Lavoie, M.A.; et al. Effects of reconstituted high-density lipoprotein infusions on coronary atherosclerosis: A randomized controlled trial. Jama 2007, 297, 1675–1682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, C.M.; Kerneis, M.; Yee, M.K.; Daaboul, Y.; Korjian, S.; Mehr, A.P.; Tricoci, P.; Alexander, J.H.; Kastelein, J.J.P.; Mehran, R.; et al. The CSL112-2001 trial: Safety and tolerability of multiple doses of CSL112 (apolipoprotein A-I [human]), an intravenous formulation of plasma-derived apolipoprotein A-I, among subjects with moderate renal impairment after acute myocardial infarction. Am. Heart J. 2019, 208, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Liu, J.; Wang, W.; Wang, M.; Zhao, F.; Sun, J.; Liu, J.; Zhao, D. Apolipoprotein E-containing high-density lipoprotein (HDL) modifies the impact of cholesterol-overloaded HDL on incident coronary heart disease risk: A community-based cohort study. J. Clin. Lipidol. 2018, 12, 89–98.e82. [Google Scholar] [CrossRef]
- White, C.R.; Garber, D.W.; Anantharamaiah, G.M. Anti-inflammatory and cholesterol-reducing properties of apolipoprotein mimetics: A review. J. Lipid Res. 2014, 55, 2007–2021. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Liu, H.; Liu, M.; Li, F.; Liu, L.; Du, F.; Fan, D.; Yu, H. A human apolipoprotein E mimetic peptide reduces atherosclerosis in aged apolipoprotein E null mice. Am. J. Transl. Res. 2016, 8, 3482–3492. [Google Scholar]
- Anantharamaiah, G.M.; Garber, D.W.; Goldberg, D.; Morrel, E.; Datta, G.; Palgunachari, M.N.; Register, T.C.; Appt, S.E.; White, C.R. Novel fatty acyl apoE mimetic peptides have increased potency to reduce plasma cholesterol in mice and macaques. J. Lipid Res. 2018, 59, 2075–2083. [Google Scholar] [CrossRef] [Green Version]
- Datta, G.; Chaddha, M.; Handattu, S.P.; Palgunachari, M.N.; Nayyar, G.; Garber, D.W.; Gupta, H.; White, C.R.; Anantharamaiah, G.M. ApoE mimetic peptide reduces plasma lipid hydroperoxide content with a concomitant increase in HDL paraoxonase activity. Adv. Exp. Med. Biol. 2010, 660, 1–4. [Google Scholar] [CrossRef]
- White, C.R.; Goldberg, D.I.; Anantharamaiah, G.M. Recent developments in modulating atherogenic lipoproteins. Curr. Opin. Lipidol. 2015, 26, 369–375. [Google Scholar] [CrossRef]
- Capstone Therapeutics. Capstone Therapeutics Announces Phase 1b/2a Study Results for AEM-28 Showing Safety and Biomarker Efficacy Signals. Available online: https://www.globenewswire.com/news-release/2014/12/16/691690/10112426/en/Capstone-Therapeutics-Announces-Phase-1b-2a-Study-Results-for-AEM-28-Showing-Safety-and-Biomarker-Efficacy-Signals.html (accessed on 3 July 2022).
- Garber, D.W.; Goldberg, D.; Anantharamaiah, G.M. Apolipoprotein E Mimetic Peptides: Cholesterol-Dependent and Cholesterol-Independent Properties. In Apolipoprotein Mimetics in the Management of Human Disease; Anantharamaiah, G.M., Goldberg, D., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 135–156. [Google Scholar]
- Le Lay, J.E.; Du, Q.; Mehta, M.B.; Bhagroo, N.; Hummer, B.T.; Falloon, J.; Carlson, G.; Rosenbaum, A.I.; Jin, C.; Kimko, H.; et al. Blocking endothelial lipase with monoclonal antibody MEDI5884 durably increases high density lipoprotein in nonhuman primates and in a phase 1 trial. Sci. Transl. Med. 2021, 13, eabb0602. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Millar, J.S.; Broedl, U.; Glick, J.M.; Rader, D.J. Inhibition of endothelial lipase causes increased HDL cholesterol levels in vivo. J. Clin. Investig. 2003, 111, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Ruff, C.T.; Koren, M.J.; Grimsby, J.; Rosenbaum, A.I.; Tu, X.; Karathanasis, S.K.; Falloon, J.; Hsia, J.; Guan, Y.; Conway, J.; et al. LEGACY: Phase 2a Trial to Evaluate the Safety, Pharmacokinetics, and Pharmacodynamic Effects of the Anti-EL (Endothelial Lipase) Antibody MEDI5884 in Patients With Stable Coronary Artery Disease. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 3005–3014. [Google Scholar] [CrossRef] [PubMed]
- Morgado-Pascual, J.L.; Rayego-Mateos, S.; Tejedor, L.; Suarez-Alvarez, B.; Ruiz-Ortega, M. Bromodomain and Extraterminal Proteins as Novel Epigenetic Targets for Renal Diseases. Front. Pharmacol. 2019, 10, 1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilham, D.; Wasiak, S.; Tsujikawa, L.M.; Halliday, C.; Norek, K.; Patel, R.G.; Kulikowski, E.; Johansson, J.; Sweeney, M.; Wong, N.C.; et al. RVX-208, a BET-inhibitor for treating atherosclerotic cardiovascular disease, raises ApoA-I/HDL and represses pathways that contribute to cardiovascular disease. Atherosclerosis 2016, 247, 48–57. [Google Scholar] [CrossRef] [Green Version]
- Tsujikawa, L.M.; Fu, L.; Das, S.; Halliday, C.; Rakai, B.D.; Stotz, S.C.; Sarsons, C.D.; Gilham, D.; Daze, E.; Wasiak, S.; et al. Apabetalone (RVX-208) reduces vascular inflammation in vitro and in CVD patients by a BET-dependent epigenetic mechanism. Clin. Epigenet. 2019, 11, 102. [Google Scholar] [CrossRef]
- Shishikura, D.; Kataoka, Y.; Honda, S.; Takata, K.; Kim, S.W.; Andrews, J.; Psaltis, P.J.; Sweeney, M.; Kulikowski, E.; Johansson, J.; et al. The Effect of Bromodomain and Extra-Terminal Inhibitor Apabetalone on Attenuated Coronary Atherosclerotic Plaque: Insights from the ASSURE Trial. Am. J. Cardiovasc. Drugs 2019, 19, 49–57. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Schwartz, G.G.; Buhr, K.A.; Ginsberg, H.N.; Johansson, J.O.; Kalantar-Zadeh, K.; Kulikowski, E.; Toth, P.P.; Wong, N.; Sweeney, M.; et al. Apabetalone and hospitalization for heart failure in patients following an acute coronary syndrome: A prespecified analysis of the BETonMACE study. Cardiovasc. Diabetol. 2021, 20, 13. [Google Scholar] [CrossRef]
- Manthei, K.A.; Patra, D.; Wilson, C.J.; Fawaz, M.V.; Piersimoni, L.; Shenkar, J.C.; Yuan, W.; Andrews, P.C.; Engen, J.R.; Schwendeman, A.; et al. Structural analysis of lecithin:cholesterol acyltransferase bound to high density lipoprotein particles. Commun. Biol. 2020, 3, 28. [Google Scholar] [CrossRef] [Green Version]
- Alam, K.; Meidell, R.S.; Spady, D.K. Effect of up-regulating individual steps in the reverse cholesterol transport pathway on reverse cholesterol transport in normolipidemic mice. J. Biol. Chem. 2001, 276, 15641–15649. [Google Scholar] [CrossRef] [Green Version]
- Shamburek, R.D.; Bakker-Arkema, R.; Shamburek, A.M.; Freeman, L.A.; Amar, M.J.; Auerbach, B.; Krause, B.R.; Homan, R.; Adelman, S.J.; Collins, H.L.; et al. Safety and Tolerability of ACP-501, a Recombinant Human Lecithin:Cholesterol Acyltransferase, in a Phase 1 Single-Dose Escalation Study. Circ Res. 2016, 118, 73–82. [Google Scholar] [CrossRef] [Green Version]
- George, R.T.; Abuhatzira, L.; Stoughton, S.M.; Karathanasis, S.K.; She, D.; Jin, C.; Buss, N.; Bakker-Arkema, R.; Ongstad, E.L.; Koren, M.; et al. MEDI6012: Recombinant Human Lecithin Cholesterol Acyltransferase, High-Density Lipoprotein, and Low-Density Lipoprotein Receptor-Mediated Reverse Cholesterol Transport. J. Am. Heart Assoc. 2021, 10, e014572. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Delawary, M.; Sakurai, H.; Kobayashi, H.; Nakao, N.; Tsuru, H.; Fukushima, Y.; Honzumi, S.; Moriyama, S.; Wada, N.; et al. Novel LCAT (Lecithin:Cholesterol Acyltransferase) Activator DS-8190a Prevents the Progression of Plaque Accumulation in Atherosclerosis Models. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 360–376. [Google Scholar] [CrossRef] [PubMed]
- Pavanello, C.; Ossoli, A.; Turri, M.; Strazzella, A.; Simonelli, S.; Laurenzi, T.; Kono, K.; Yamada, K.; Kiyosawa, N.; Eberini, I.; et al. Activation of Naturally Occurring Lecithin:Cholesterol Acyltransferase Mutants by a Novel Activator Compound. J. Pharmacol. Exp. Ther. 2020, 375, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wang, S.P.; Krsmanovic, M.L.; Castro-Perez, J.; Gagen, K.; Mendoza, V.; Rosa, R.; Shah, V.; He, T.; Stout, S.J.; et al. Small molecule activation of lecithin cholesterol acyltransferase modulates lipoprotein metabolism in mice and hamsters. Metabolism 2012, 61, 470–481. [Google Scholar] [CrossRef] [PubMed]
- Li, A.C.; Glass, C.K. PPAR- and LXR-dependent pathways controlling lipid metabolism and the development of atherosclerosis. J. Lipid Res. 2004, 45, 2161–2173. [Google Scholar] [CrossRef] [Green Version]
- Baranowski, M. Biological role of liver X receptors. J. Physiol. Pharmacol. 2008, 59 (Suppl. S7), 31–55. [Google Scholar]
- Sato, M.; Kawata, Y.; Erami, K.; Ikeda, I.; Imaizumi, K. LXR agonist increases the lymph HDL transport in rats by promoting reciprocally intestinal ABCA1 and apo A-I mRNA levels. Lipids 2008, 43, 125–131. [Google Scholar] [CrossRef]
- Katz, A.; Udata, C.; Ott, E.; Hickey, L.; Burczynski, M.E.; Burghart, P.; Vesterqvist, O.; Meng, X. Safety, pharmacokinetics, and pharmacodynamics of single doses of LXR-623, a novel liver X-receptor agonist, in healthy participants. J. Clin. Pharmacol. 2009, 49, 643–649. [Google Scholar] [CrossRef]
- Kirchgessner, T.G.; Sleph, P.; Ostrowski, J.; Lupisella, J.; Ryan, C.S.; Liu, X.; Fernando, G.; Grimm, D.; Shipkova, P.; Zhang, R.; et al. Beneficial and Adverse Effects of an LXR Agonist on Human Lipid and Lipoprotein Metabolism and Circulating Neutrophils. Cell Metab. 2016, 24, 223–233. [Google Scholar] [CrossRef] [Green Version]
- van Eenige, R.; Ying, Z.; Tambyrajah, L.; Pronk, A.C.M.; Blomberg, N.; Giera, M.; Wang, Y.; Coskun, T.; van der Stelt, M.; Rensen, P.C.N.; et al. Cannabinoid type 1 receptor inverse agonism attenuates dyslipidemia and atherosclerosis in APOE∗3-Leiden.CETP mice. J. Lipid Res. 2021, 62, 100070. [Google Scholar] [CrossRef] [PubMed]
- Després, J.P.; Ross, R.; Boka, G.; Alméras, N.; Lemieux, I. Effect of rimonabant on the high-triglyceride/low-HDL-cholesterol dyslipidemia, intraabdominal adiposity, and liver fat: The ADAGIO-Lipids trial. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 416–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, R.; Kristensen, P.K.; Bartels, E.M.; Bliddal, H.; Astrup, A. Efficacy and safety of the weight-loss drug rimonabant: A meta-analysis of randomised trials. Lancet 2007, 370, 1706–1713. [Google Scholar] [CrossRef]
- European Medicines Agency. The European Medicines Agency Recommends Suspension of the Marketing Authorisation of Acomplia. Available online: https://www.ema.europa.eu/en/news/european-medicines-agency-recommends-suspension-marketing-authorisation-acomplia (accessed on 3 July 2022).
- Leite, C.E.; Mocelin, C.A.; Petersen, G.O.; Leal, M.B.; Thiesen, F.V. Rimonabant: An antagonist drug of the endocannabinoid system for the treatment of obesity. Pharmacol. Rep. 2009, 61, 217–224. [Google Scholar] [CrossRef]
- Wilensky, R.L.; Shi, Y.; Mohler, E.R., 3rd; Hamamdzic, D.; Burgert, M.E.; Li, J.; Postle, A.; Fenning, R.S.; Bollinger, J.G.; Hoffman, B.E.; et al. Inhibition of lipoprotein-associated phospholipase A2 reduces complex coronary atherosclerotic plaque development. Nat. Med. 2008, 14, 1059–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donoghue, M.L.; Braunwald, E.; White, H.D.; Lukas, M.A.; Tarka, E.; Steg, P.G.; Hochman, J.S.; Bode, C.; Maggioni, A.P.; Im, K.; et al. Effect of darapladib on major coronary events after an acute coronary syndrome: The SOLID-TIMI 52 randomized clinical trial. Jama 2014, 312, 1006–1015. [Google Scholar] [CrossRef] [Green Version]
- White, H.D.; Held, C.; Stewart, R.; Tarka, E.; Brown, R.; Davies, R.Y.; Budaj, A.; Harrington, R.A.; Steg, P.G.; Ardissino, D.; et al. Darapladib for preventing ischemic events in stable coronary heart disease. N. Engl. J. Med. 2014, 370, 1702–1711. [Google Scholar] [CrossRef] [Green Version]
- Zhuo, S.; Yuan, C. Active site competition is the mechanism for the inhibition of lipoprotein-associated phospholipase A(2) by detergent micelles or lipoproteins and for the efficacy reduction of darapladib. Sci. Rep. 2020, 10, 17232. [Google Scholar] [CrossRef]
- Maity, S.; Wairkar, S. Dietary polyphenols for management of rheumatoid arthritis: Pharmacotherapy and novel delivery systems. Phytother. Res. 2022, 36, 2324–2341. [Google Scholar] [CrossRef]
- Millar, C.L.; Duclos, Q.; Blesso, C.N. Effects of Dietary Flavonoids on Reverse Cholesterol Transport, HDL Metabolism, and HDL Function. Adv. Nutr. 2017, 8, 226–239. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.Z.; Chen, J.F.; Shen, L.M.; Zhou, J.; Wang, C.F. Anti-atherosclerotic effect of hesperidin in LDLr−/− mice and its possible mechanism. Eur. J. Pharmacol. 2017, 815, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Li, S.S.; Cao, H.; Shen, D.Z.; Chen, C.; Xing, S.L.; Dou, F.F.; Jia, Q.L. Effect of Quercetin on Atherosclerosis Based on Expressions of ABCA1, LXR-α and PCSK9 in ApoE−/− Mice. Chin. J. Integr. Med. 2020, 26, 114–121. [Google Scholar] [CrossRef]
- Amengual-Cladera, E.; Nadal-Casellas, A.; Gómez-Pérez, Y.; Gomila, I.; Prieto, R.M.; Proenza, A.M.; Lladó, I. Phytotherapy in a rat model of hyperoxaluria: The antioxidant effects of quercetin involve serum paraoxonase 1 activation. Exp. Biol. Med. 2011, 236, 1133–1138. [Google Scholar] [CrossRef] [PubMed]
- Susanti, E.; Ratnawati, R.; Aulanni’am, A.; Rudijanto, A. Catechins green tea upregulates the expression of ABCA1, ABCG1, and SRB1 in rats induced atherogenic diet. J. Appl. Pharm. Sci. 2019, 9, 091–097. [Google Scholar] [CrossRef] [Green Version]
- Tas, S.; Sarandol, E.; Ziyanok, S.; Aslan, K.; Dirican, M. Effects of green tea on serum paraoxonase/arylesterase activities in streptozotocin-induced diabetic rats. Nutr. Res. 2005, 25, 1061–1074. [Google Scholar] [CrossRef]
- Millar, C.L.; Norris, G.H.; Jiang, C.; Kry, J.; Vitols, A.; Garcia, C.; Park, Y.K.; Lee, J.Y.; Blesso, C.N. Long-Term Supplementation of Black Elderberries Promotes Hyperlipidemia, but Reduces Liver Inflammation and Improves HDL Function and Atherosclerotic Plaque Stability in Apolipoprotein E-Knockout Mice. Mol. Nutr. Food Res. 2018, 62, e1800404. [Google Scholar] [CrossRef] [PubMed]
- Berrougui, H.; Grenier, G.; Loued, S.; Drouin, G.; Khalil, A. A new insight into resveratrol as an atheroprotective compound: Inhibition of lipid peroxidation and enhancement of cholesterol efflux. Atherosclerosis 2009, 207, 420–427. [Google Scholar] [CrossRef]
- Zhou, L.; Long, J.; Sun, Y.; Chen, W.; Qiu, R.; Yuan, D. Resveratrol ameliorates atherosclerosis induced by high-fat diet and LPS in ApoE−/− mice and inhibits the activation of CD4+ T cells. Nutr. Metab. 2020, 17, 41. [Google Scholar] [CrossRef]
- Xu, L.; Wang, R.; Liu, H.; Wang, J.; Mang, J.; Xu, Z. Resveratrol Treatment Is Associated with Lipid Regulation and Inhibition of Lipoprotein-Associated Phospholipase A2 (Lp-PLA2) in Rabbits Fed a High-Fat Diet. Evid. Based Complement. Alternat. Med. 2020, 2020, 9641582. [Google Scholar] [CrossRef]
- Li, X.; Zhou, Y.; Yu, C.; Yang, H.; Zhang, C.; Ye, Y.; Xiao, S. Paeonol suppresses lipid accumulation in macrophages via upregulation of the ATP-binding cassette transporter A1 and downregulation of the cluster of differentiation 36. Int. J. Oncol. 2015, 46, 764–774. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.K.; Ha, T.Y.; McGregor, R.A.; Choi, M.S. Long-term curcumin administration protects against atherosclerosis via hepatic regulation of lipoprotein cholesterol metabolism. Mol. Nutr. Food Res. 2011, 55, 1829–1840. [Google Scholar] [CrossRef] [PubMed]
- Chuengsamarn, S.; Rattanamongkolgul, S.; Phonrat, B.; Tungtrongchitr, R.; Jirawatnotai, S. Reduction of atherogenic risk in patients with type 2 diabetes by curcuminoid extract: A randomized controlled trial. J. Nutr. Biochem. 2014, 25, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Panahi, Y.; Khalili, N.; Sahebi, E.; Namazi, S.; Reiner, Ž.; Majeed, M.; Sahebkar, A. Curcuminoids modify lipid profile in type 2 diabetes mellitus: A randomized controlled trial. Complement. Ther. Med. 2017, 33, 1–5. [Google Scholar] [CrossRef]
- Panahi, Y.; Kianpour, P.; Mohtashami, R.; Jafari, R.; Simental-Mendía, L.E.; Sahebkar, A. Curcumin Lowers Serum Lipids and Uric Acid in Subjects with Nonalcoholic Fatty Liver Disease: A Randomized Controlled Trial. J. Cardiovasc. Pharmacol. 2016, 68, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Boscá, A.; Soler, A.; Carrión, M.A.; Díaz-Alperi, J.; Bernd, A.; Quintanilla, C.; Quintanilla Almagro, E.; Miquel, J. An hydroalcoholic extract of curcuma longa lowers the apo B/apo A ratio. Implications for atherogenesis prevention. Mech. Ageing Dev. 2000, 119, 41–47. [Google Scholar] [CrossRef]
- Li, D.; Zhang, Y.; Liu, Y.; Sun, R.; Xia, M. Purified anthocyanin supplementation reduces dyslipidemia, enhances antioxidant capacity, and prevents insulin resistance in diabetic patients. J. Nutr. 2015, 145, 742–748. [Google Scholar] [CrossRef] [Green Version]
- Qin, Y.; Xia, M.; Ma, J.; Hao, Y.; Liu, J.; Mou, H.; Cao, L.; Ling, W. Anthocyanin supplementation improves serum LDL- and HDL-cholesterol concentrations associated with the inhibition of cholesteryl ester transfer protein in dyslipidemic subjects. Am. J. Clin. Nutr. 2009, 90, 485–492. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Xie, J.; Zhang, H.; Pang, J.; Li, Q.; Wang, X.; Xu, H.; Sun, X.; Zhao, H.; Yang, Y.; et al. Anthocyanin supplementation at different doses improves cholesterol efflux capacity in subjects with dyslipidemia-a randomized controlled trial. Eur. J. Clin. Nutr. 2021, 75, 345–354. [Google Scholar] [CrossRef]
- Hsu, S.P.; Wu, M.S.; Yang, C.C.; Huang, K.C.; Liou, S.Y.; Hsu, S.M.; Chien, C.T. Chronic green tea extract supplementation reduces hemodialysis-enhanced production of hydrogen peroxide and hypochlorous acid, atherosclerotic factors, and proinflammatory cytokines. Am. J. Clin. Nutr. 2007, 86, 1539–1547. [Google Scholar] [CrossRef] [Green Version]
- Kurowska, E.M.; Spence, J.D.; Jordan, J.; Wetmore, S.; Freeman, D.J.; Piché, L.A.; Serratore, P. HDL-cholesterol-raising effect of orange juice in subjects with hypercholesterolemia. Am. J. Clin. Nutr. 2000, 72, 1095–1100. [Google Scholar] [CrossRef] [Green Version]
- Tresserra-Rimbau, A.; Rimm, E.B.; Medina-Remón, A.; Martínez-González, M.A.; de la Torre, R.; Corella, D.; Salas-Salvadó, J.; Gómez-Gracia, E.; Lapetra, J.; Arós, F.; et al. Inverse association between habitual polyphenol intake and incidence of cardiovascular events in the PREDIMED study. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Ren, K.; Chen, Q.; Li, H.; Yao, R.; Hu, H.; Lv, Y.C.; Zhao, G.J. Leonurine Prevents Atherosclerosis Via Promoting the Expression of ABCA1 and ABCG1 in a Pparγ/Lxrα Signaling Pathway-Dependent Manner. Cell Physiol. Biochem. 2017, 43, 1703–1717. [Google Scholar] [CrossRef] [PubMed]
- Reyes, B.A.S.; Dufourt, E.C.; Ross, J.; Warner, M.J.; Tanquilut, N.C.; Leung, A.B. Chapter 4—Selected Phyto and Marine Bioactive Compounds: Alternatives for the Treatment of Type 2 Diabetes. In Studies in Natural Products Chemistry; Attaur, R., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; Volume 55, pp. 111–143. [Google Scholar]
- Prasad, K.; Laxdal, V.A.; Yu, M.; Raney, B.L. Antioxidant activity of allicin, an active principle in garlic. Mol. Cell Biochem. 1995, 148, 183–189. [Google Scholar] [CrossRef] [PubMed]
- García-Trejo, E.M.; Arellano-Buendía, A.S.; Argüello-García, R.; Loredo-Mendoza, M.L.; García-Arroyo, F.E.; Arellano-Mendoza, M.G.; Castillo-Hernández, M.C.; Guevara-Balcázar, G.; Tapia, E.; Sánchez-Lozada, L.G.; et al. Effects of Allicin on Hypertension and Cardiac Function in Chronic Kidney Disease. Oxid. Med. Cell Longev. 2016, 2016, 3850402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, Y.; Azuma, K. Evaluation of the in vitro antifungal activity of allicin. Antimicrob. Agents Chemother. 1977, 11, 743–749. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.L.; Hu, H.J.; Liu, Y.B.; Hu, X.M.; Fan, X.J.; Zou, W.W.; Pan, Y.Q.; Zhou, W.Q.; Peng, M.W.; Gu, C.H. Allicin induces the upregulation of ABCA1 expression via PPARγ/LXRα signaling in THP-1 macrophage-derived foam cells. Int. J. Mol. Med. 2017, 39, 1452–1460. [Google Scholar] [CrossRef] [Green Version]
- Nazeri, Z.; Azizidoost, S.; Cheraghzadeh, M.; Mohammadi, A.; Kheirollah, A. Increased protein expression of ABCA1, HMG-CoA reductase, and CYP46A1 induced by garlic and allicin in the brain mouse and astrocytes-isolated from C57BL/6J. Avicenna J. Phytomed. 2021, 11, 473–483. [Google Scholar] [CrossRef]
- Gong-chen, W.; Lu-lu, H.; Jing, W.; Wan-nan, L.; Chuan-yi, P.; Yan-fei, L. Effects of Allicin on Lipid Metabolism and Antioxidant Activity in Chickens. J. Northeast Agric. Univ. (Engl. Ed.) 2014, 21, 46–49. [Google Scholar] [CrossRef]
- Gonen, A.; Harats, D.; Rabinkov, A.; Miron, T.; Mirelman, D.; Wilchek, M.; Weiner, L.; Ulman, E.; Levkovitz, H.; Ben-Shushan, D.; et al. The antiatherogenic effect of allicin: Possible mode of action. Pathobiology 2005, 72, 325–334. [Google Scholar] [CrossRef]
- Liu, D.S.; Wang, S.L.; Li, J.M.; Liang, E.S.; Yan, M.Z.; Gao, W. Allicin improves carotid artery intima-media thickness in coronary artery disease patients with hyperhomocysteinemia. Exp. Ther. Med. 2017, 14, 1722–1726. [Google Scholar] [CrossRef] [Green Version]
- McEneny, J.; Wade, L.; Young, I.S.; Masson, L.; Duthie, G.; McGinty, A.; McMaster, C.; Thies, F. Lycopene intervention reduces inflammation and improves HDL functionality in moderately overweight middle-aged individuals. J. Nutr. Biochem. 2013, 24, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.M.; Koutsidis, G.; Lodge, J.K.; Ashor, A.; Siervo, M.; Lara, J. Tomato and lycopene supplementation and cardiovascular risk factors: A systematic review and meta-analysis. Atherosclerosis 2017, 257, 100–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Bhandari, U.; Tripathi, C.D.; Khanna, G. Anti-obesity effect of Gymnema sylvestre extract on high fat diet-induced obesity in Wistar rats. Drug Res. 2013, 63, 625–632. [Google Scholar] [CrossRef]
- Pothuraju, R.; Sharma, R.K.; Chagalamarri, J.; Jangra, S.; Kumar Kavadi, P. A systematic review of Gymnema sylvestre in obesity and diabetes management. J. Sci. Food Agric. 2014, 94, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Soliman, G.A. Dietary Fiber, Atherosclerosis, and Cardiovascular Disease. Nutrients 2019, 11, 1155. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, S.; Yamashita, K. Effect of high-fiber diet on plasma high density lipoprotein (HDL) cholesterol level in streptozotocin-induced diabetic rats. Endocrinol. Jpn. 1980, 27, 671–673. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Wu, J.; Tang, J.; Wang, J.J.; Lu, C.H.; Wang, P.X. Beneficial Effect of Higher Dietary Fiber Intake on Plasma HDL-C and TC/HDL-C Ratio among Chinese Rural-to-Urban Migrant Workers. Int. J. Environ Res. Public Health 2015, 12, 4726–4738. [Google Scholar] [CrossRef] [Green Version]
- Moss, J.W.E.; Williams, J.O.; Ramji, D.P. Nutraceuticals as therapeutic agents for atherosclerosis. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1562–1572. [Google Scholar] [CrossRef]
- Asleh, R.; Blum, S.; Kalet-Litman, S.; Alshiek, J.; Miller-Lotan, R.; Asaf, R.; Rock, W.; Aviram, M.; Milman, U.; Shapira, C.; et al. Correction of HDL dysfunction in individuals with diabetes and the haptoglobin 2-2 genotype. Diabetes 2008, 57, 2794–2800. [Google Scholar] [CrossRef] [Green Version]
- Morton, A.M.; Furtado, J.D.; Mendivil, C.O.; Sacks, F.M. Dietary unsaturated fat increases HDL metabolic pathways involving apoE favorable to reverse cholesterol transport. JCI Insight 2019, 4, e124620. [Google Scholar] [CrossRef] [Green Version]
- Mathew, A.V.; Li, L.; Byun, J.; Guo, Y.; Michailidis, G.; Jaiswal, M.; Chen, Y.E.; Pop-Busui, R.; Pennathur, S. Therapeutic Lifestyle Changes Improve HDL Function by Inhibiting Myeloperoxidase-Mediated Oxidation in Patients with Metabolic Syndrome. Diabetes Care 2018, 41, 2431–2437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, M.; Qian, Z.; Yin, J.; Xu, W.; Zhou, X. The role of intestinal microbiota in cardiovascular disease. J. Cell Mol. Med. 2019, 23, 2343–2350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Li, H. The Role of Gut Microbiota in Atherosclerosis and Hypertension. Front. Pharmacol. 2018, 9, 1082. [Google Scholar] [CrossRef] [PubMed]
- Kanmani, P.; Ansari, A.; Villena, J.; Kim, H. Immunobiotics Beneficially Modulate TLR4 Signaling Triggered by Lipopolysaccharide and Reduce Hepatic Steatosis In Vitro. J. Immunol. Res. 2019, 2019, 3876896. [Google Scholar] [CrossRef]
- Alipour, B.; Homayouni-Rad, A.; Vaghef-Mehrabany, E.; Sharif, S.K.; Vaghef-Mehrabany, L.; Asghari-Jafarabadi, M.; Nakhjavani, M.R.; Mohtadi-Nia, J. Effects of Lactobacillus casei supplementation on disease activity and inflammatory cytokines in rheumatoid arthritis patients: A randomized double-blind clinical trial. Int. J. Rheum. Dis. 2014, 17, 519–527. [Google Scholar] [CrossRef]
- Vaghef-Mehrabany, E.; Alipour, B.; Homayouni-Rad, A.; Sharif, S.K.; Asghari-Jafarabadi, M.; Zavvari, S. Probiotic supplementation improves inflammatory status in patients with rheumatoid arthritis. Nutrition 2014, 30, 430–435. [Google Scholar] [CrossRef]
- O’Morain, V.L.; Ramji, D.P. The Potential of Probiotics in the Prevention and Treatment of Atherosclerosis. Mol. Nutr. Food Res. 2020, 64, e1900797. [Google Scholar] [CrossRef]
- Roselli, M.; Finamore, A.; Brasili, E.; Rami, R.; Nobili, F.; Orsi, C.; Zambrini, A.V.; Mengheri, E. Beneficial effects of a selected probiotic mixture administered to high fat-fed mice before and after the development of obesity. J. Funct. Foods 2018, 45, 321–329. [Google Scholar] [CrossRef]
- Chan, Y.K.; El-Nezami, H.; Chen, Y.; Kinnunen, K.; Kirjavainen, P.V. Probiotic mixture VSL#3 reduce high fat diet induced vascular inflammation and atherosclerosis in ApoE−/− mice. AMB Express 2016, 6, 61. [Google Scholar] [CrossRef] [Green Version]
- Chan, Y.K.; Brar, M.S.; Kirjavainen, P.V.; Chen, Y.; Peng, J.; Li, D.; Leung, F.C.; El-Nezami, H. High fat diet induced atherosclerosis is accompanied with low colonic bacterial diversity and altered abundances that correlates with plaque size, plasma A-FABP and cholesterol: A pilot study of high fat diet and its intervention with Lactobacillus rhamnosus GG (LGG) or telmisartan in ApoE−/− mice. BMC Microbiol. 2016, 16, 264. [Google Scholar] [CrossRef] [Green Version]
- Kocsis, T.; Molnár, B.; Németh, D.; Hegyi, P.; Szakács, Z.; Bálint, A.; Garami, A.; Soós, A.; Márta, K.; Solymár, M. Probiotics have beneficial metabolic effects in patients with type 2 diabetes mellitus: A meta-analysis of randomized clinical trials. Sci. Rep. 2020, 10, 11787. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, S.; Xue, P.; Yu, L.; Tian, F.; Zhao, J.; Chen, W.; Xue, Y.; Zhai, Q. The effect of probiotic supplementation on lipid profiles in adults with overweight or obesity: A meta-analysis of randomized controlled trials. J. Funct. Foods 2021, 86, 104711. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, Q.; Ren, Y.; Ruan, Z. Effect of probiotic Lactobacillus on lipid profile: A systematic review and meta-analysis of randomized, controlled trials. PLoS ONE 2017, 12, e0178868. [Google Scholar] [CrossRef] [PubMed]
- Shakeri, H.; Hadaegh, H.; Abedi, F.; Tajabadi-Ebrahimi, M.; Mazroii, N.; Ghandi, Y.; Asemi, Z. Consumption of synbiotic bread decreases triacylglycerol and VLDL levels while increasing HDL levels in serum from patients with type-2 diabetes. Lipids 2014, 49, 695–701. [Google Scholar] [CrossRef]
- Hong, Y.F.; Kim, H.; Kim, H.S.; Park, W.J.; Kim, J.Y.; Chung, D.K. Lactobacillus acidophilus K301 Inhibits Atherogenesis via Induction of 24 (S), 25-Epoxycholesterol-Mediated ABCA1 and ABCG1 Production and Cholesterol Efflux in Macrophages. PLoS ONE 2016, 11, e0154302. [Google Scholar] [CrossRef] [Green Version]
- Ooi, L.G.; Ahmad, R.; Yuen, K.H.; Liong, M.T. Lactobacillus gasseri [corrected] CHO-220 and inulin reduced plasma total cholesterol and low-density lipoprotein cholesterol via alteration of lipid transporters. J. Dairy Sci. 2010, 93, 5048–5058. [Google Scholar] [CrossRef] [Green Version]
- Navab, M.; Anantharamaiah, G.M.; Reddy, S.T.; Van Lenten, B.J.; Wagner, A.C.; Hama, S.; Hough, G.; Bachini, E.; Garber, D.W.; Mishra, V.K.; et al. An oral apoJ peptide renders HDL antiinflammatory in mice and monkeys and dramatically reduces atherosclerosis in apolipoprotein E-null mice. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1932–1937. [Google Scholar] [CrossRef] [Green Version]
- Rivas-Urbina, A.; Rull, A.; Aldana-Ramos, J.; Santos, D.; Puig, N.; Farre-Cabrerizo, N.; Benitez, S.; Perez, A.; de Gonzalo-Calvo, D.; Escola-Gil, J.C.; et al. Subcutaneous Administration of Apolipoprotein J-Derived Mimetic Peptide d-[113–122]apoJ Improves LDL and HDL Function and Prevents Atherosclerosis in LDLR-KO Mice. Biomolecules 2020, 10, 829. [Google Scholar] [CrossRef]
- Amar, M.J.; Sakurai, T.; Sakurai-Ikuta, A.; Sviridov, D.; Freeman, L.; Ahsan, L.; Remaley, A.T. A novel apolipoprotein C-II mimetic peptide that activates lipoprotein lipase and decreases serum triglycerides in apolipoprotein E-knockout mice. J. Pharmacol. Exp. Ther. 2015, 352, 227–235. [Google Scholar] [CrossRef] [Green Version]
- Wolska, A.; Lo, L.; Sviridov, D.O.; Pourmousa, M.; Pryor, M.; Ghosh, S.S.; Kakkar, R.; Davidson, M.; Wilson, S.; Pastor, R.W.; et al. A dual apolipoprotein C-II mimetic-apolipoprotein C-III antagonist peptide lowers plasma triglycerides. Sci. Transl. Med. 2020, 12, eaaw7905. [Google Scholar] [CrossRef]
- Bell, T.A., 3rd; Graham, M.J.; Lee, R.G.; Mullick, A.E.; Fu, W.; Norris, D.; Crooke, R.M. Antisense oligonucleotide inhibition of cholesteryl ester transfer protein enhances RCT in hyperlipidemic, CETP transgenic, LDLr−/− mice. J. Lipid Res. 2013, 54, 2647–2657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rayner, K.J.; Esau, C.C.; Hussain, F.N.; McDaniel, A.L.; Marshall, S.M.; van Gils, J.M.; Ray, T.D.; Sheedy, F.J.; Goedeke, L.; Liu, X.; et al. Inhibition of miR-33a/b in non-human primates raises plasma HDL and lowers VLDL triglycerides. Nature 2011, 478, 404–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najafi-Shoushtari, S.H.; Kristo, F.; Li, Y.; Shioda, T.; Cohen, D.E.; Gerszten, R.E.; Näär, A.M. MicroRNA-33 and the SREBP host genes cooperate to control cholesterol homeostasis. Science 2010, 328, 1566–1569. [Google Scholar] [CrossRef]
- Masson, D.; Koseki, M.; Ishibashi, M.; Larson, C.J.; Miller, S.G.; King, B.D.; Tall, A.R. Increased HDL cholesterol and apoA-I in humans and mice treated with a novel SR-BI inhibitor. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 2054–2060. [Google Scholar] [CrossRef] [Green Version]
- Syder, A.J.; Lee, H.; Zeisel, M.B.; Grove, J.; Soulier, E.; Macdonald, J.; Chow, S.; Chang, J.; Baumert, T.F.; McKeating, J.A.; et al. Small molecule scavenger receptor BI antagonists are potent HCV entry inhibitors. J. Hepatol. 2011, 54, 48–55. [Google Scholar] [CrossRef]
- Song, G.; Li, M.; Sang, H.; Zhang, L.; Li, X.; Yao, S.; Yu, Y.; Zong, C.; Xue, Y.; Qin, S. Hydrogen-rich water decreases serum LDL-cholesterol levels and improves HDL function in patients with potential metabolic syndrome. J. Lipid Res. 2013, 54, 1884–1893. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Zhao, M.; Xue, J.; Gu, Q.; Zhang, X.; Qin, S. Hydrogen influences HDL-associated enzymes and reduces oxidized phospholipids levels in rats fed with a high-fat diet. Life Sci. 2021, 267, 118945. [Google Scholar] [CrossRef] [PubMed]
- Louala, S.; Hamza-Reguig, S.; Benyahia-Mostefaoui, A.; Boualga, A.; Lamri-Senhadji, M.Y. Effects of highly purified sardine proteins on lipid peroxidation and reverse cholesterol transport in rats fed a cholesterol-rich diet. J. Funct. Foods 2011, 3, 321–328. [Google Scholar] [CrossRef]
- Affane, F.; Louala, S.; El Imane Harrat, N.; Bensalah, F.; Chekkal, H.; Allaoui, A.; Lamri-Senhadji, M. Hypolipidemic, antioxidant and antiatherogenic property of sardine by-products proteins in high-fat diet induced obese rats. Life Sci. 2018, 199, 16–22. [Google Scholar] [CrossRef]
- Houthoofd, S.; Vuylsteke, M.; Mordon, S.; Fourneau, I. Photodynamic therapy for atherosclerosis. The potential of indocyanine green. Photodiagn. Photodyn. Ther. 2020, 29, 101568. [Google Scholar] [CrossRef]
- Han, X.B.; Li, H.X.; Jiang, Y.Q.; Wang, H.; Li, X.S.; Kou, J.Y.; Zheng, Y.H.; Liu, Z.N.; Li, H.; Li, J.; et al. Upconversion nanoparticle-mediated photodynamic therapy induces autophagy and cholesterol efflux of macrophage-derived foam cells via ROS generation. Cell Death Dis. 2017, 8, e2864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.; Wei, D.; Liu, R.; Zhang, C.; Jiang, S.; Wang, W.; Hu, H.; Shen, L.; Liang, X. A novel therapeutic strategy for atherosclerosis: Autophagy-dependent cholesterol efflux. J. Physiol. Biochem. 2022, 78, 557–572. [Google Scholar] [CrossRef] [PubMed]
- Song, J.W.; Lee, M.W.; Kim, H.J.; Joo, Y.D.; Choi, J.Y.; Oh, W.-Y.; Yoo, H.; Park, K.; Kim, J.W. Abstract 14558: Macrophage Targeted Theranostic Photoactivation Attenuates Plaque Inflammation and Regresses the Atheroma via Autophagy-Induced Cholesterol Efflux Assessed by Serial in vivo Imaging. Circulation 2018, 138, A14558. [Google Scholar] [CrossRef]
- Hayashi, J.; Sato, H.; Saito, T.; Kuroiwa, Y.; Aizawa, K.; Fujiwara, T.; Hosoda, Y. Percutaneous Transluminal Photodynamic Therapy of Atheroma Using Mono-L-Aspartyl Chlorin e6. In Proceedings of the 5th International Photodynamic Association Biennial Meeting, Amelia Island, FL, USA, 21–24 September 1994; Volume 2371. [Google Scholar]




{kind=link}
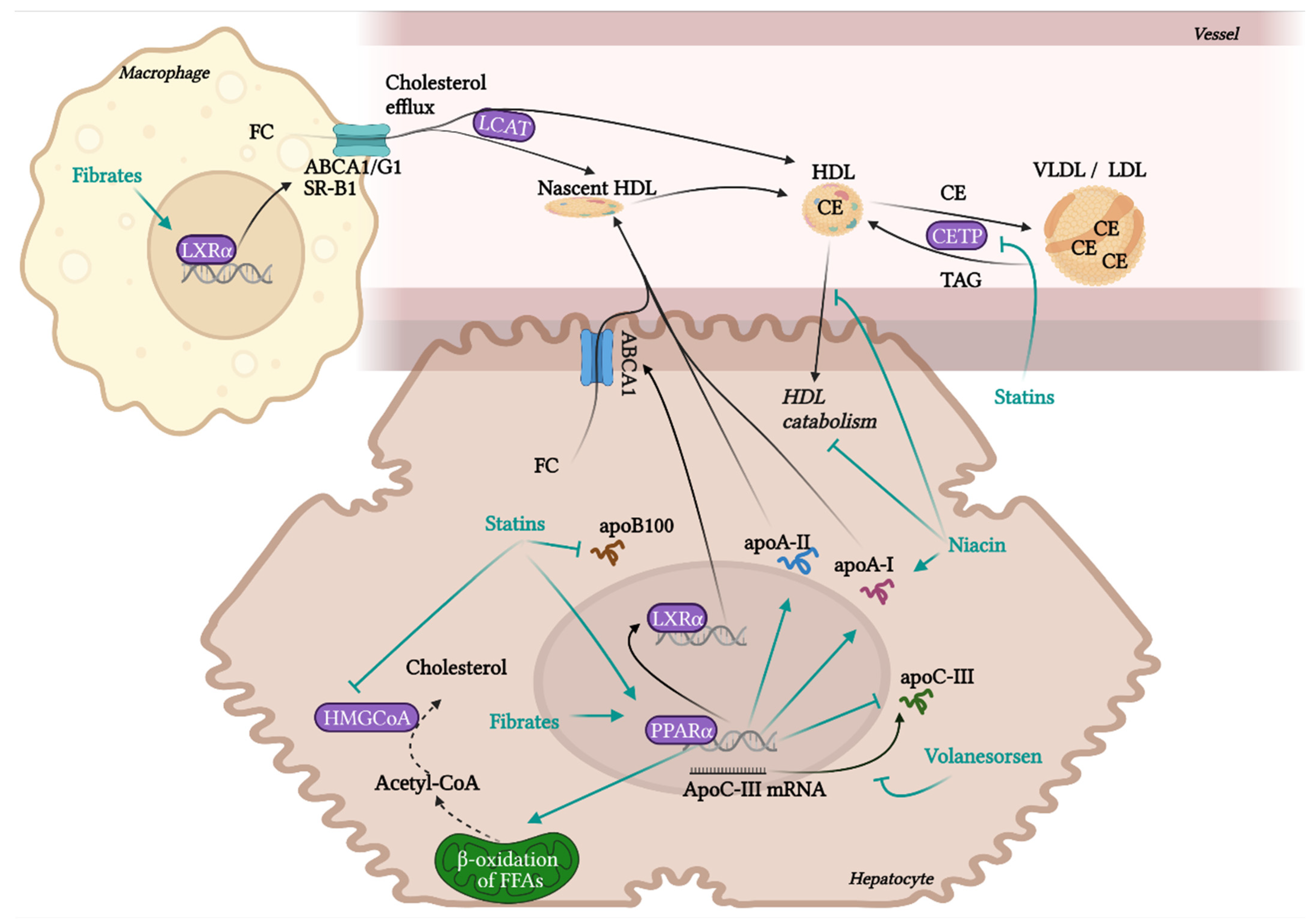{kind=link}
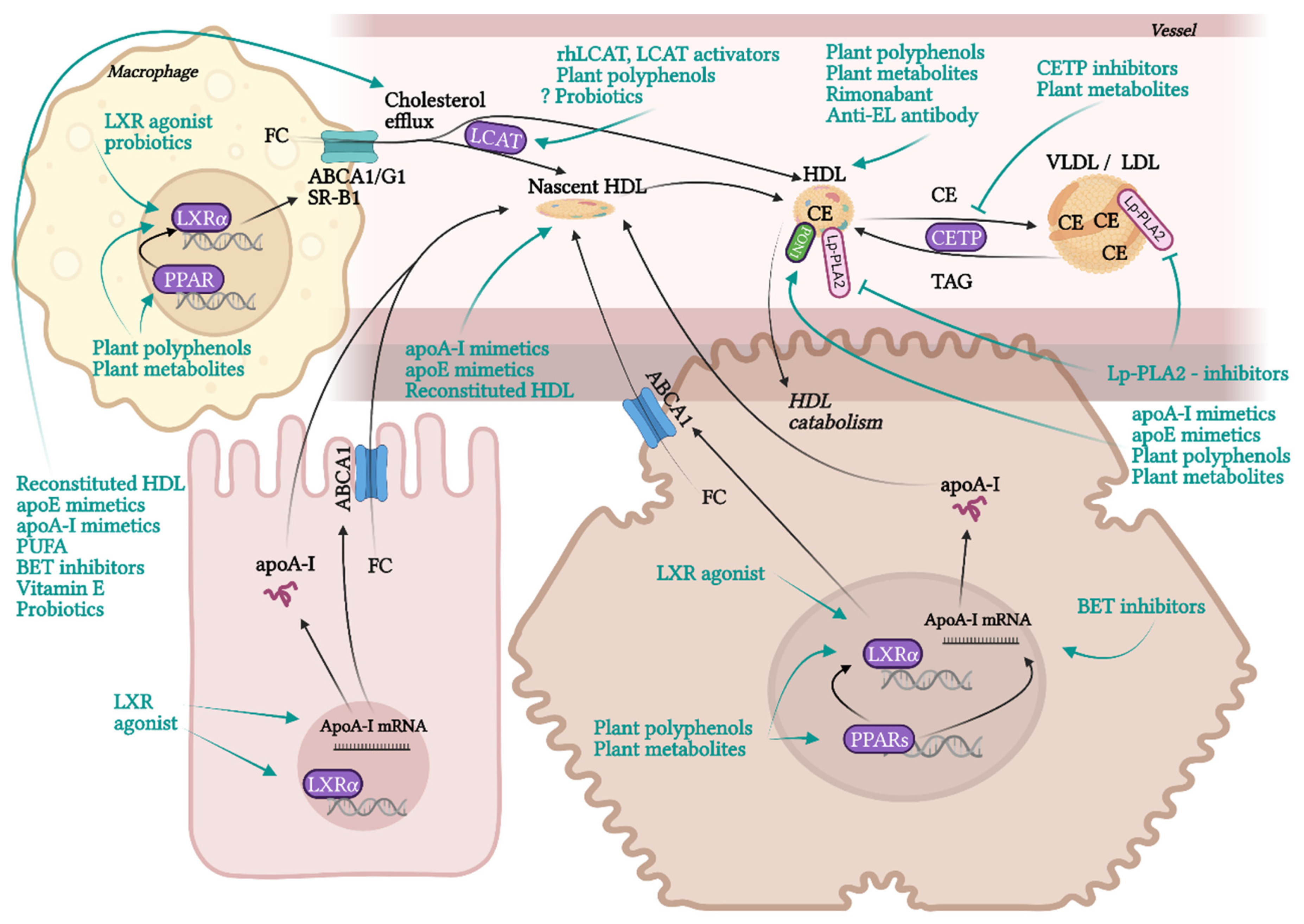{kind=link}
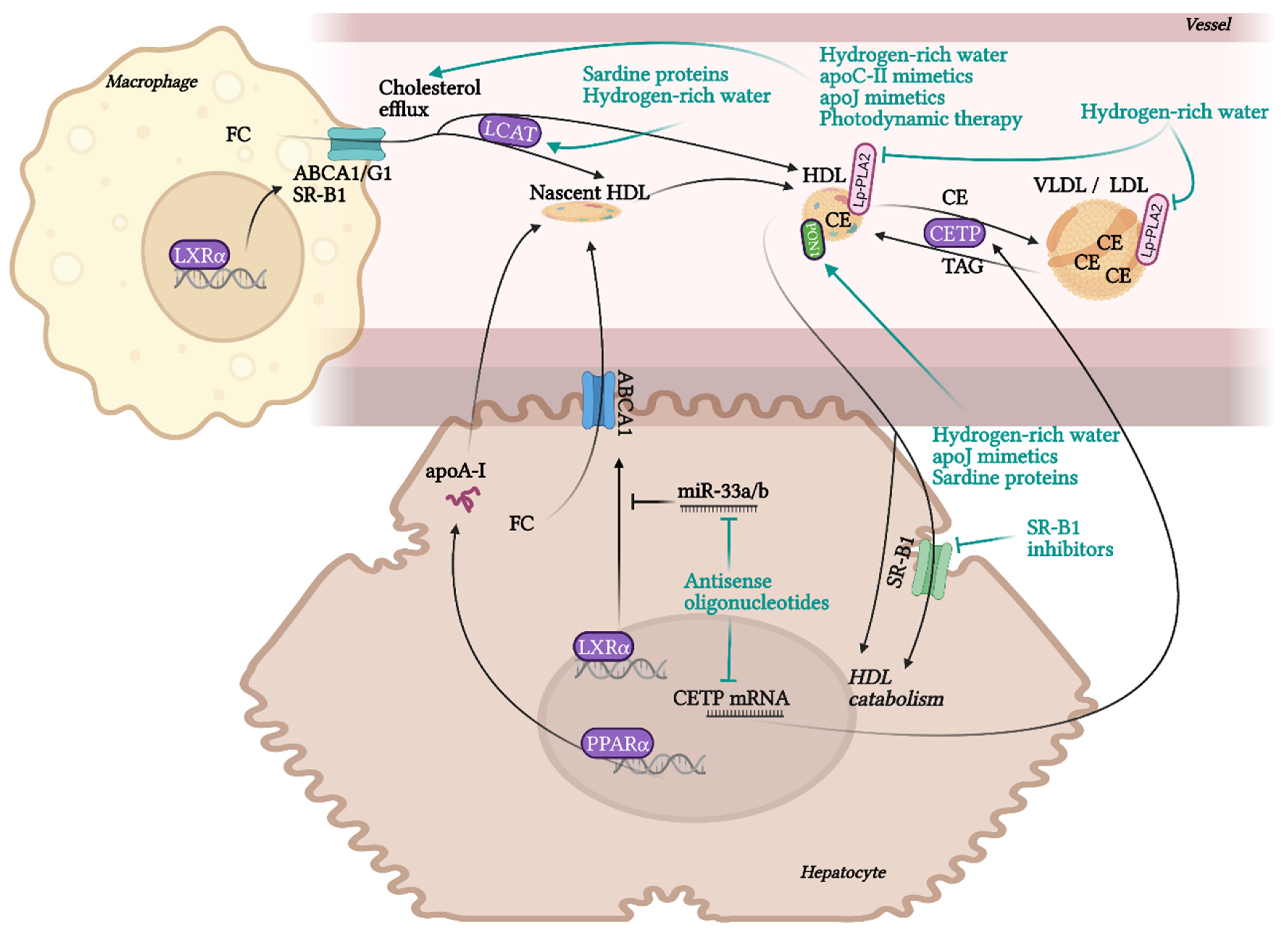{kind=link}
| Acute Inflammation | Chronic Inflammation | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| RA [9] | SLE [333] | Psoriasis [330] | PD [5] | T2DM [321] | CKD1 [334] | CKD2 [335] | CKD3 [336] | NAFLD [337] | Other | ||
| apoA-I | Mice 1: ↓ [338] Rab 2: ↓ [22] hSurg 3: ↓ [22] | ↓ ns | ↓ | ↓ | ↑ ns | ↓ | ↓ | SLE: ↓ [339] | |||
| apoA-II | Mice 1: ↓ [338] | ↓ ns | ↑ | ↑ | ↑ | ↓ | ↓ | ↑ | ↓ | ||
| apoA-IV | Mice 1: ↑ [338] | ↑ ns | ↓ | ↑ | ↑ | ↑ | |||||
| apoE | Mice 1: ↑ [338] | ↓ | ↑ | ↑ | SLE: ↑ [339] | ||||||
| apoC-I | Mice 1: ↓ [338] | ↓ ns | ↑ | ↓ ns | ↓ | ↓ | ↑ | ||||
| apoC-II | ↑ | ↑ | ↑ | ↑ | ↑ | ||||||
| apoC-III | Mice 1: ↓ [338] | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | ||||
| apoJ | Mice 4: ↑ [326] | ↑ | ↓ | ↓ | ↓ | ||||||
| apoM | ↑ | ↓ | ↓ | ↓ | ↓ | ↑ | |||||
| Lp-PLA2 | ↑ | ||||||||||
| PLTP | Mice 1: ↓ [338] | ↓ | ↓ | ||||||||
| LBP | Mice 1: ↑ [338] | ||||||||||
| LCAT | ↓ ns | ↓ | ↑ | ||||||||
| PON1 | Mice 1: ↓ [338] | ↓ ns | ↓ | ↓ | ↑ | ↓ | |||||
| Transferrin | ↓ | ↑ | ↑ | ↓ | |||||||
| SAA | Mice 1: ↑ [338] Rab 2: ↑ [22] hSurg 3: ↑ [22] hEtox 5: ↑ [338] | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | RA: ↑ [106] | |||
| Ceruloplasmin | Rab 2: ↑ [22] hSurg 3: ↑ [22] Mice 4: ↑ [326] | ↑ | |||||||||
| Fibrinogen | Mice 1: ↑ [338] | ↑ | ↑ ns | ↑ | ↓ | ||||||
| Hp | Mice 1: ↑ [338] | ↑ | ↑ | ↑ ns | ↓ | ||||||
| Hb | Mice 1: ↓ [338] | ↑ | |||||||||
| Hx | Mice 1: ↑ [338] | ↑ ns | ↑ ns | ↑ | ↓ | ||||||
| AAT | Mice 1: ↓ [338] | ↑ | ↑ | ↓ ns | ↑ | ↑ | |||||
| AAG | Mice 1: ↑ [338] | ↑ ns | ↓ | ↑ | ↓ | ↑ | |||||
| α2M | ↓ | ↓ | |||||||||
| B2M | Mice 1: ↓ [338] | ↓ | ↑ | ↑ | |||||||
| C3 complement | ↑ | ↑ | ↓ | ↓ | ↓ | ↓ | ↑ | ||||
| Enzyme | Acute Inflammation | Chronic Inflammation |
|---|---|---|
| CETP | hSeps 1: ↓ pl [340] | RA: ↑ HDL [341] SLE: ↑ pl [342] Psoriasis: ↓ pl [343] T2DM: ↓ pl [344,345] CKD: ↑ pl [346] NAFLD: ↑ pl [347,348] |
| Lp-PLA2 | Rab 2: ↓ HDL [22] hSurg 3: ↓ HDL [22] Mice 4: ↓ HDL [326] | RA: ↓ pl [331] ↑ pl [332] SLE 7: ↑ pl [328] Psoriasis: ↑ HDL [329,330] CKD: ↑ pl [349] |
| LCAT | hSeps 1: ↓ pl [340] Ham 5: ↓ HDL [350] hEtox 6: ↓ pl [351] | RA: ↓ pl [106,352] Psoriasis: ↓ pl [353,354] T2DM: ↑ pl [355] ↓ pl [356] CKD: ↓ pl [357] NAFLD: ↑ pl [358] |
| Gpx3 | RA: ↑ pl [359] ↓ pl [360,361] SLE: ↓ pl [362,363,364] Psoriasis: ↓ pl [365] PD: ↑ pl [366] ↓ pl [367,368] T2DM: ↑ pl [369] ↓ pl [370] CKD: ↓ pl [371,372] NAFLD: ↑ pl [373] | |
| PON1 | Rab 2: ↓ HDL [22] hSurg 3: ↓ HDL [22] Mice 4: ↓ HDL [326] | RA: ↓ HDL [341] ↓ pl [352] SLE: ↓ pl [342] Psoriasis: ↓ HDL [329] ↓ pl [353] PD: ↓ pl [374] T2DM: ↓ HDL [375] ↓ pl [376,377] CKD: ↓ pl [346,378] NAFLD: ↓ pl [347] |
| sPLA2 | RA, SLE: ↑ pl [379] |
| Drug | Effect | Additional Information | Reference |
|---|---|---|---|
| Fibrates | PPAR ligands, ↑apoA-I, ↑apoA-II → ↑HDL biogenesis, ↓apoC-III, ↑ABCA1/G1 and SR-B1 expression → ↑RCT | some trials showed lack of efficacy, better effect was observed in the combination with statins, recent meta-analyses showed promising reduction in CVD risk in population with specific metabolic conditions | [423] |
| Niacin | liver: ↑apoA-I, ↓HDL uptake, ↓HDL catabolism → ↑HDLc | trials show lack of benefit in patients with CVD concomitantly treated with statins | [443] |
| Statins | ↑PPARα → ↑ApoA-I mRNA, ↓CETP | beneficial effect on CVD risk was confirmed in meta-analyses, effect on HDL is not the primary mechanism of statins’ therapy, but it could contribute to its efficacy, observed raised incidence of DM, muscular side effects | [452,463] |
| Volanesorsen | antisense oligonucleotid against ApoC-III mRNA → ↓apoC-III synthesis | increase in plasmatic HDL and decrease in TAGs and non-HDL levels was observed in clinical trials | [468] |
| Drug | Effect | Additional Information | |
|---|---|---|---|
| CETP inhibitors | ↓CETP → ↑HDLc | in most clinical trials, CETP-inhibitors failed to decrease CVD risk, trial with anacetrapib showed efficacy in reduction in coronary events but with side effects—liver steatosis, IR | [471] |
| ApoA-I mimetics | small peptides mimicking apoA-I → ↑pre-β HDLs, ↑CEC, anti-inflammatory properties of HDL | oral apoA-I mimetic D-4F was well tolerated and effectively reduced the inflammatory index of HDL particles in patients | [487] |
| Reconstituted HDL | recombinant apoA-I with phospholipids → ↑CEC | some molecules were clinically ineffective, some showed high incidence of adverse effects. Second-generation molecules with good safety profile are under investigation | [491,493,494,495] |
| ApoE mimetics | ↑clearance of cholesterol by SR-B1 and LDLR → ↑CEC and RCT, ↑PON1, ↓SAA | AEM-28 was promisingly tested in Phase I/II clinical trial as treatment for refractory hypercholesterolemia | [503,504] |
| Anti-EL monoclonal antibody | ↓EL → ↑HDLc, ↑HDL particle size, ↑cholesterol efflux, improved anti-inflammatory HDL function | MEDI5884 was succefully tested in Phase IIa study on patients with stable coronary artery disease | [508] |
| Epigenetic therapy (bromodomain and extraterminal domain (BET) inhibitor apabetalon) | ↑ApoA-I mRNA/apoA-I expression, ↑CEC | clinical trials show effective modulation of lipid profile, improvement of vascular inflammation and reduction in CVD events in T2DM patients | [510,511] |
| rhLCAT | recombinant human LCAT → ↑formation of HDL and RCT | in patients with CHD, rhLCAT increased HDLc, favourably altered HDL metabolism and trial showed also good tolerability and safety of treatment | [516] |
| LXR agonist | ↑ABCA1/G1, ↑ApoA-I mRNA, ↑RCT | the effect on ABCA1/ABCG1 expression was confirmed in a clinical trial, however, CNS-associated side effect and risk for steatosis was observed in animal models or humans | [523,524,525] |
| Rimonabant | inverse agonist of CB1 → ↑apoA-I, ↑HDL particles size, ↑HDLc | effect on HDL was confirmed in clinical practice, serious psychiatric side effects led to discontinuation of clinical usage | [527,528] |
| Lp-PLA2-inhibitors | ↓Lp-PLA2 | darapladib failed to reduce CVD events in patients | [532,533] |
| Plant polyphenols | LXR or PPAR activation, ↑apoA-I, ↑HDLc, ↑ABCA1/G1, SR-B1 → ↑RCT, ↑PON1, ↑LCAT and others… | positive effect on HDLc and properties was confirmed in many clinical trials | [536,546,547] |
| Other plant metabolites and compounds | LXR or PPARγ activation, ↑HDLc, ↓SAA, ↑ABCA1/G1, → ↑RCT ↓CETP, ↑Gpx-3, ↑PON1 and others… | positive effects on cardiovascular system were observed in many animal and human studies | [558,567] |
| Vitamin E | ↑CEC, ↓HDL dysfunction and peroxidation | effect observed only in Hp 2-2 but not Hp1-1 T2DM individuals | [576] |
| PUFAs | ↑RCT in apoE-HDL, ↓MPO oxidation products | Human | [577,578] |
| Probiotics and synbiotics | ↑HDLc, ↑ABCA1/G1, ↑RCT, ↑CEs in HDL = ↑LCAT? | mice, human | [588,592,593] |
| Drug | Effect | Tested Subjects | |
|---|---|---|---|
| ApoJ mimetic peptides | ↑RCT, ↓lipoprotein lipid peroxides, ↑PON1, improved anti-inflammatory HDL function | Mice, monkeys | [594,595] |
| ApoC-II mimetic peptides | ↑ABCA1-mediated cholesterol efflux | mice | [596,597] |
| Antisense oligonucleotides | altering expression of specific genes (CETP) | mice | [598] |
| inhibiting miRNAs involved in HDL metabolism and function (miR-33 a/b) | primate, mice | [599,600] | |
| SR-B1 inhibitors | ↓SR-B1-mediated uptake of HDL to liver → ↑HDLc | Humans, mice | [443,601] |
| Hydrogen-rich water | ↓Lp-PLA2, ↑LCAT, antioxidant activity (↑PON1) → improved HDL function, ↑CEC | humans, rats, hamsters | [603,604] |
| Purified sardine proteins | ↑LCAT → ↑RCT, ↑PON1 → ↓HDL oxidation | rats | [605,606] |
| Photodynamic therapy with photosensitisers | ↑ABCA1-mediated cholesterol eflux | rabbits, mice | [610,611] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vyletelová, V.; Nováková, M.; Pašková, Ľ. Alterations of HDL’s to piHDL’s Proteome in Patients with Chronic Inflammatory Diseases, and HDL-Targeted Therapies. Pharmaceuticals 2022, 15, 1278. https://doi.org/10.3390/ph15101278
Vyletelová V, Nováková M, Pašková Ľ. Alterations of HDL’s to piHDL’s Proteome in Patients with Chronic Inflammatory Diseases, and HDL-Targeted Therapies. Pharmaceuticals. 2022; 15(10):1278. https://doi.org/10.3390/ph15101278
Chicago/Turabian StyleVyletelová, Veronika, Mária Nováková, and Ľudmila Pašková. 2022. "Alterations of HDL’s to piHDL’s Proteome in Patients with Chronic Inflammatory Diseases, and HDL-Targeted Therapies" Pharmaceuticals 15, no. 10: 1278. https://doi.org/10.3390/ph15101278
APA StyleVyletelová, V., Nováková, M., & Pašková, Ľ. (2022). Alterations of HDL’s to piHDL’s Proteome in Patients with Chronic Inflammatory Diseases, and HDL-Targeted Therapies. Pharmaceuticals, 15(10), 1278. https://doi.org/10.3390/ph15101278