
Computational and Biological Evaluation of β-Adrenoreceptor Blockers as Promising Bacterial Anti-Virulence Agents

 , ,
, ,  ,
,  and
and 
Abstract
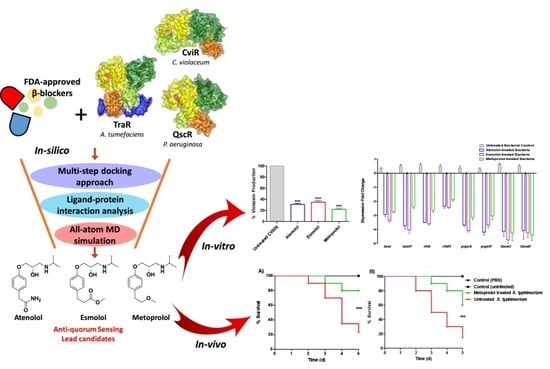
:
1. Introduction
2. Results
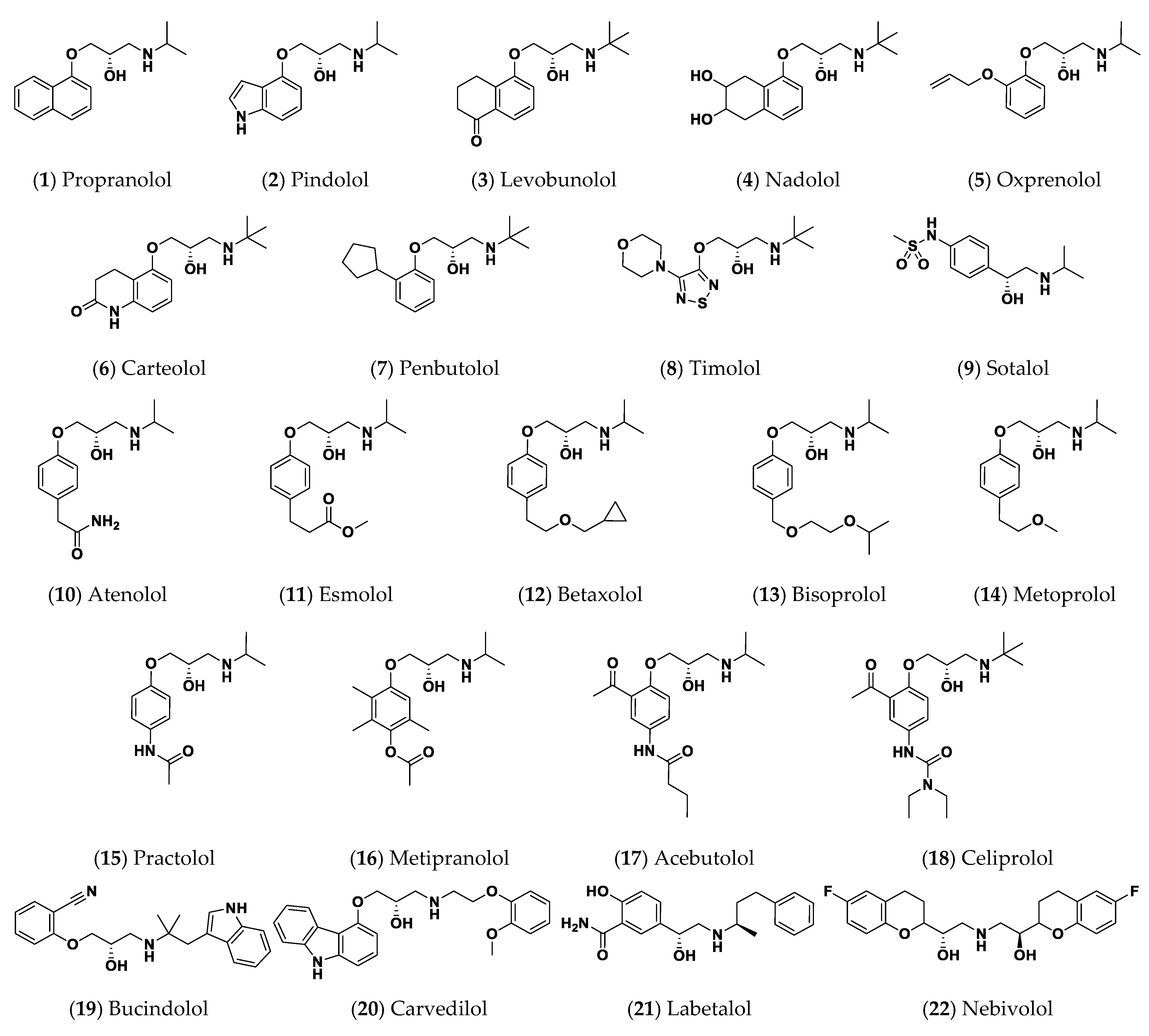
2.1. Two-Stage Multi-Target Docking Analysis
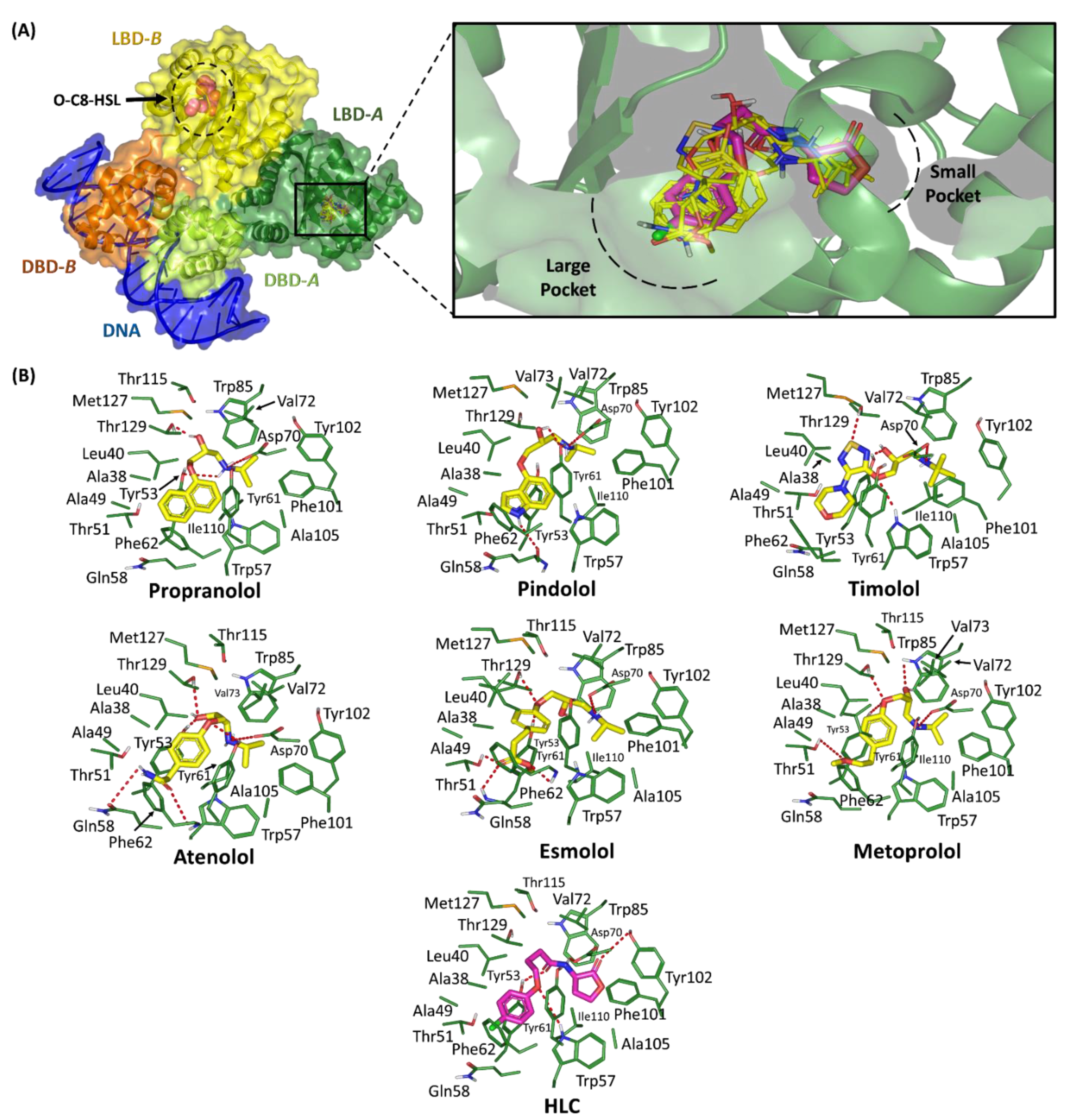
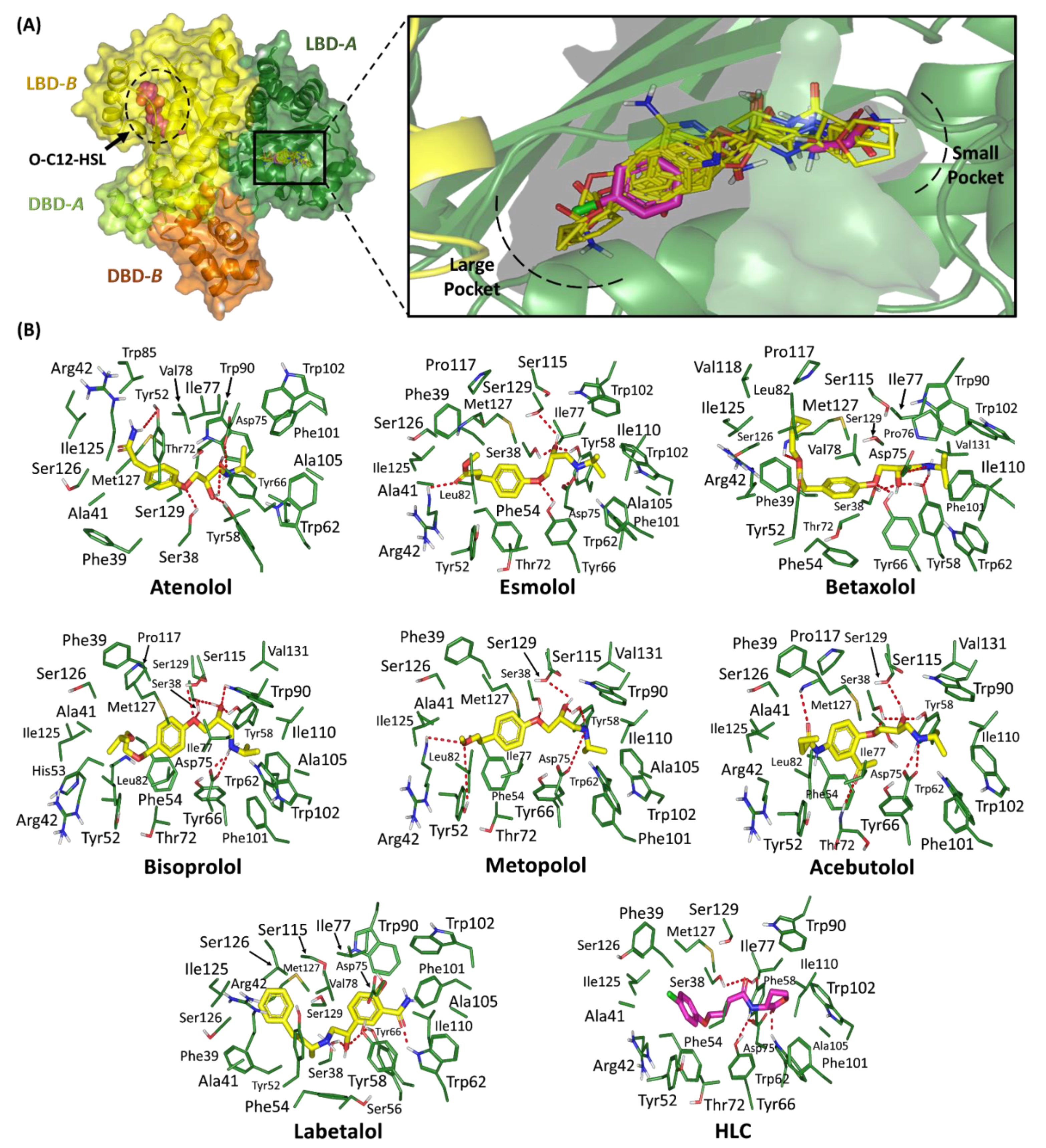
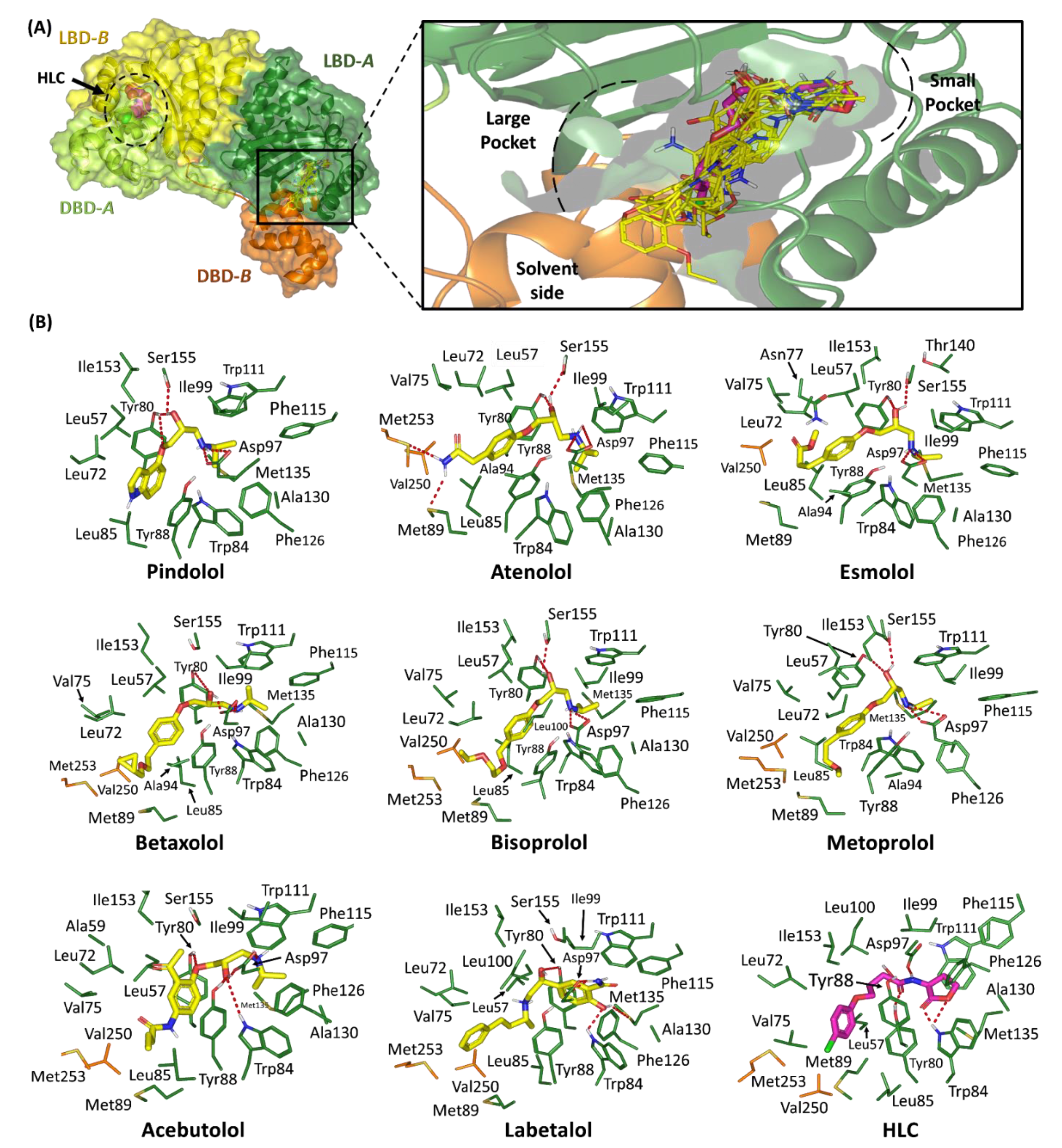
2.1.1. Binding Interaction Analysis of Ligand–TraR A. tumefaciens Complexes
2.1.2. Binding Interaction Analysis of Ligand–QscR P. aeruginosa Complexes
2.1.3. Binding Interaction Analysis of Ligand–CviR C. violaceum Complexes
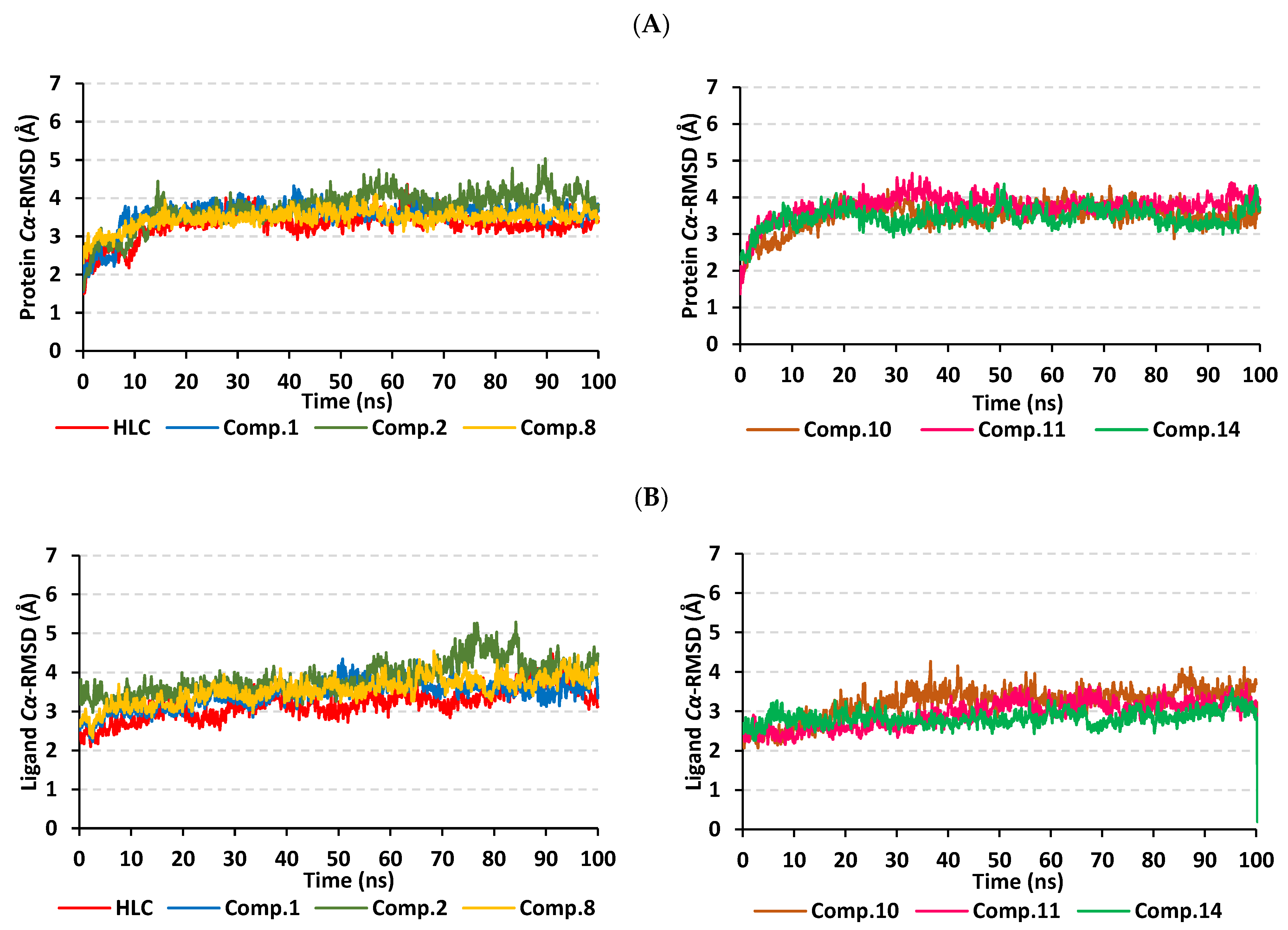
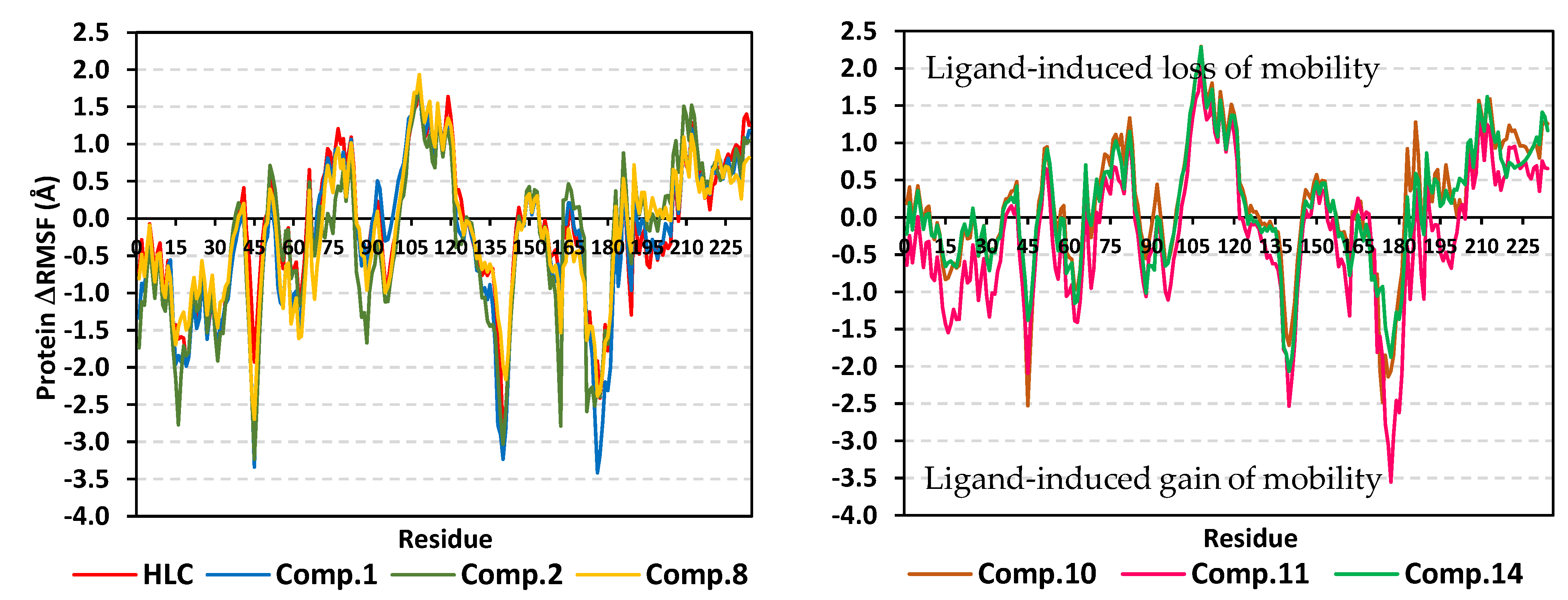
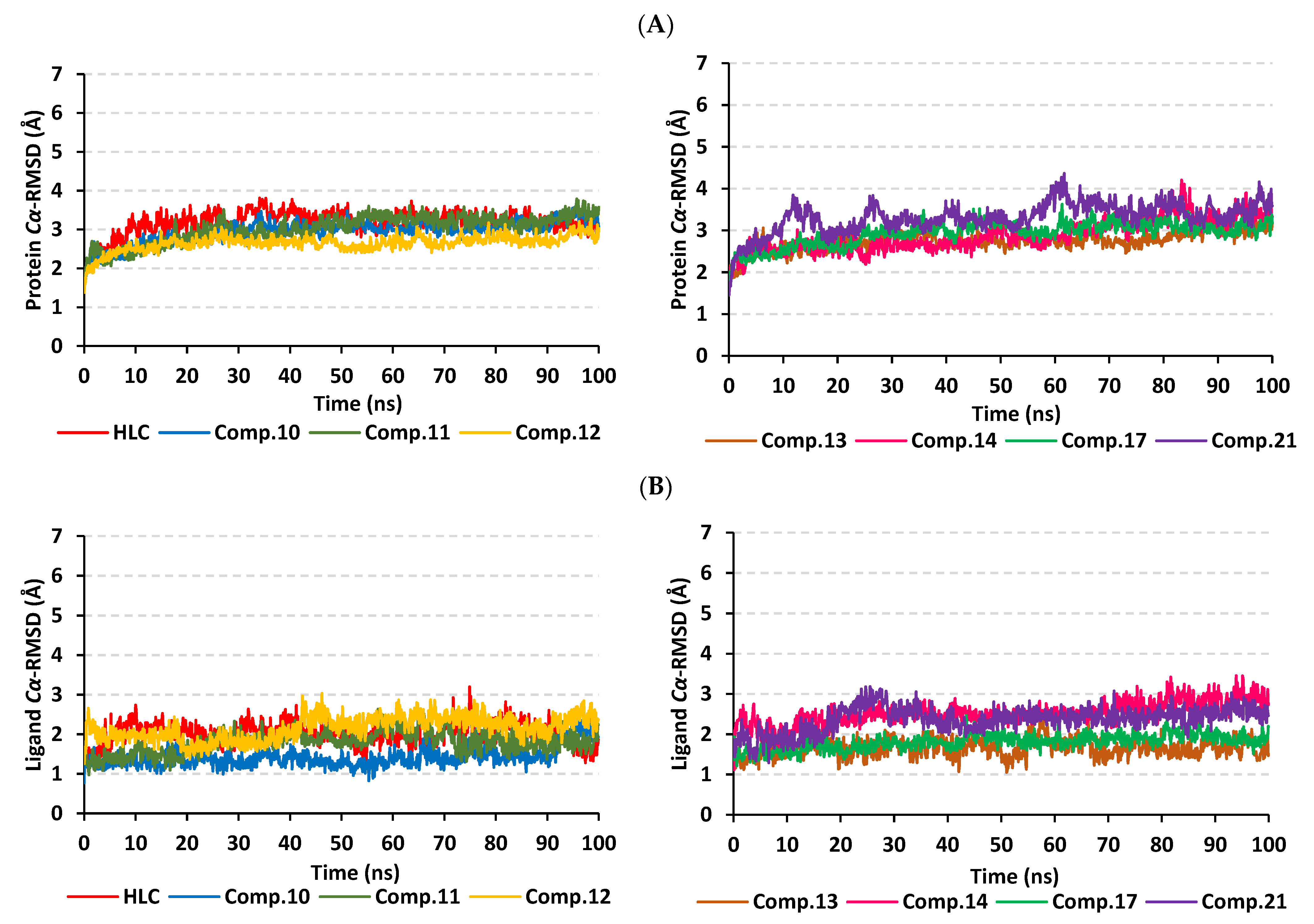
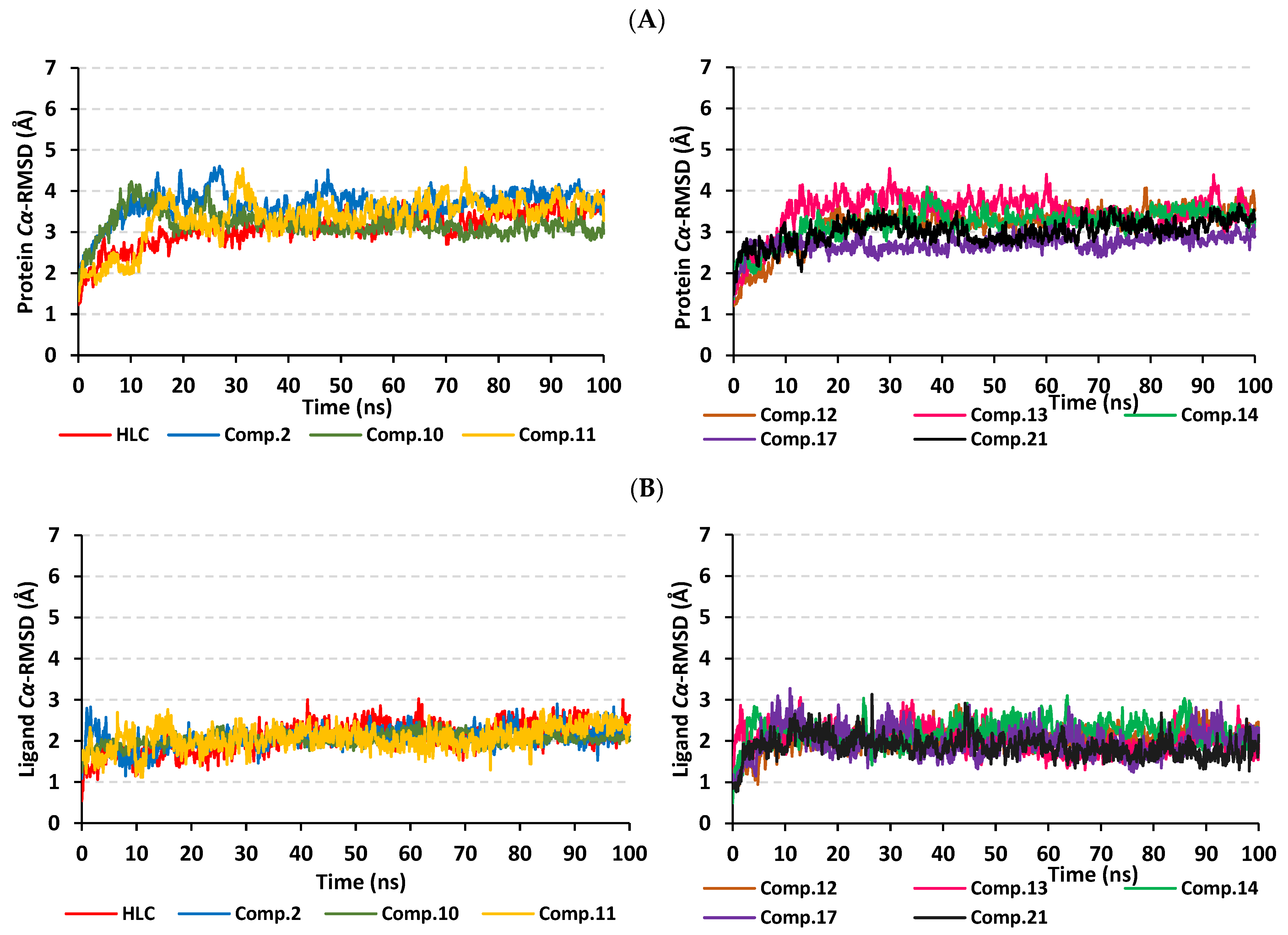
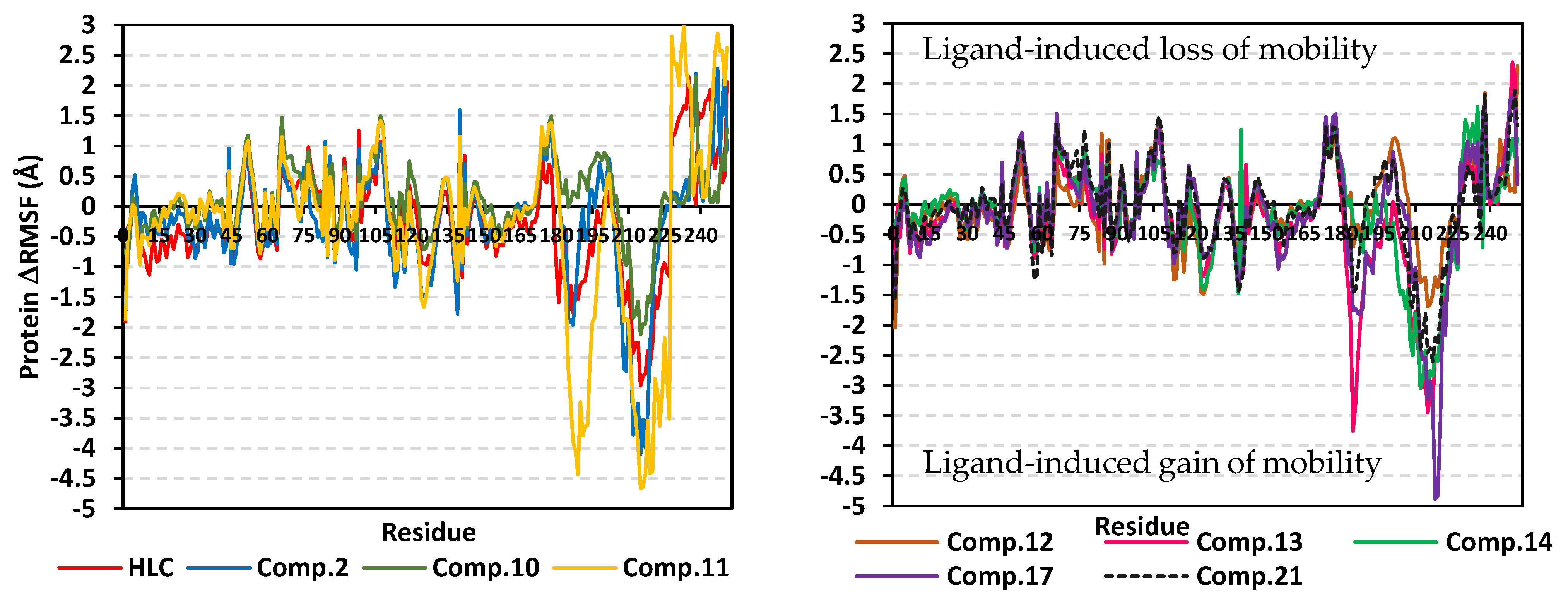
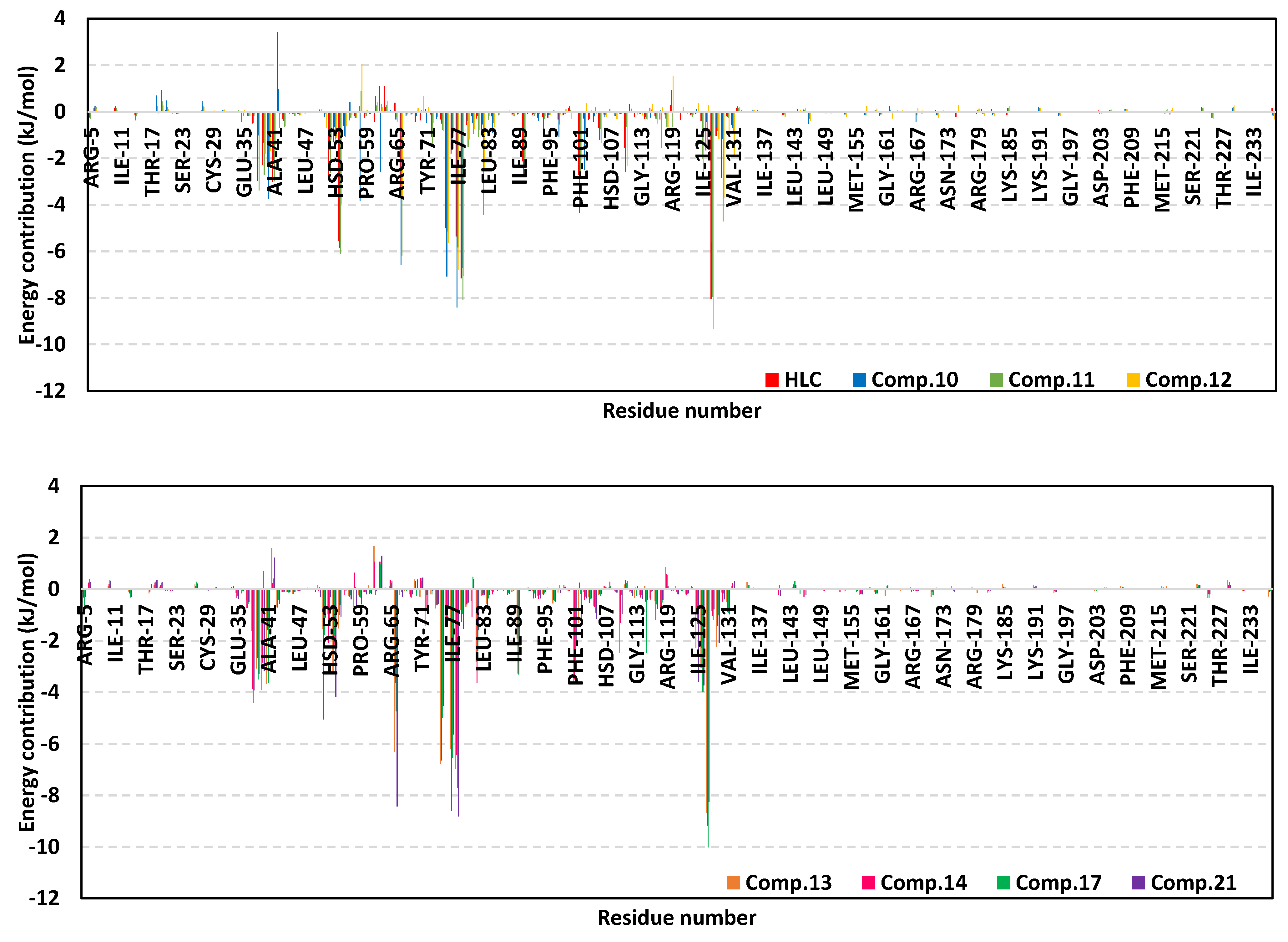
2.2. Molecular Dynamics Simulation
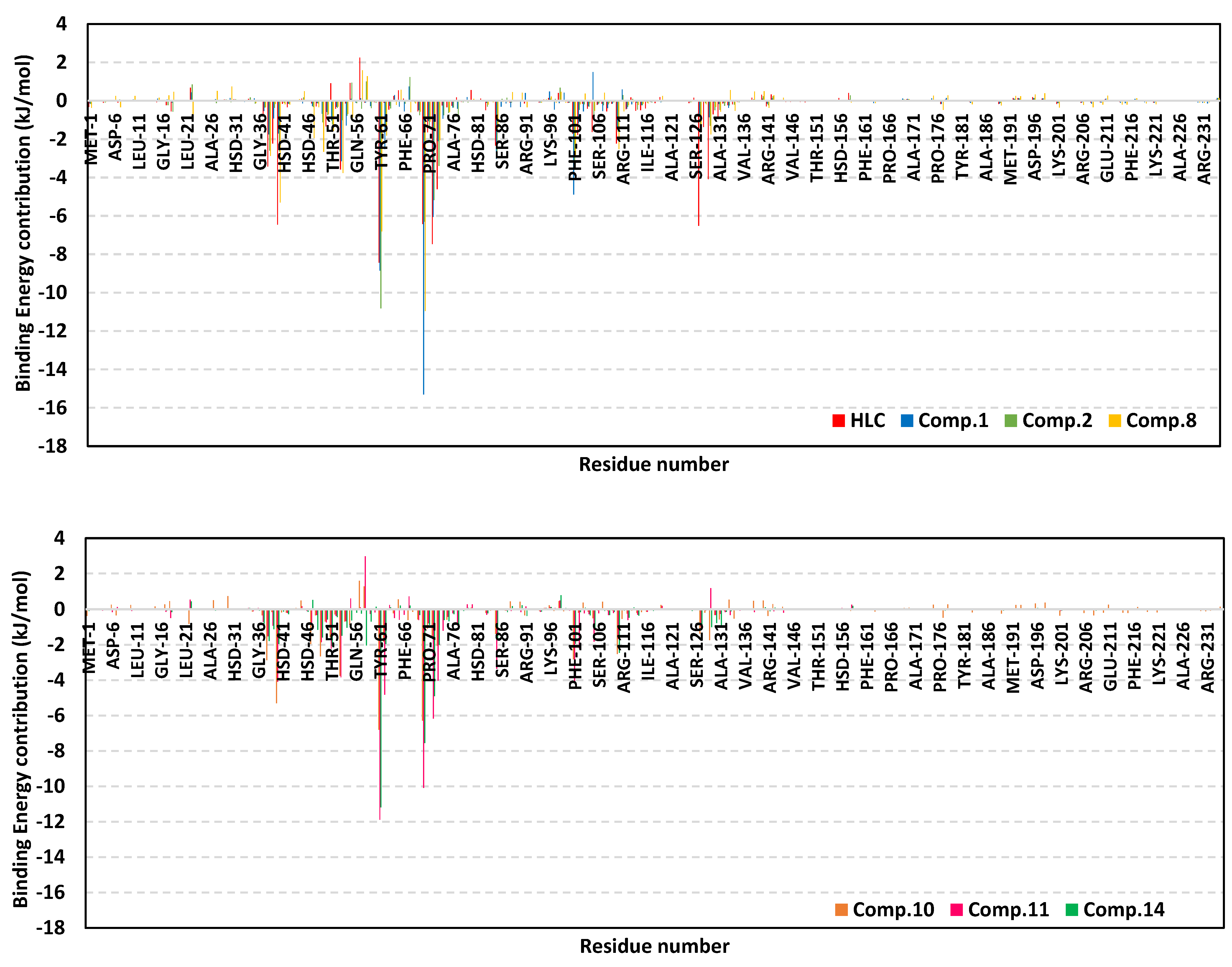
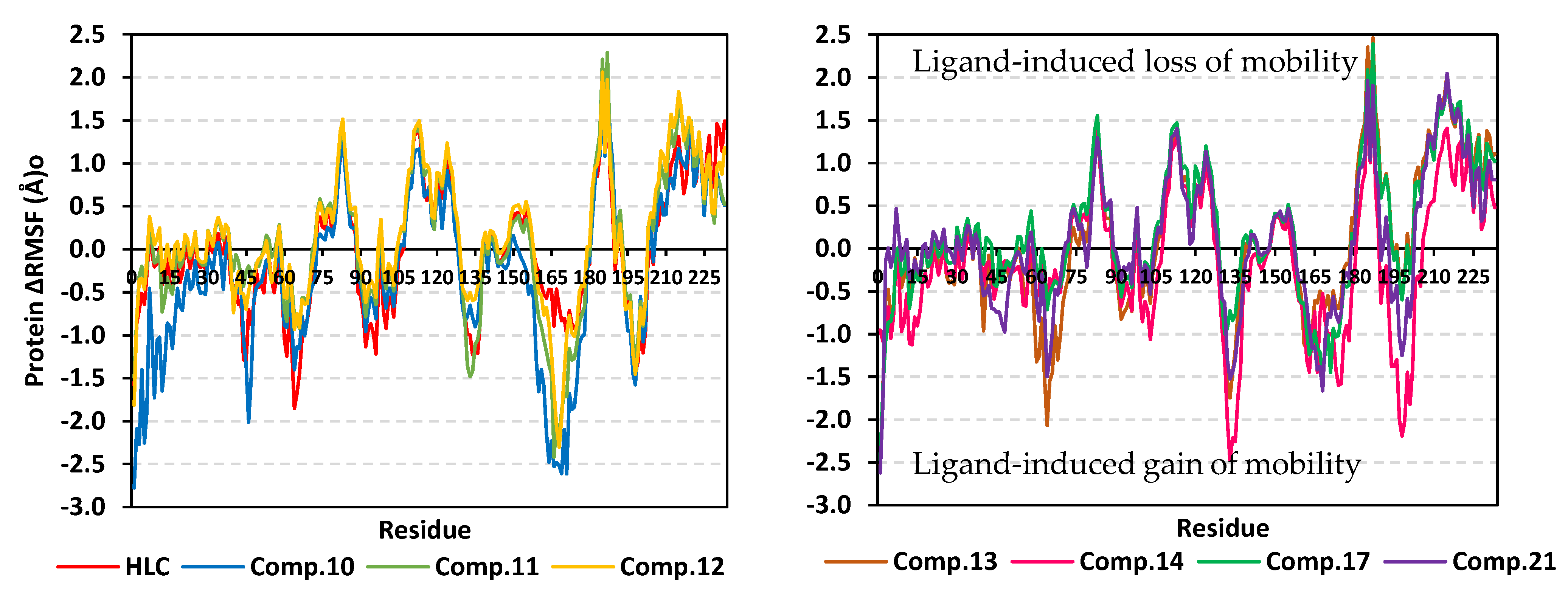
2.2.1. Analysis of Ligand–TraR A. tumefaciens Complexes
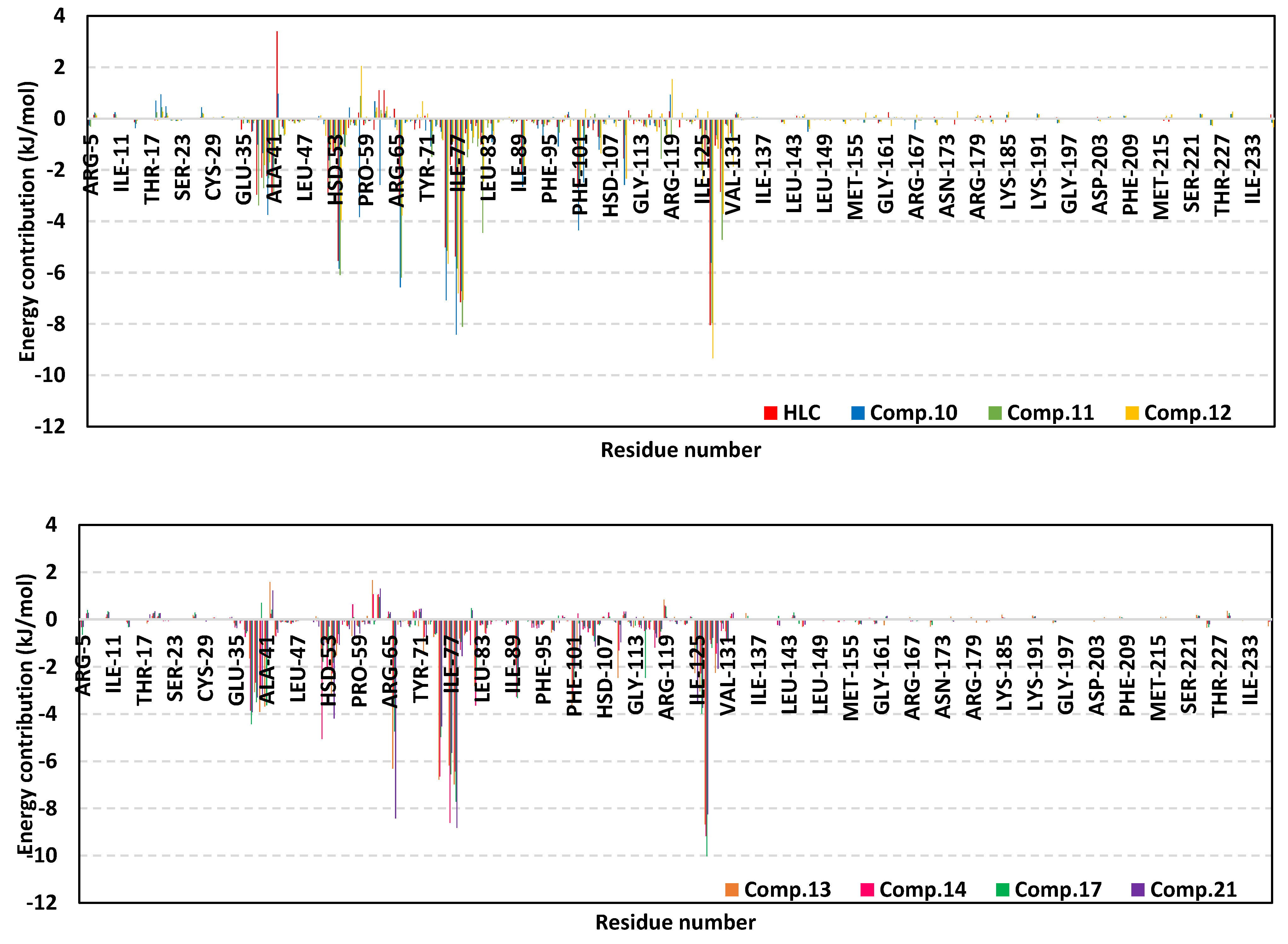
2.2.2. Analysis of Ligand–QscR P. aeruginosa Complexes
2.2.3. Analysis of Ligand–CviR C. violaceum Complexes
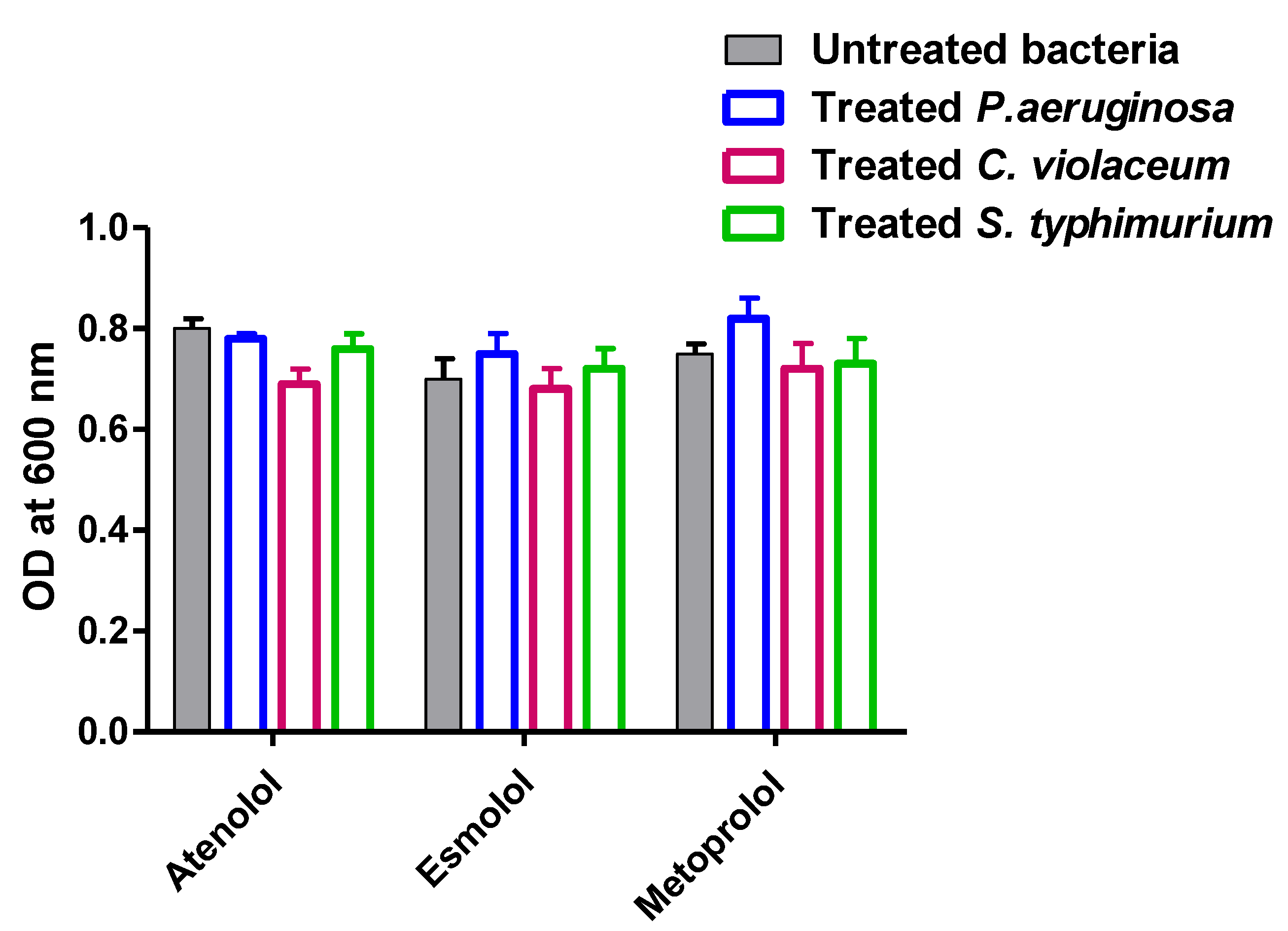
2.3. Determination of Selected B-Blockers’ Minimum Inhibitory Concentrations (MICs) against P. aeruginosa, C. violaceum and S. typhimurium
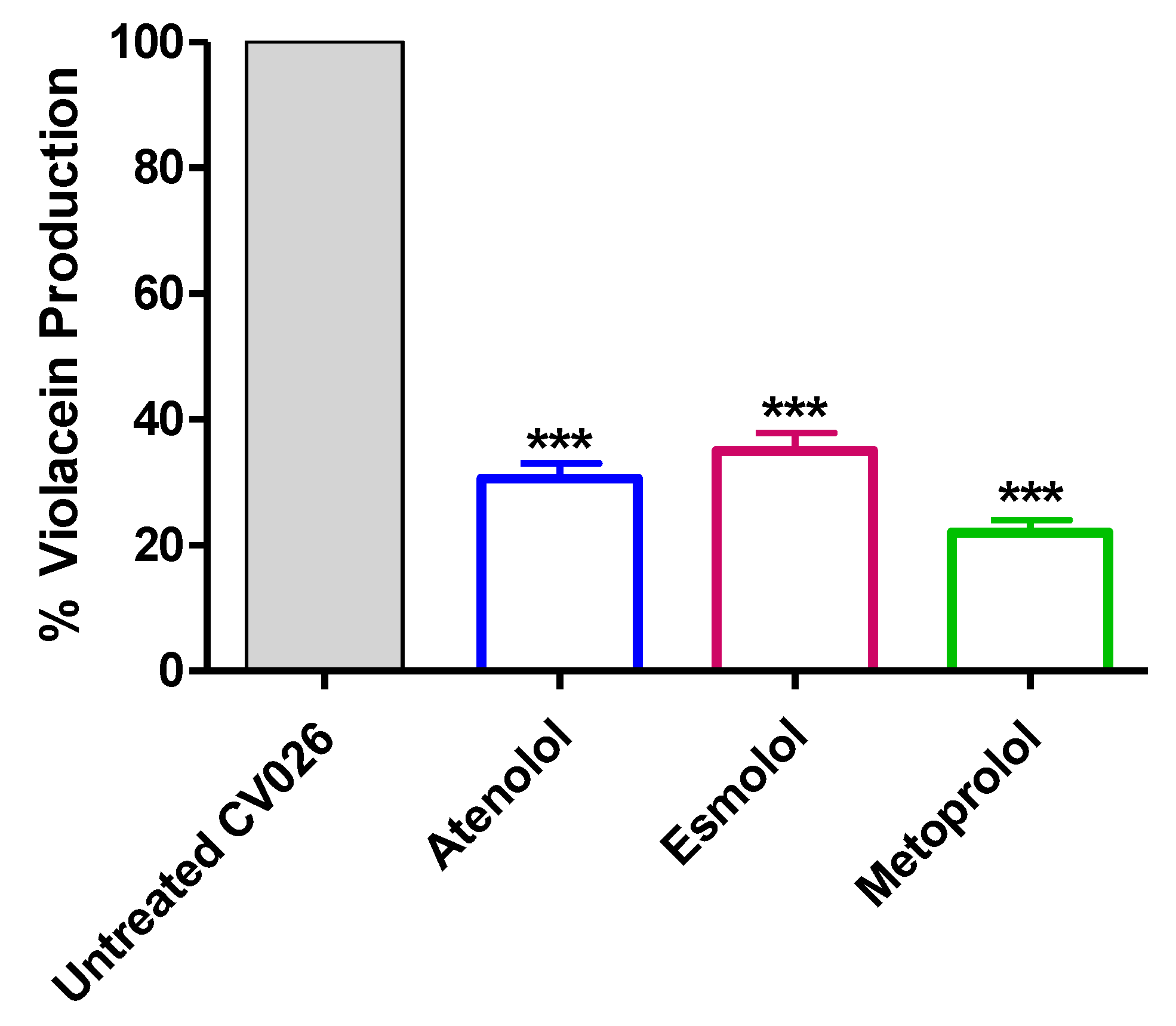
2.4. Inhibition of Violacein Production
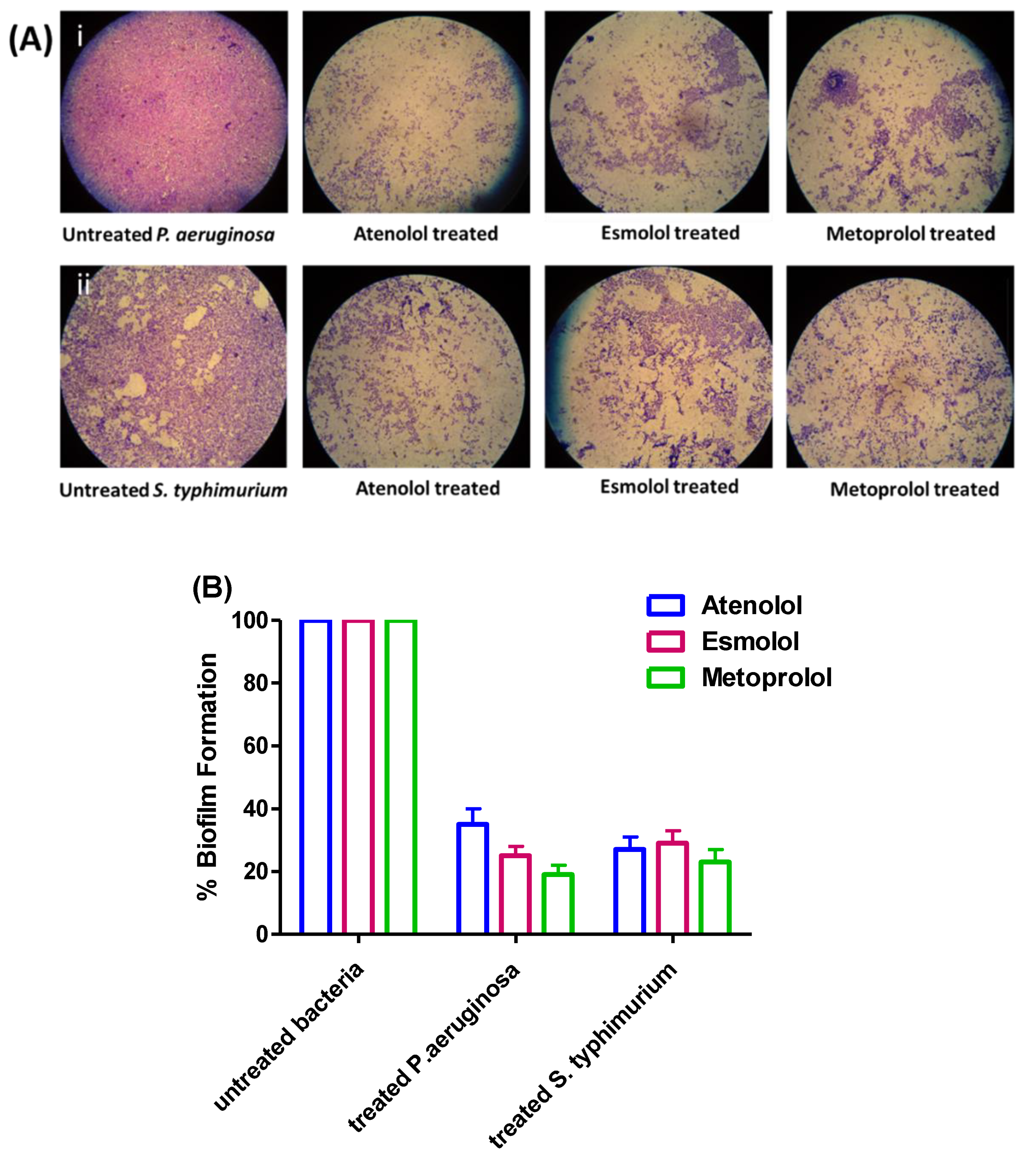
2.5. Anti-Biofilm Activities of β-Blockers in P. aeruginosa and S. typhimurium
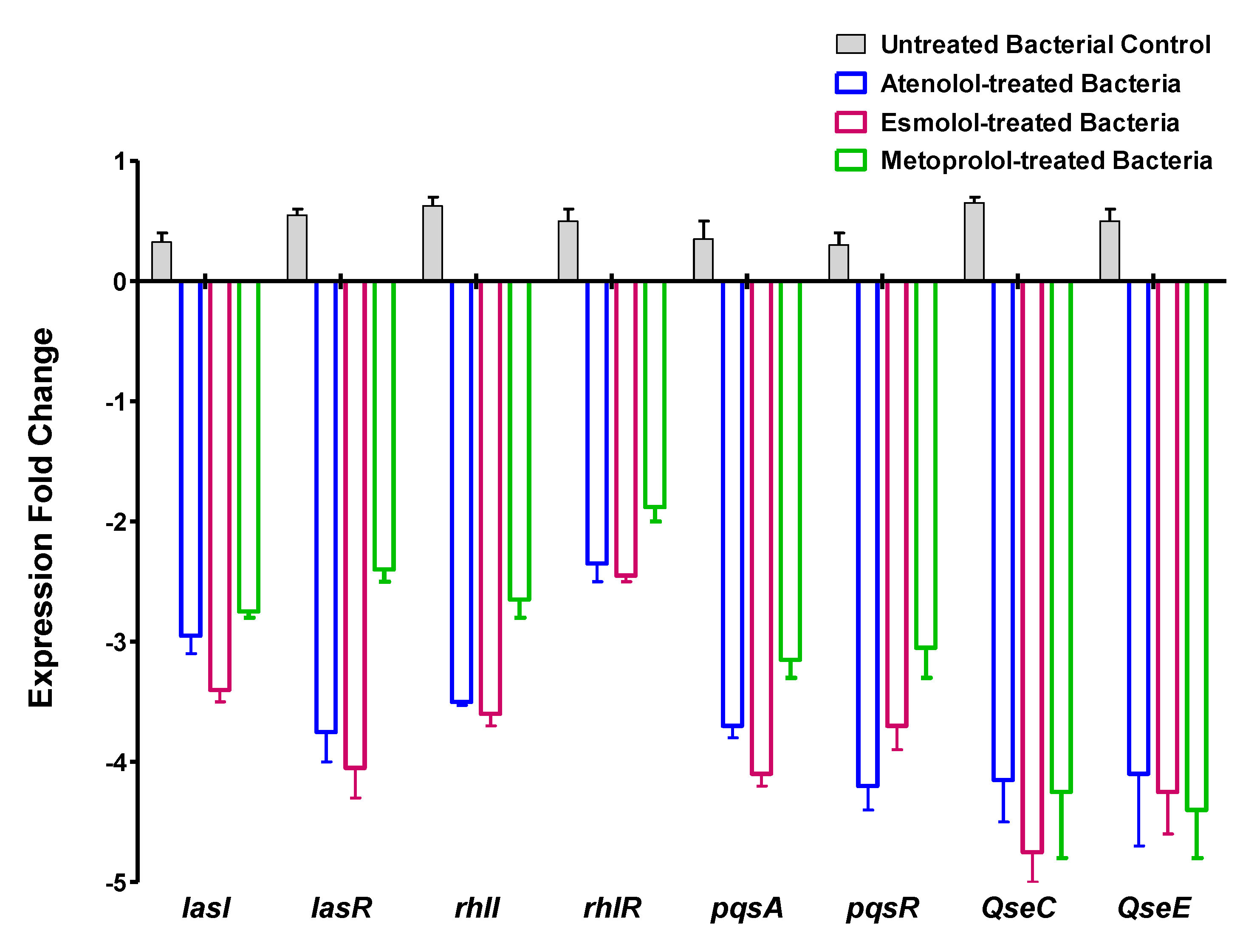
2.6. Effect of β-Blockers on the Expression of Virulence and QS-Encoding Genes in P. aeruginosa and S. typhimurium
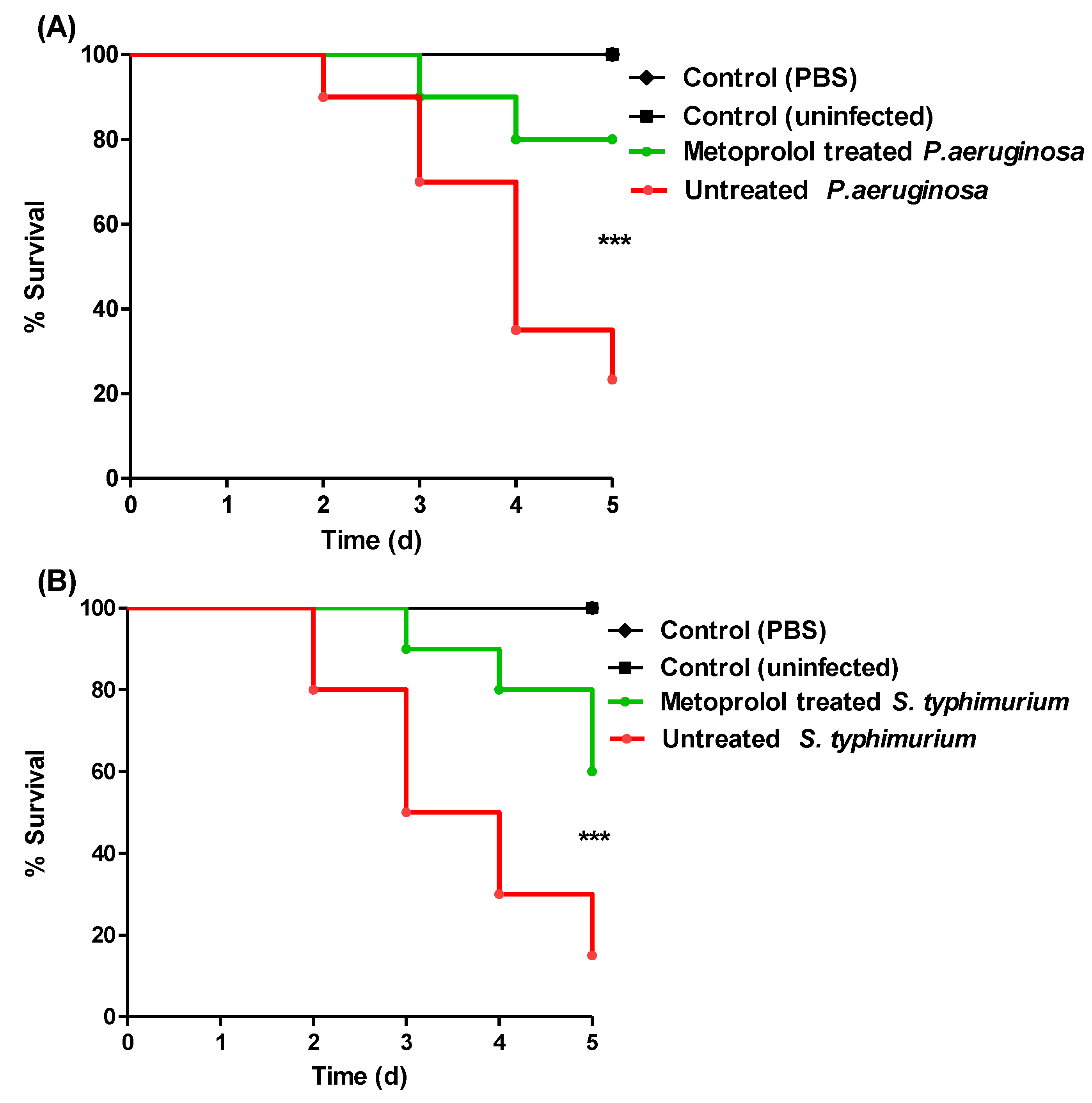
2.7. Metoprolol Protects Mice against P. aeruginosa and S. typhimurium
3. Discussion
4. Materials and Methods
4.1. Target Preparation and Ligand Construction for Docking Analysis
4.2. Two-Stage Multi-Target Docking Protocol
4.3. Molecular Dynamics Simulations
4.4. Chemicals, Microbiological Media and Bacterial Strains
4.5. Determination of MICs of β-Blockers, and the Effect of β-Blockers at Sub-MIC on Bacterial Growth
4.6. Assy of Violacein Production
4.7. Assay of Biofilm Formation
4.8. Quantitative RT-PCR of P. aeruginosa QS-Encoding Genes and S. typhimurium Sensor Kinase Encoding Genes
4.9. In-Vivo Mice Protection Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mohr, K.I. History of Antibiotics Research. Curr. Top. Microbiol. Immunol. 2016, 398, 237–272. [Google Scholar] [CrossRef] [PubMed]
- Blair, J.M.; Webber, M.A.; Baylay, A.J.; Ogbolu, D.O.; Piddock, L.J. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 2015, 13, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Rasko, D.A.; Sperandio, V. Anti-virulence strategies to combat bacteria-mediated disease. Nat. Rev. Drug Discov. 2010, 9, 117–128. [Google Scholar] [CrossRef]
- Cegelski, L.; Marshall, G.R.; Eldridge, G.R.; Hultgren, S.J. The biology and future prospects of antivirulence therapies. Nat. Rev. Microbiol. 2008, 6, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Hegazy, W.A.H.; Khayat, M.T.; Ibrahim, T.S.; Nassar, M.S.; Bakhrebah, M.A.; Abdulaal, W.H.; Alhakamy, N.A.; Bendary, M.M. Repurposing Anti-diabetic Drugs to Cripple Quorum Sensing in Pseudomonas aeruginosa. Microorganisms 2020, 8, 1285. [Google Scholar] [CrossRef]
- Muhlen, S.; Dersch, P. Anti-virulence Strategies to Target Bacterial Infections. Curr. Top. Microbiol. Immunol. 2016, 398, 147–183. [Google Scholar] [CrossRef] [PubMed]
- Abisado, R.G.; Benomar, S.; Klaus, J.R.; Dandekar, A.A.; Chandler, J.R. Bacterial Quorum Sensing and Microbial Community Interactions. mBio 2018, 9, e02331-17. [Google Scholar] [CrossRef] [Green Version]
- LaSarre, B.; Federle, M.J. Exploiting quorum sensing to confuse bacterial pathogens. Microbiol. Mol. Biol. Rev. 2013, 77, 73–111. [Google Scholar] [CrossRef] [Green Version]
- Rutherford, S.T.; Bassler, B.L. Bacterial quorum sensing: Its role in virulence and possibilities for its control. Cold Spring Harb. Perspect. Med. 2012, 2, a012427. [Google Scholar] [CrossRef]
- Papenfort, K.; Bassler, B.L. Quorum sensing signal-response systems in Gram-negative bacteria. Nat. Rev. Microbiol. 2016, 14, 576–588. [Google Scholar] [CrossRef]
- Stevens, A.M.; Dolan, K.M.; Greenberg, E.P. Synergistic binding of the Vibrio fischeri LuxR transcriptional activator domain and RNA polymerase to the lux promoter region. Proc. Natl. Acad. Sci. USA 1994, 91, 12619–12623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, D.T.; Sperandio, V. Inter-kingdom signalling: Communication between bacteria and their hosts. Nat. Rev. Microbiol. 2008, 6, 111–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kendall, M.M.; Sperandio, V. What a Dinner Party! Mechanisms and Functions of Interkingdom Signaling in Host-Pathogen Associations. mBio 2016, 7, e01748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karavolos, M.H.; Winzer, K.; Williams, P.; Khan, C.M. Pathogen espionage: Multiple bacterial adrenergic sensors eavesdrop on host communication systems. Mol. Microbiol. 2013, 87, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Hentzer, M.; Wu, H.; Andersen, J.B.; Riedel, K.; Rasmussen, T.B.; Bagge, N.; Kumar, N.; Schembri, M.A.; Song, Z.; Kristoffersen, P.; et al. Attenuation of Pseudomonas aeruginosa virulence by quorum sensing inhibitors. EMBO J. 2003, 22, 3803–3815. [Google Scholar] [CrossRef] [PubMed]
- Brothers, K.M.; Stella, N.A.; Romanowski, E.G.; Kowalski, R.P.; Shanks, R.M. EepR Mediates Secreted-Protein Production, Desiccation Survival, and Proliferation in a Corneal Infection Model. Infect. Immun. 2015, 83, 4373–4382. [Google Scholar] [CrossRef] [Green Version]
- Aldawsari, M.F.; Khafagy, E.S.; Saqr, A.A.; Alalaiwe, A.; Abbas, H.A.; Shaldam, M.A.; Hegazy, W.A.H.; Goda, R.M. Tackling Virulence of Pseudomonas aeruginosa by the Natural Furanone Sotolon. Antibiotics 2021, 10, 871. [Google Scholar] [CrossRef]
- Saqr, A.A.; Aldawsari, M.F.; Khafagy, E.-S.; Shaldam, M.A.; Hegazy, W.A.H.; Abbas, H.A. A Novel Use of Allopurinol as A Quorum-Sensing Inhibitor in Pseudomonas aeruginosa. Antibiotics 2021, 10, 1385. [Google Scholar] [CrossRef]
- Hegazy, W.A.H.; Rajab, A.A.H.; Abu Lila, A.S.; Abbas, H.A. Anti-diabetics and antimicrobials: Harmony of mutual interplay. World J. Diabetes 2021, 12, 1832–1855. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Szentmiklosi, A.J.; Szentandrassy, N.; Hegyi, B.; Horvath, B.; Magyar, J.; Banyasz, T.; Nanasi, P.P. Chemistry, physiology, and pharmacology of beta-adrenergic mechanisms in the heart. Why are beta-blocker antiarrhythmics superior? Curr. Pharm. Des. 2015, 21, 1030–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siebert, C.D.; Hansicke, A.; Nagel, T. Stereochemical comparison of nebivolol with other beta-blockers. Chirality 2008, 20, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.G.; Pappas, K.M.; Brace, J.L.; Miller, P.C.; Oulmassov, T.; Molyneaux, J.M.; Anderson, J.C.; Bashkin, J.K.; Winans, S.C.; Joachimiak, A. Structure of a bacterial quorum-sensing transcription factor complexed with pheromone and DNA. Nature 2002, 417, 971–974. [Google Scholar] [CrossRef]
- Lintz, M.J.; Oinuma, K.; Wysoczynski, C.L.; Greenberg, E.P.; Churchill, M.E. Crystal structure of QscR, a Pseudomonas aeruginosa quorum sensing signal receptor. Proc. Natl. Acad. Sci. USA 2011, 108, 15763–15768. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Swem, L.R.; Swem, D.L.; Stauff, D.L.; O’Loughlin, C.T.; Jeffrey, P.D.; Bassler, B.L.; Hughson, F.M. A strategy for antagonizing quorum sensing. Mol. Cell 2011, 42, 199–209. [Google Scholar] [CrossRef] [Green Version]
- Kolb, P.; Irwin, J.J. Docking screens: Right for the right reasons? Curr. Top. Med. Chem. 2009, 9, 755–770. [Google Scholar] [CrossRef] [Green Version]
- Pantsar, T.; Poso, A. Binding Affinity via Docking: Fact and Fiction. Molecules 2018, 23, 1899. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Lynn, A.M.; Gupta, V. Standardization of virtual-screening and post-processing protocols relevant to in-silico drug discovery. 3 Biotech 2018, 8, 504. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hospital, A.; Goñi, J.R.; Orozco, M.; Gelpí, J.L. Molecular dynamics simulations: Advances and applications. Adv. Appl. Bioinform. Chem. 2015, 8, 37–47. [Google Scholar] [CrossRef] [Green Version]
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002, 9, 646–652. [Google Scholar] [CrossRef]
- Arnittali, M.; Rissanou, A.N.; Harmandaris, V. Structure of Biomolecules through Molecular Dynamics Simulations. Procedia Comput. Sci. 2019, 156, 69–78. [Google Scholar] [CrossRef]
- Liu, K.; Watanabe, E.; Kokubo, H. Exploring the stability of ligand binding modes to proteins by molecular dynamics simulations. J. Comput.-Aided Mol. Des. 2017, 31, 201–211. [Google Scholar] [CrossRef]
- Benson, N.C.; Daggett, V. A comparison of multiscale methods for the analysis of molecular dynamics simulations. J. Phys. Chem. B 2012, 116, 8722–8731. [Google Scholar] [CrossRef] [Green Version]
- Cavasotto, C.N. Binding Free Energy Calculation Using Quantum Mechanics Aimed for Drug Lead Optimization. Methods Mol. Biol. 2020, 2114, 257–268. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Harrison, A.M.; Soby, S.D. Reclassification of Chromobacterium violaceum ATCC 31532 and its quorum biosensor mutant CV026 to Chromobacterium subtsugae. AMB Express 2020, 10, 202. [Google Scholar] [CrossRef]
- Abbas, H.A.; Hegazy, W.A.H. Repurposing anti-diabetic drug “Sitagliptin” as a novel virulence attenuating agent in Serratia marcescens. PLoS ONE 2020, 15, e0231625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegazy, W.A.H.; Khayat, M.T.; Ibrahim, T.S.; Youns, M.; Mosbah, R.; Soliman, W.E. Repurposing of antidiabetics as Serratia marcescens virulence inhibitors. Braz. J. Microbiol. 2021, 52, 627–638. [Google Scholar] [CrossRef]
- Pacheco, A.R.; Sperandio, V. Inter-kingdom signaling: Chemical language between bacteria and host. Curr. Opin. Microbiol. 2009, 12, 192–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreira, C.G.; Russell, R.; Mishra, A.A.; Narayanan, S.; Ritchie, J.M.; Waldor, M.K.; Curtis, M.M.; Winter, S.E.; Weinshenker, D.; Sperandio, V. Bacterial Adrenergic Sensors Regulate Virulence of Enteric Pathogens in the Gut. mBio 2016, 7, e00826-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sperandio, V.; Torres, A.G.; Jarvis, B.; Nataro, J.P.; Kaper, J.B. Bacteria-host communication: The language of hormones. Proc. Natl. Acad. Sci. USA 2003, 100, 8951–8956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandrini, S.M.; Shergill, R.; Woodward, J.; Muralikuttan, R.; Haigh, R.D.; Lyte, M.; Freestone, P.P. Elucidation of the mechanism by which catecholamine stress hormones liberate iron from the innate immune defense proteins transferrin and lactoferrin. J. Bacteriol. 2010, 192, 587–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flierl, M.A.; Rittirsch, D.; Nadeau, B.A.; Sarma, J.V.; Day, D.E.; Lentsch, A.B.; Huber-Lang, M.S.; Ward, P.A. Upregulation of phagocyte-derived catecholamines augments the acute inflammatory response. PLoS ONE 2009, 4, e4414. [Google Scholar] [CrossRef] [Green Version]
- Swem, L.R.; Swem, D.L.; O’Loughlin, C.T.; Gatmaitan, R.; Zhao, B.; Ulrich, S.M.; Bassler, B.L. A quorum-sensing antagonist targets both membrane-bound and cytoplasmic receptors and controls bacterial pathogenicity. Mol. Cell 2009, 35, 143–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geske, G.D.; O’Neill, J.C.; Miller, D.M.; Mattmann, M.E.; Blackwell, H.E. Modulation of bacterial quorum sensing with synthetic ligands: Systematic evaluation of N-acylated homoserine lactones in multiple species and new insights into their mechanisms of action. J. Am. Chem. Soc. 2007, 129, 13613–13625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kontoyianni, M.; McClellan, L.M.; Sokol, G.S. Evaluation of Docking Performance: Comparative Data on Docking Algorithms. J. Med. Chem. 2004, 47, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Kim, J.; Park, H.Y.; Park, H.J.; Kim, C.K.; Yoon, J.; Lee, J.H. Development of inhibitors against TraR quorum-sensing system in Agrobacterium tumefaciens by molecular modeling of the ligand-receptor interaction. Mol. Cells 2009, 28, 447–453. [Google Scholar] [CrossRef]
- Kim, C.; Kim, J.; Park, H.Y.; McLean, R.J.; Kim, C.K.; Jeon, J.; Yi, S.S.; Kim, Y.G.; Lee, Y.S.; Yoon, J. Molecular modeling, synthesis, and screening of new bacterial quorumsensing antagonists. J. Microbiol. Biotechnol. 2007, 17, 1598–1606. [Google Scholar] [PubMed]
- Qin, X.; Vila-Sanjurjo, C.; Singh, R.; Philipp, B.; Goycoolea, F.M. Screening of Bacterial Quorum Sensing Inhibitors in a Vibrio fischeri LuxR-Based Synthetic Fluorescent, E. coli Biosensor. Pharmaceuticals 2020, 13, 263. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Yin, B.; Qian, L.; Zeng, Z.; Yang, Z.; Li, H.; Lu, Y.; Zhou, S. Screening for novel quorum-sensing inhibitors to interfere with the formation of Pseudomonas aeruginosa biofilm. J. Med. Microbiol. 2011, 60, 1827–1834. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Qian, L.; Cao, L.; Tan, H.; Huang, Y.; Xue, X.; Shen, Y.; Zhou, S. Virtual screening for novel quorum sensing inhibitors to eradicate biofilm formation of Pseudomonas aeruginosa. Appl. Microbiol. Biotechnol. 2008, 79, 119–126. [Google Scholar] [CrossRef]
- Xu, Y.; Tong, X.; Sun, P.; Bi, L.; Lin, K. Virtual screening and biological evaluation of biofilm inhibitors on dual targets in quorum sensing system. Future Med. Chem. 2017, 9, 1983–1994. [Google Scholar] [CrossRef]
- Sadiq, S.; Rana, N.F.; Zahid, M.A.; Zargaham, M.K.; Tanweer, T.; Batool, A.; Naeem, A.; Nawaz, A.; Rizwan Ur, R.; Muneer, Z.; et al. Virtual Screening of FDA-Approved Drugs against LasR of Pseudomonas aeruginosa for Antibiofilm Potential. Molecules 2020, 25, 3723. [Google Scholar] [CrossRef]
- Vetrivel, A.; Natchimuthu, S.; Subramanian, V.; Murugesan, R. High-Throughput Virtual Screening for a New Class of Antagonist Targeting LasR of Pseudomonas aeruginosa. ACS Omega 2021, 6, 18314–18324. [Google Scholar] [CrossRef]
- Askoura, M.; Almalki, A.J.; Lila, A.S.A.; Almansour, K.; Alshammari, F.; Khafagy, E.-S.; Ibrahim, T.S.; Hegazy, W.A.H. Alteration of Salmonella enterica Virulence and Host Pathogenesis through Targeting sdiA by Using the CRISPR-Cas9 System. Microorganisms 2021, 9, 2564. [Google Scholar] [CrossRef]
- Martins, F.G.; Melo, A.; Sousa, S.F. Identification of New Potential Inhibitors of Quorum Sensing through a Specialized Multi-Level Computational Approach. Molecules 2021, 26, 2600. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, V.; Zhong, L.; Wang, H.; Yu, G.; Zhang, Y.; Li, A. Virtual Screening and Biomolecular Interactions of CviR-Based Quorum Sensing Inhibitors against Chromobacterium violaceum. Front. Cell. Infect. Microbiol. 2018, 8, 292. [Google Scholar] [CrossRef]
- Herrera-Arizmendi, J.L.; Curiel-Quesada, E.; Correa-Basurto, J.; Bello, M.; Reyes-Arellano, A. Effect of New Analogs of Hexyloxy Phenyl Imidazoline on Quorum Sensing in Chromobacterium violaceum and In Silico Analysis of Ligand-Receptor Interactions. J. Chem. 2020, 2020, 8735190. [Google Scholar] [CrossRef] [Green Version]
- Venkatramanan, M.; Sankar Ganesh, P.; Senthil, R.; Akshay, J.; Veera Ravi, A.; Langeswaran, K.; Vadivelu, J.; Nagarajan, S.; Rajendran, K.; Shankar, E.M. Inhibition of Quorum Sensing and Biofilm Formation in Chromobacterium violaceum by Fruit Extracts of Passiflora edulis. ACS Omega 2020, 5, 25605–25616. [Google Scholar] [CrossRef] [PubMed]
- Mellini, M.; Di Muzio, E.; D’Angelo, F.; Baldelli, V.; Ferrillo, S.; Visca, P.; Leoni, L.; Polticelli, F.; Rampioni, G. In silico Selection and Experimental Validation of FDA-Approved Drugs as Anti-quorum Sensing Agents. Front. Microbiol. 2019, 10, 2355. [Google Scholar] [CrossRef] [PubMed]
- Passos da Silva, D.; Patel, H.K.; González, J.F.; Devescovi, G.; Meng, X.; Covaceuszach, S.; Lamba, D.; Subramoni, S.; Venturi, V. Studies on synthetic LuxR solo hybrids. Front. Cell. Infect. Microbiol. 2015, 5, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCready, A.R.; Paczkowski, J.E.; Henke, B.R.; Bassler, B.L. Structural determinants driving homoserine lactone ligand selection in the Pseudomonas aeruginosa LasR quorum-sensing receptor. Proc. Natl. Acad. Sci. USA 2019, 116, 245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Askoura, M.; Hegazy, W.A.H. Ciprofloxacin interferes with Salmonella Typhimurium intracellular survival and host virulence through repression of Salmonella pathogenicity island-2 (SPI-2) genes expression. Pathog. Dis. 2020, 78, 78. [Google Scholar] [CrossRef] [PubMed]
- Hegazy, W.A.H.; Abbas, H.A. Evaluation of the role of SsaV Salmonella pathogenicity island-2 dependent type III secretion system components on the virulence behavior of Salmonella enterica serovar Typhimurium. Afr. J. Biotechnol. 2017, 16, 718–726. [Google Scholar] [CrossRef] [Green Version]
- Michael, B.; Smith, J.N.; Swift, S.; Heffron, F.; Ahmer, B.M. SdiA of Salmonella enterica is a LuxR homolog that detects mixed microbial communities. J. Bacteriol. 2001, 183, 5733–5742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmer, B.M. Cell-to-cell signalling in Escherichia coli and Salmonella enterica. Mol. Microbiol. 2004, 52, 933–945. [Google Scholar] [CrossRef]
- Moreira, C.G.; Sperandio, V. Interplay between the QseC and QseE bacterial adrenergic sensor kinases in Salmonella enterica serovar Typhimurium pathogenesis. Infect. Immun. 2012, 80, 4344–4353. [Google Scholar] [CrossRef] [Green Version]
- Hoiby, N.; Bjarnsholt, T.; Givskov, M.; Molin, S.; Ciofu, O. Antibiotic resistance of bacterial biofilms. Int. J. Antimicrob. Agents 2010, 35, 322–332. [Google Scholar] [CrossRef] [Green Version]
- Khayyat, A.N.; Hegazy, W.A.H.; Shaldam, M.A.; Mosbah, R.; Almalki, A.J.; Ibrahim, T.S.; Khayat, M.T.; Khafagy, E.S.; Soliman, W.E.; Abbas, H.A. Xylitol Inhibits Growth and Blocks Virulence in Serratia marcescens. Microorganisms 2021, 9, 1083. [Google Scholar] [CrossRef]
- Vishwa, B.; Moin, A.; Gowda, D.V.; Rizvi, S.M.D.; Hegazy, W.A.H.; Abu Lila, A.S.; Khafagy, E.S.; Allam, A.N. Pulmonary Targeting of Inhalable Moxifloxacin Microspheres for Effective Management of Tuberculosis. Pharmaceutics 2021, 13, 79. [Google Scholar] [CrossRef]
- Arabi, Y.M.; Shalhoub, S.; Mandourah, Y.; Al-Hameed, F.; Al-Omari, A.; Al Qasim, E.; Jose, J.; Alraddadi, B.; Almotairi, A.; Al Khatib, K.; et al. Ribavirin and Interferon Therapy for Critically Ill Patients With Middle East Respiratory Syndrome: A Multicenter Observational Study. Clin. Infect. Dis. 2020, 70, 1837–1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, J.; Edelsbrunner, H.; Woodward, C. Anatomy of protein pockets and cavities: Measurement of binding site geometry and implications for ligand design. Protein Sci. A Publ. Protein Soc. 1998, 7, 1884–1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef]
- Wojciechowski, M.; Lesyng, B. Generalized Born Model: Analysis, Refinement, and Applications to Proteins. J. Phys. Chem. B 2004, 108, 18368–18376. [Google Scholar] [CrossRef]
- Labute, P. The generalized Born/volume integral implicit solvent model: Estimation of the free energy of hydration using London dispersion instead of atomic surface area. J. Comput. Chem. 2008, 29, 1693–1698. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, 2.0.6; Schrödinger, LLC: New York, NY, USA, 2020.
- de Souza, A.S.; Pacheco, B.D.C.; Pinheiro, S.; Muri, E.M.F.; Dias, L.R.S.; Lima, C.H.S.; Garrett, R.; de Moraes, M.B.M.; de Souza, B.E.G.; Puzer, L. 3-Acyltetramic acids as a novel class of inhibitors for human kallikreins 5 and 7. Bioorganic Med. Chem. Lett. 2019, 29, 1094–1098. [Google Scholar] [CrossRef]
- Albuquerque, S.O.; Barros, T.G.; Dias, L.R.; Lima, C.H.D.S.; Pedro, H.D.A.; Flores-Junior, L.A.; Dos Santos, E.G.; Loponte, H.F.; Pinheiro, S.; Dias, W.B.; et al. Biological evaluation and molecular modeling of peptidomimetic compounds as inhibitors for O-GlcNAc transferase (OGT). Eur. J. Pharm. Sci. 2020, 154, 105510. [Google Scholar] [CrossRef]
- Páll, S.; Abraham, M.J.; Kutzner, C.; Hess, B.; Lindahl, E. Tackling Exascale Software Challenges in Molecular Dynamics Simulations with GROMACS. In Solving Software Challenges for Exascale; Springer: Cham, Switzerland, 2015; pp. 3–27. [Google Scholar]
- Saleh, A.H.; Abdelwaly, A.; Darwish, K.M.; Eissa, A.; Chittiboyina, A.; Helal, M.A. Deciphering the molecular basis of the kappa opioid receptor selectivity: A Molecular Dynamics study. J. Mol. Graph. Model. 2021, 106, 107940. [Google Scholar] [CrossRef]
- Ross, G.A.; Rustenburg, A.S.; Grinaway, P.B.; Fass, J.; Chodera, J.D. Biomolecular Simulations under Realistic Macroscopic Salt Conditions. J. Phys. Chem. B 2018, 122, 5466–5486. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Páll, S.; Hess, B. A flexible algorithm for calculating pair interactions on SIMD architectures. Comput. Phys. Commun. 2013, 184, 2641–2650. [Google Scholar] [CrossRef] [Green Version]
- Khayyat, A.N.; Abbas, H.A.; Khayat, M.T.; Shaldam, M.A.; Askoura, M.; Asfour, H.Z.; Khafagy, E.-S.; Abu Lila, A.S.; Allam, A.N.; Hegazy, W.A.H. Secnidazole Is a Promising Imidazole Mitigator of Serratia marcescens Virulence. Microorganisms 2021, 9, 2333. [Google Scholar] [CrossRef]
- Khayyat, A.N.; Abbas, H.A.; Mohamed, M.F.A.; Asfour, H.Z.; Khayat, M.T.; Ibrahim, T.S.; Youns, M.; Khafagy, E.-S.; Abu Lila, A.S.; Safo, M.K.; et al. Not Only Antimicrobial: Metronidazole Mitigates the Virulence of Proteus mirabilis Isolated from Macerated Diabetic Foot Ulcer. Appl. Sci. 2021, 11, 6847. [Google Scholar] [CrossRef]
- Youns, M.; Askoura, M.; Abbas, H.A.; Attia, G.H.; Khayyat, A.N.; Goda, R.M.; Almalki, A.J.; Khafagy, E.S.; Hegazy, W.A.H. Celastrol Modulates Multiple Signaling Pathways to Inhibit Proliferation of Pancreatic Cancer via DDIT3 and ATF3 Up-Regulation and RRM2 and MCM4 Down-Regulation. Onco Targets Ther. 2021, 14, 3849–3860. [Google Scholar] [CrossRef] [PubMed]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Name | Binding Energy (Kcal/mol) a | ||
|---|---|---|---|---|
| 1L3L | 3SZT | 3QP5 | ||
| 1 | Propranolol | −6.9518 | −6.8120 | −6.9592 |
| 2 | Pindolol | −6.7337 | −6.7648 | −7.2291 |
| 3 | Levobunolol | −5.8300 | −5.3392 | −5.2812 |
| 4 | Nadolol | −5.3036 | −6.6720 | −6.0193 |
| 5 | Oxprenolol | −5.5981 | −6.7365 | −6.7374 |
| 6 | Carteolol | −5.3073 | −6.7183 | −7.0493 |
| 7 | Penbutolol | −4.0056 | −5.2514 | −5.1763 |
| 8 | Timolol | −6.8266 | −6.9061 | −6.6453 |
| 9 | Sotalol | −6.1145 | −6.3450 | −6.8093 |
| 10 | Atenolol | −7.9923 | −8.2452 | −8.2351 |
| 11 | Esmolol | −6.9957 | −8.2803 | −7.4849 |
| 12 | Betaxolol | −5.0198 | −7.6193 | −7.7426 |
| 13 | Bisoprolol | −6.1495 | −9.2750 | −7.3777 |
| 14 | Metoprolol | −7.2391 | −8.7438 | −7.8018 |
| 15 | Practolol | −5.6051 | −6.2557 | −6.9717 |
| 16 | Metipranolol | −4.0760 | −5.8692 | −6.7320 |
| 17 | Acebutolol | −5.7674 | −8.4568 | −8.4732 |
| 18 | Celiprolol | −3.8698 | −5.1621 | −5.3801 |
| 19 | Bucindolol | −2.2153 | −5.8371 | −6.2381 |
| 20 | Carvedilol | −1.8936 | −3.8607 | −4.2513 |
| 21 | Labetalol | −4.7226 | −7.7411 | −7.8677 |
| 22 | Nebivolol | −1.9527 | −5.2294 | −4.8969 |
| 1L3L Reference | O-C8-HSL | −6.2977 | – | – |
| 3SZT Reference | O-C12-HSL | – | −7.5547 | – |
| 3QP5 Reference | HLC | −6.6245 | −7.6488 | −7.2051 |
| Compound | Docking Energy (Kcal/mol) a | H-Bond Interactions | Hydrophobic Interactions | π-Interactions | Van Der Waal with Side Chain Carbons | |
|---|---|---|---|---|---|---|
| Preliminary (Rigid) | Induced-Fit (Flexible) | |||||
| Propranolol | −6.9518 | −7.5013 | Tyr53, Asp70, Thr129 | Ala38, Leu40, Ala49, Tyr53, Trp57, Phe62, Val72, Trp85, Phe101, Tyr102, Ala105, Ile110, Met127 | Tyr61 (π-π) | Gln58 (Cβ, Cδ) |
| Pindolol | −6.7337 | −7.4013 | Gln58, Tyr61, Asp70 | Ala38, Leu40, Ala49, Tyr53, Trp57, Phe62, Val72, Val73, Trp85, Phe101, Tyr102, Ile110, Met127 | Tyr61 (π-π) | - |
| Timolol | −6.8266 | −7.4812 | Tyr53, Trp57, Asp70, Thr129 | Ala38, Leu40, Ala49, Tyr53, Trp57, Val72, Trp85, Phe101, Tyr102, Ala105, Ile110, Met127 | Tyr61 (H-π) | - |
| Atenolol | −7.9923 | −8.5142 | Tyr53, Gln85, Tyr61, Phe62, Asp70, Thr129 | Ala38, Leu40, Ala49, Tyr53, Trp57, Val72, Val73, Trp85, Phe101, Tyr102, Ala105, Ile110, Met127 | Tyr61 (π-π) | - |
| Esmolol | −6.9957 | −7.5913 | Thr51, Tyr53 *, Phe62, Asp70, Thr129 | Ala38, Leu40, Ala49, Tyr53, Trp57, Val72, Trp85, Phe101, Tyr102, Ala105, Ile110, Met127 | Tyr61 (π-π) | - |
| Metoprolol | −7.2391 | −8.1923 | Thr51, Tyr53, Asp70, Trp85, Thr129 | Ala38, Leu40, Ala49, Tyr53, Trp57, Val72, Val73, Trp85, Phe101, Tyr102, Ala105, Ile110, Met127 | Tyr61 (π-π) | - |
| HLC | −6.6245 | −7.1612 | Tyr53, Trp57, Asp70, Tyr102 | Ala38, Leu40, Ala49, Tyr53, Trp57, Val72, Trp85, Phe101, Tyr102, Ala105, Ile110, Met127 | Tyr61 (π-π) | Gln58 (Cβ, Cδ) |
| Compound | Docking Energy (Kcal/mol) a | H-Bond Interactions | Hydrophobic Interactions | π-Interactions | Van Der Waal with Side Chain Carbons | |
|---|---|---|---|---|---|---|
| Preliminary (Rigid) | Induced-Fit (Flexible) | |||||
| Atenolol | −8.2452 | −8.9102 | Ser38, Tyr52, Tyr58, Tyr66, Asp75 | Ala41, Tyr52, Tyr58, Trp62, Tyr66, Ile77, Val78, Leu82, Phe101, Trp102, Ala105, Ile110, Ile125, Met127, Val131 | Trp90 (π-H) Trp102 (π-H) | Arg42 (Cβ) |
| Esmolol | −8.2803 | −8.9182 | Ser38, Arg42, Tyr58, Trp66, Ser129, Asp75 | Phe39, Ala41, Tyr52, His53, Phe54, Tyr58, Trp62, Pro76, Ile77, Val78, Leu82, Trp90, Phe101, Trp102, Ala105, Ile110, Pro117, Ile125, Met127, Val131 | Phe54 (π-π) Trp90 (π-H) | - |
| Betaxolol | −7.6193 | −8.2918 | Ser38, Tyr58 *, Trp66, Asp75, Met127 | Phe39, Ala41, Tyr52, His53, Tyr58, Trp62, Ile77, Val78, Leu82, Trp90, Phe101, Trp102, Ala105, Ile110, Pro117, Ile125, Met127, Val131 | Phe54 (π-π) Trp90 (π-H) | - |
| Bisoprolol | −9.2750 | −9.7616 | Ser38 *, Tyr58, Trp90, Asp75 *, Leu82, Ser129 | Phe39, Ala41, Tyr52, His53, Tyr58, Trp62, Ile77, Val78, Leu82, Trp90, Phe101, Trp102, Ile110, Pro117, Ile125, Met127, Val131 | Phe54 (π-π) Trp90 (π-H) Tyr66 (π-H) | Arg42 (Cβ, Cδ) |
| Metoprolol | −8.7438 | −9.5953 | Ser38, Arg42, Tyr52, Tyr58 *, Asp75, Ser129 | Phe39, Ala41, Tyr52, Tyr58, Trp62, Ile77, Val78, Leu82, Trp90, Phe101, Trp102, Ala105, Ile110, Ile125, Met127, Val131 | Phe54 (π-π) Trp90 (π-H) Trp102 (π-H) | Arg42 (Cβ, Cδ) |
| Acebutolol | −8.4568 | −9.3164 | Ser38 *, Tyr58, Thr72, Asp75 *, Met127, Ser129 | Phe39, Ala41, Tyr52, His53, Tyr58, Trp62, Ile77, Val78, Leu82, Trp90, Phe101, Trp102, Ile110, Pro117, Ile125, Met127, Val131 | Phe54 (π-π) Tyr66 (π-H) Trp90 (π-H) | Arg42 (Cβ, Cδ) |
| Labetalol | −7.7411 | −8.7880 | Ser38, Tyr58, Trp62, Trp90 | Phe39, Ala41, Tyr52, His53, Phe54, Tyr58, Trp62, Ile77, Val78, Leu82, Phe101, Trp102, Ala105, Ile110, Ile125, Met127 | Tyr52 (π-π) Phe54 (π-H) Tyr58 (π-H) Tyr66 (π-H) | Arg42 (Cβ) |
| HLC | −7.6488 | −7.9912 | Ser38, Tyr58, Trp62, Tyr66, Asp75 | Phe39, Ala41, Tyr52, Tyr58, Trp62, Ile77, Val78, Phe101, Trp102, Ala105, Ile110, Ile125, Met127 | Phe54 (π-π) Trp90 (π-H) | Arg42 (Cβ) |
| Compound | Docking Energy (Kcal/mol) a | H-Bond Interactions | Hydrophobic Interactions | π-Interactions | Van Der Waal with Side Chain Carbons | |
|---|---|---|---|---|---|---|
| Preliminary (Rigid) | Induced-Fit (Flexible) | |||||
| Pindolol | −7.2291 | −8.0192 | Tyr80 *, Asp97 *, Ser155 | Leu57, Leu72, Trp84, Leu85, Ala94, Ile99, Leu100, Phe115, Phe126, Ala130, Met135, Ile153 | Tyr80 (π-π) Tyr88 (π-π) Trp111 (π-H) | - |
| Atenolol | −8.2351 | −8.9128 | Tyr80, Met89, Asp97 *, Ser155, Met253 | Leu57, Leu72, Val75, Trp84, Leu85, Tyr88, Met89, Ala94, Ile99, Leu100, Phe115, Phe126, Ala130, Met135, Ile153, Val250, Met253 | Tyr80 (π-π) Trp111 (π-H) | - |
| Esmolol | −7.4849 | −8.2830 | Tyr80, Asp97 *, Ser155 | Leu57, Leu72, Val75, Tyr80, Trp84, Leu85, Met89, Ala94, Ile99, Leu100, Phe115, Phe126, Ala130, Met135, Ile153, Val250, Met253 | Tyr88 (π-π) Trp111 (π-H) | - |
| Betaxolol | −7.3777 | −8.1034 | Tyr80, Asp97 * | Leu57, Leu72, Val75, Tyr80, Trp84, Leu85, Met89, Ala94, Ile99, Leu100, Phe115, Phe126, Ala130, Met135, Ile153, Val250, Met253 | Tyr88 (π-π) Trp111 (π-H) | - |
| Bisoprolol | −7.8018 | −8.8064 | Tyr80, Asp97 *, Ser155 | Leu57, Leu72, Val75, Tyr80, Trp84, Leu85, Met89, Ala94, Ile99, Leu100, Phe115, Phe126, Ala130, Met135, Ile153, Val250, Met253 | Tyr88 (π-π) Trp111 (π-H) | - |
| Metoprolol | −7.7426 | −8.7912 | Tyr80, Asp97 *, Ser155 | Leu57, Leu72, Val75, Tyr80, Trp84, Leu85, Met89, Ala94, Ile99, Leu100, Phe115, Phe126, Ala130, Met135, Ile153, Val250, Met253 | Tyr88 (π-π) Trp111 (π-H) | - |
| Acebutolol | −8.4732 | −9.0849 | Tyr80, Trp84, Tyr88, Asp97 * | Leu57, Ala59, Leu72, Val75, Trp84, Leu85, Met89, Ala94, Ile99, Leu100, Phe115, Phe126, Ala130, Met135, Ile153, Val250, Met253 | Tyr80 (π-π) Tyr88 (π-π) Trp111 (π-H) | - |
| Labetalol | −7.8677 | −8.8048 | Tyr80, Trp84, Asp97 *, Met135 | Leu57, Ala59, Leu72, Val75, Tyr80, Trp84, Leu85, Met89, Ala94, Pro98, Ile99, Leu100, Phe115, Phe126, Ala130, Met135, Ile153, Val250, Met253 | Tyr88 (π-H) Trp111 (π-π) | - |
| HLC | −7.2051 | −8.08374 | Tyr80, Trp84 *, Asp97 | Leu57, Leu72, Val75, Trp84, Leu85, Met89, Ala94, Ile99, Leu100, Phe115, Phe126, Ala130, Met135, Ile153, Val250, Met253 | Tyr80 (π-H) Tyr88 (π-π) Trp111 (π-H) | - |
| Energy (kJ/mol ± SD) | Ligand–Protein Complex | ||||||
|---|---|---|---|---|---|---|---|
| HLC | Comp.1 | Comp.2 | Comp.8 | Comp.10 | Comp.11 | Comp.14 | |
| ΔGvan der Waals | −122.79 ± 14.13 | −140.41 ± 3.50 | −145.10 ± 10.69 | −141.05 ± 25.17 | −154.95 ± 28.56 | −178.84 ± 11.78 | −157.27 ± 15.93 |
| ΔGElectrostatic | −46.75 ± 2.55 | −39.64 ± 2.46 | −47.33 ± 2.69 | −42.92 ± 3.70 | −52.74 ± 8.07 | −47.20 ± 3.46 | −36.65 ± 9.51 |
| ΔGSolvation; Polar | 120.42 ± 1.28 | 112.99 ± 5.10 | 135.90 ± 12.55 | 132.66 ± 16.36 | 155.59 ± 9.55 | 156.12 ± 5.68 | 122.03 ± 20.69 |
| ΔGSolvation; non-polar; SASA | −18.75 ± 0.04 | −17.26 ± 0.19 | −16.34 ± 0.39 | −18.21 ± 0.37 | −18.21 ± 0.04 | −19.79 ± 0.93 | −17.17 ± 0.05 |
| ΔGTotal binding | −67.87 ± 10.34 | −84.32 ± 0.66 | −72.87 ± 4.16 | −69.52 ± 12.88 | −70.31 ± 27.13 | −89.71 ± 3.56 | −89.05 ± 4.71 |
| Energy (kJ/mol ± SD) | Ligand–Protein Complex | |||||||
|---|---|---|---|---|---|---|---|---|
| HLC | Comp.10 | Comp.11 | Comp.12 | Comp.13 | Comp.14 | Comp.17 | Comp.21 | |
| ΔGvan der Waals | −156.93 ± 22.35 | −186.11 ± 6.21 | −215.32 ± 5.77 | −202.48 ± 6.06 | −188.95 ± 0.89 | −219.53 ± 1.04 | −194.57 ± 5.13 | −218.16 ± 8.55 |
| ΔGElectrostatic | −67.65 ± 16.73 | −109.52 ± 7.45 | −71.01 ± 9.23 | −56.97 ± 1.48 | −48.43 ± 0.51 | −58.31 ± 6.72 | −54.54 ± 0.91 | −46.50 ± 6.78 |
| ΔGSolvation; Polar | 162.60 ± 5.19 | 195.45 ± 7.07 | 188.52 ± 1.51 | 170.69 ± 7.56 | 160.40 ± 6.81 | 185.98 ± 8.38 | 185.73 ± 8.25 | 192.86 ± 7.42 |
| ΔGSolvation; non-polar; SASA | −17.52 ± 0.71 | −17.45 ± 0.29 | −20.66 ± 0.42 | −20.28 ± 0.20 | −19.34 ± 0.11 | −22.53 ± 0.18 | −18.87 ± 0.64 | −21.20 ± 0.13 |
| ΔGTotal binding | −79.50 ± 1.14 | −117.63 ± 6.12 | −118.47 ± 13.07 | −109.04 ± 2.77 | −96.33 ± 7.31 | −114.40 ± 13.87 | −82.25 ± 1.57 | −93.00 ± 9.32 |
| Energy (kJ/mol ± SD) | Ligand–Protein Complex | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| HLC | Comp.2 | Comp.10 | Comp.11 | Comp.12 | Comp.13 | Comp.14 | Comp.17 | Comp.21 | |
| ΔGvan der Waals | −120.62 ± 8.69 | −173.26 ± 2.38 | −170.77 ± 4.23 | −181.41 ± 4.10 | −147.37 ± 23.03 | −171.40 ± 29.61 | −182.07 ± 9.12 | −184.75 ± 15.74 | −168.13 ± 6.62 |
| ΔGElectrostatic | −52.81 ± 15.96 | −31.78 ± 8.88 | −99.14 ± 7.53 | −60.14 ± 4.73 | −31.87 ± 9.69 | −35.48 ± 12.10 | −39.67 ± 1.36 | −39.57 ± 22.05 | −39.02 ± 16.73 |
| ΔGSolvation; Polar | 120.50 ± 15.88 | 149.10 ± 1.64 | 176.95 ± 9.60 | 157.83 ± 4.39 | 126.96 ± 13.52 | 142.19 ± 47.71 | 144.95 ± 2.54 | 144.14 ± 34.16 | 150.94 ± 2.65 |
| ΔGSolvation; non-polar; SASA | −18.00 ± 0.27 | −17.06 ± 0.21 | −18.11 ± 0.27 | −20.92 ± 1.04 | −18.83 ± 0.70 | −20.96 ± 1.82 | −21.72 ± 0.24 | −21.28 ± 0.77 | −20.30 ± 0.63 |
| ΔGTotal binding | −70.93 ± 9.05 | −73.00 ± 9.41 | −111.07 ± 6.04 | −104.64 ± 4.79 | −71.10 ± 19.89 | −85.66 ± 4.18 | −98.51 ± 13.26 | −101.46 ± 4.40 | −76.51 ± 21.33 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almalki, A.J.; Ibrahim, T.S.; Elhady, S.S.; Hegazy, W.A.H.; Darwish, K.M. Computational and Biological Evaluation of β-Adrenoreceptor Blockers as Promising Bacterial Anti-Virulence Agents. Pharmaceuticals 2022, 15, 110. https://doi.org/10.3390/ph15020110
Almalki AJ, Ibrahim TS, Elhady SS, Hegazy WAH, Darwish KM. Computational and Biological Evaluation of β-Adrenoreceptor Blockers as Promising Bacterial Anti-Virulence Agents. Pharmaceuticals. 2022; 15(2):110. https://doi.org/10.3390/ph15020110
Chicago/Turabian StyleAlmalki, Ahmad J., Tarek S. Ibrahim, Sameh S. Elhady, Wael A. H. Hegazy, and Khaled M. Darwish. 2022. "Computational and Biological Evaluation of β-Adrenoreceptor Blockers as Promising Bacterial Anti-Virulence Agents" Pharmaceuticals 15, no. 2: 110. https://doi.org/10.3390/ph15020110
APA StyleAlmalki, A. J., Ibrahim, T. S., Elhady, S. S., Hegazy, W. A. H., & Darwish, K. M. (2022). Computational and Biological Evaluation of β-Adrenoreceptor Blockers as Promising Bacterial Anti-Virulence Agents. Pharmaceuticals, 15(2), 110. https://doi.org/10.3390/ph15020110