2. Acridines
The class of acridine derivatives (
Figure 2) constitutes an interesting group of nitrogen-containing tricyclic compounds that caught the scientific community’s attention, mainly due to its wide range of pharmaceutical properties [
18,
19]. This type of compound’s unique physical and chemical properties allows several derivatives to have been associated with numerous bioactivities, such as anti-inflammatory, anti-cancer, antimicrobial, antitubercular, antiparasitic, antimalarial, antiviral, and fungicidal activities. Furthermore, several methodologies have been developed to obtain a more extensive range of acridine-based compounds throughout the years. These methodologies include the Bernthsen synthesis, the first synthetic method for the obtention of an acridine, the Ullmann reaction, and the Friedländer synthesis [
20,
21]. Acridine derivatives can also be obtained by the manipulation of acylated diphenylamines, reduction of acridones, or the functionalization of 9-chloroacridines [
20].
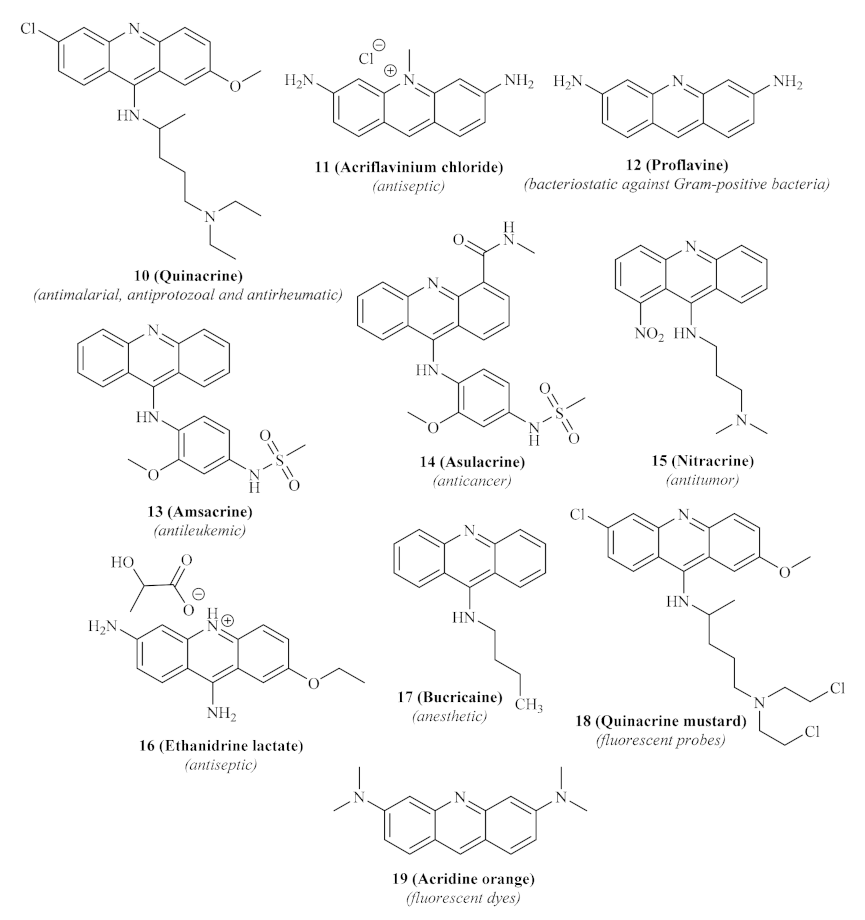
Until today, numerous acridine derivatives have been used as pharmaceutical agents, such as quinacrine
10, acriflavinium chloride
11, proflavine
12, amsacrine
13, asulacrine
14, nitracrine
15, ethacridine lactate
16, and bucricaine
17 (
Figure 3). Additionally, this family of compounds has been widely used as fluorescent dyes, such as acridine orange
19, as well as fluorescent probes, such as quinacrine mustard
18 [
20].
Despite using acridine derivatives for pharmaceutical purposes, their potential use as antileishmanial agents remains unclear. However, next, we intend to demonstrate that there are already some reports of acridine derivatives with interesting antileishmanial effects. Thus, this might serve as proof that the acridine scaffold may be used for the development of novel acridine-based antileishmanial drugs and incentivize more studies involving this scaffold.
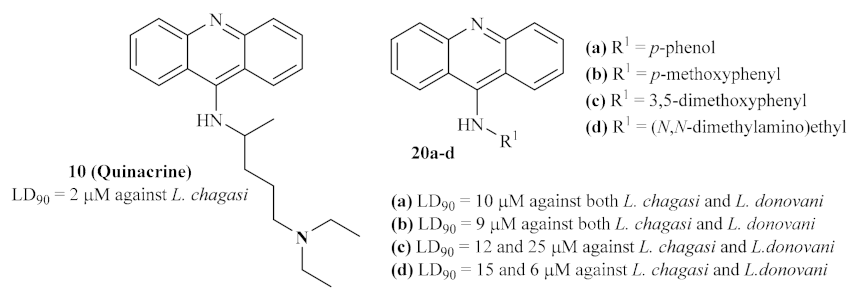
In the last decade of the 20th century, the acridine scaffold emerged as an exciting structural core for developing novel antileishmanial agents. Focusing their efforts on the leishmanial DNA topoisomerase II (TPII) as a promising molecular target, Werbovetz et al. assessed the efficiency of quinacrine
10 and several other 9-aminoacridines
20 (
Figure 4) against DNA TPII of two
Leishmania species,
L. donovani and
L. chagasi [
22]. DNA topoisomerases are a widely distributed family of enzymes crucial for many essential biological processes, including DNA replication, transcription, recombination, and repair. Throughout the years, these enzymes have been indicated as chemotherapeutic targets for developing antibacterial and antiparasitic treatments, suggesting that the inhibition of these enzymes might consequently promote antileishmanial properties [
23].
Their results demonstrated that several 9-substituted acridine derivatives, namely p-phenol 20a, p-methoxyphenyl 20b, 3,5-dimethoxyphenyl 20c, (N,N-dimethylamino)ethyl 9-aminoacridine 20d, and also the well-known antileishmanial agent quinacrine 10, presented high levels of antileishmanial activity against the promastigote stage of the parasite, with lethal dose capable of killing 90% of parasite (LD90) values below 25 µM. Interestingly, except for p-phenol 9-aminoacridine 20a, all of these active antileishmanial acridine derivatives also showed weak to no mammalian TPII inhibition, suggesting a selective mechanism of action solely against the enzyme of the Leishmania promastigotes. However, in our opinion, the authors must corroborate this hypothesis with an assay that shows these compounds’ effect on leishmanial TPII.
Following the promising results of the previously mentioned 9-aminated acridines active against leishmanial DNA topoisomerase II, a series of 9-anilinoacridines was screened against macrophage-infected
L. major [
24]. By comparing the effect of some of these compounds on promastigotes and intracellular parasites, it becomes clear that derivatives active against the free form of the parasite are also able to act against the parasites inside the macrophages, being even more active against the intracellular microorganisms. Their results demonstrated that several 9-anilinoacridines
21 (
Figure 5) show high levels of antileishmanial activity, killing more than 80% of intracellular parasites at 1 µM while maintaining moderate to low levels of mammalian toxicity. From this series of 9-anilinoacridines, the 4′-NHhexyl derivative
21.b has been identified as the most promising antileishmanial compound, being one of the most active compounds while having the lowest level of toxicity. Interestingly, some structural features can be associated with both high levels of antileishmanial activity and low levels of toxicity, such as its lipophilicity or the presence of specific substituents, such as 3′-methoxy groups, with more promising values of toxicity.
In the final years of the 20th century, Gamage et al. continued their previous work with 9-anilinoacridines by synthesizing and evaluating against intracellular
L. major amastigotes several other 4′-substituted 9-anilinoacridines
22 [
25]. This study’s results corroborated some of the evidence already stated by this research group, such as the importance of a lipophilic electron-donating group at 4′-position of this type of derivatives for higher selectivity indexes (SI). Contrarily, some other conclusions of the previous work have now been clarified, for example, the presence of a 3′-substitution does not always promote an increase in SI by decreasing the derivative’s toxicity levels, or the introduction of a 3,6-diNCH
3 pattern being crucial to the antileishmanial activity of the compounds (
Figure 5).
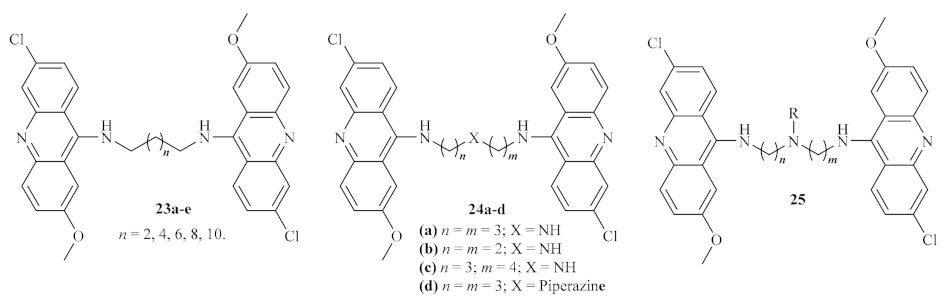
At the beginning of the century, a research group synthesized a huge series of forty bis (9-amino-6-chloro-2-methoxyacridine) derivatives using several linkers between the two acridine fragments, such as alkanediamines, polyamines, or side chain-containing polyamines (
Figure 6) [
26]. These series of compounds were further evaluated for their activity against some different protozoan parasites, with particular interest in the amastigote stage
of L. infantum. The variety of linkers evaluated allows a deeper understanding of the optimal type and length to be introduced between the two acridine fragments in order to enhance their antileishmanial properties. Curiously, except for the piperazine-containing derivative, the first two series of compounds
23 and
24 revealed high levels of cytotoxicity, which might be associated with strong interactions with DNA, particularly characteristic of bis-intercalation [
27]. From the third series of compounds
25, only five demonstrated the absence of cytotoxicity. These compounds are characterized by the presence of a polar group, such as an amine, alcohol, or carboxylic acid, in the side chain attached to the linker. Even though some of the compounds evaluated displayed high antileishmanial activity (half maximal inhibitory concentration (IC
50) as low as 1.56 µM), the majority are not suitable as potential antileishmanial agents due to their high cytotoxicity. In turn, those with low or no levels of cytotoxicity were revealed to be only slightly effective against
L. infantum, without considerable activity.
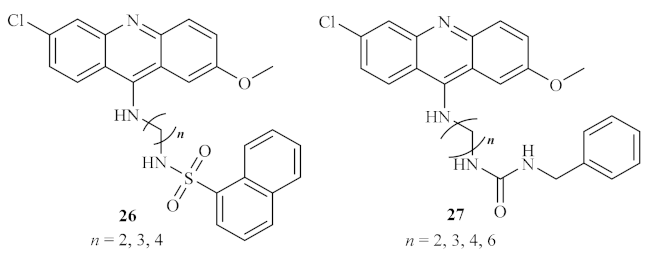
A year later, based on the proven antiprotozoal properties of quinacrine
10 (
Figure 3), some sulfonamide
26 and urea analogs
27 of quinacrine were synthesized and evaluated against a few types of protozoan, particularly against
L. donovani amastigotes [
28]. These derivatives are characterized by the presence of methylene groups with terminal sulfonamide and urea moieties between the acridine fragment and another hydrophobic scaffold (
Figure 7). Therefore, the introduction of these groups is intended to improve the solubility of this type of compound. In addition to the most common in vitro assays against the parasite amastigote form, these compounds were also evaluated for their inhibitory activity against the trypanothione reductase enzyme (TR). TR is the enzyme responsible for maintaining trypanothione in its reduced state and, consequently, guaranteeing the proper function of the trypanothione pathway, a metabolic pathway essential for parasite growth and survival by regulation of oxidative stress [
29]. Thus, this enzyme has been widely depicted as a potential molecular target for developing novel antiprotozoal agents. This research demonstrated that these derivatives could considerably inhibit the TR enzyme, with IC
50 levels ranging from 3.3 µM to 19.3 µM. Furthermore, the results also show that these compounds display significant effects against the parasite by suppressing the survival and growth of the parasite, with median effective dose (ED
50) values below 1.0 µM. From a metabolic perspective, considering that the compounds are more active against the parasite itself than against a specific enzyme (in this case, the TR enzyme), it is possible to suggest that these compounds might act against more than one metabolic pathway of the parasite. However, these compounds present high toxicity levels, which impairs their potential use to develop novel antileishmanial agents.
In 2002, another French research group performed a thorough study of the antileishmanial potential of three acridine thioethers
28 (
Figure 8) against
L. donovani, both in vitro and in vivo [
30]. The results demonstrated that the first two derivatives (
28a and
28b) are more effective against the promastigote forms of
L. donovani than the third acridine-based compound evaluated
28c, being able to considerably inhibit the parasite’s growth at the 0.39 µM (1 µg/mL), 0.27 µM (1 µg/mL), and 2.9 µM (0.1 µg/mL), respectively. In addition, considering the effect of these compounds on the in vitro growth of the amastigote forms of
L. donovani, the derivative
28b emerged as the most active derivative at the lowest concentration, leading to an inhibition of 43% at 0.27 µM (0.1 µg/mL). In contrast, the remaining derivatives appear to be virtually inactive against the amastigote form of this parasite. In turn, the in vivo studies demonstrated that these compounds could also decrease the percentage of parasitation in infected rats, with particular emphasis on derivative
28b. In conclusion, these authors, in association with the results of previous studies developed on
Trypanosoma cruzi [
31], suggest that the antiparasitic activity of these acridine derivatives is closely related to the interaction between the DNA of the parasite and the compounds, particularly when the sulfur atoms are substituted with an alkylaminoalkyl group. Additionally, from a morphological perspective, these compounds can also promote a separation of the nuclear membrane from the chromatin, leading to the death of the parasite by apoptosis.
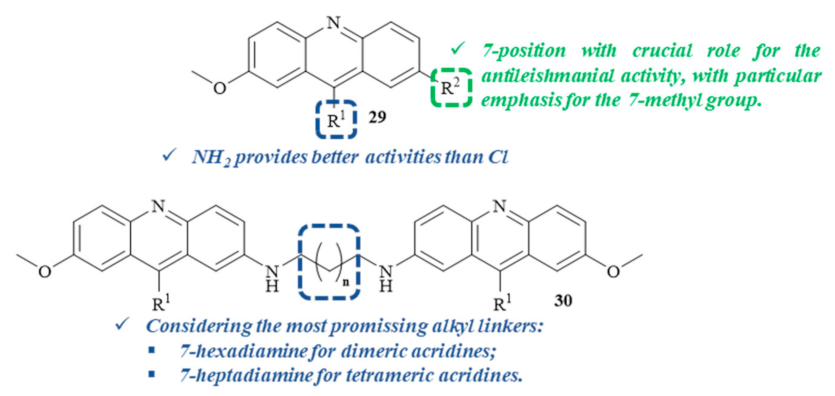
A year later, two series of 7-substituted 9-(chloro or amino)-2-methoxyacridines
29, as well as some corresponding unsubstituted dimers
30 and tetramers (
Figure 9), were evaluated for their in vitro antiproliferative activity against both forms of
L. infantum, with a focus on understanding their potential mechanisms of action [
32]. Considering the toxicity levels of these series of compounds against human monocytes, it is important to mention that, with a few exceptions, most of the compounds from both series displayed low toxicity levels. In relation to their antileishmanial activity, the results demonstrated that the 9-aminoacridines are more active than the corresponding 9-chloroacridines, with the first group being more capable of affecting parasite growth and survival. Furthermore, the 7-substituent also plays a crucial role in the inhibitory activity of these compounds, with special emphasis on the introduction of a 7-methyl group, making 9-amino-2-methoxy-7-methylacridine and 9-chloro-2-methoxy-7-methylacridine the most promising antileishmanial compounds of the single acridine series. Thus, these monomers were used to obtain the corresponding dimeric and tetrameric derivatives, using several alkanediamines as linkers. In the case of the dimeric derivatives, the compound containing a 7-hexanediamine linker demonstrated the most promising selective antileishmanial activity. In contrast, the 7-heptanediamine linker was revealed to be the optimal linker for the antileishmanial activity of tetrameric derivatives. However, dimeric and tetrameric complexes presented lower activities than their corresponding monomers.
Regarding these compounds’ potential mechanism of action, this research work suggests that the antileishmanial properties demonstrated by these acridine derivatives might be closely related to a multitarget approach. This multitarget mechanism is primarily associated with inhibiting the DNA metabolism through DNA intercalation or even the inhibition of the processes involving DNA–protein interactions and, additionally, with effects on many other metabolic pathways, such as protein and lipid metabolisms [
32].
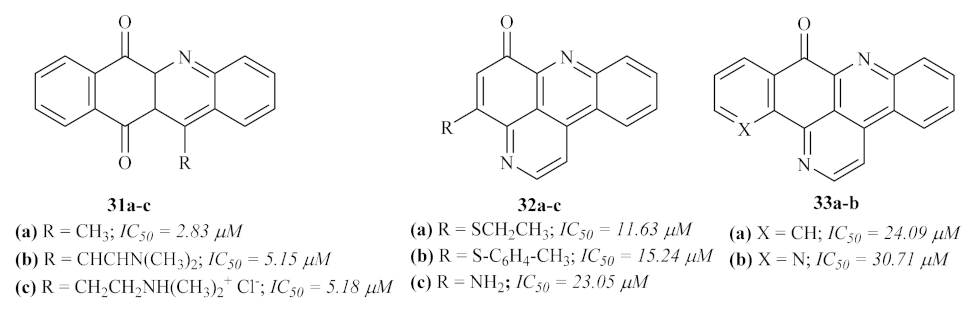
In the same year, a series of marine pyridoacridone alkaloids were evaluated for their in vitro antileishmanial activity against
L. donovani promastigotes and amastigotes, including ascididemin and other related synthetic derivatives [
33]. The results demonstrated that some of the evaluated derivatives present considerable antileishmanial activity, even if none of it reaches the values of the reference drug amphotericin B. Considering the activity against the promastigote stage, a series of eight compounds (
31,
32, and
33,
Figure 10) demonstrated significant levels of activity, with the remaining derivatives presenting moderate to no activity. However, due to its high cytotoxicity values against mammalian cells, none of these derivatives were further evaluated for their antileishmanial activity against the amastigote stage of the parasite.
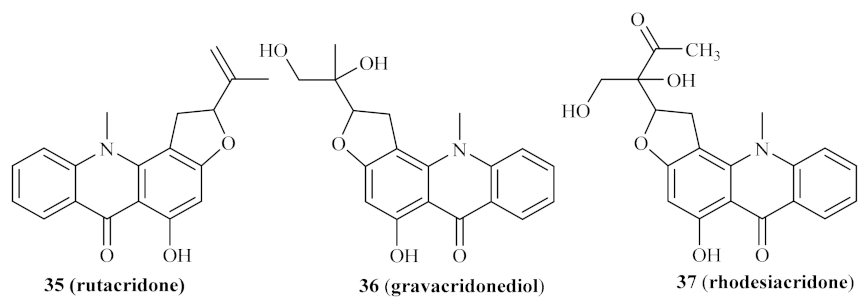
In 2004, Ahua et al. isolated several compounds from
Thamnosma rhodesica (Bak. f.) Mendonça, a plant commonly used in Zimbabwe as an ant and flea repellent or for relieving chest pain, from which we must highlight four acridones [
34]. These compounds were then evaluated against both promastigote and amastigote forms of
L. major, with the results demonstrating that some of these derivatives present promising antileishmanial properties. From the four acridones evaluated, rutacridone
35, gravacridonediol
36, and rhodesiacridone
37 (
Figure 11) demonstrated similar efficiencies against the promastigote form of the parasite, at 10 µM, by inhibiting its survival by 63.1%, 46.0%, and 69.3%, respectively. At the same concentration, none of these derivatives demonstrated considerable toxicity to mammalian macrophages, allowing its evaluation against the intracellular amastigotes. Interestingly, the effect of gravacridonediol
36 and rhodesiacridone
37 turned out to be even more pronounced against the amastigote form of
L. major by inhibiting this form by 90.5% and 93.8%, respectively.
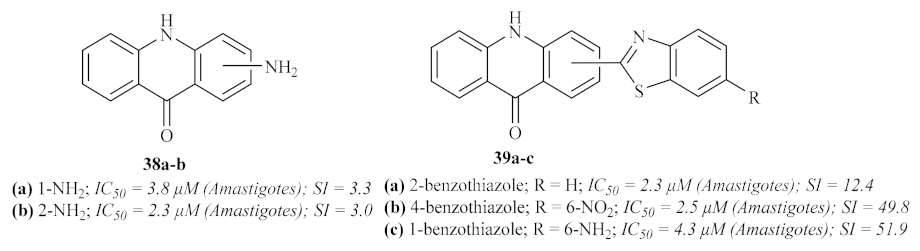
In the same year, the Delmas group synthesized a series of acridine–benzothiazole hybrids, namely acridin-9-(
10H)-ones substituted with amino or (1,3-benzothiazol-2-yl)-amino groups, and evaluated them for their in vitro antileishmanial properties against both stages of
L. infantum (
Figure 12) [
35]. This research work demonstrated that the antileishmanial activity of this type of compound is highly dependent on the nature and position of the substituted benzothiazole fragment. Furthermore, the addition of a benzothiazole group is also responsible for decreasing the compounds’ cytotoxicity, which contributes to more suitable SI. Considering the type of benzothiazole introduced in the acridone scaffold, this series of compounds may be further divided into four smaller groups: the simple amino acridones and the unsubstituted benzothiazole, nitrobenzothiazole, and aminobenzothiazole.
The first group of compounds 38, those without any benzothiazole group, demonstrated low to no antileishmanial activity against the promastigote stage of the parasite (IC50 values from 21 µM to >200 µM). Against the amastigotes, these compounds were considerably more effective, with the presence of a 1-NH2 38a or a 2-NH2 38b being the optimal positions for the antileishmanial activity of this group of compounds. However, these compounds also demonstrated high toxicity levels, resulting in weak SI values of 3.3 and 3.0, respectively.
As mentioned above, the introduction of the benzothiazole promoted better SI levels, regardless of its position, and enhanced efficiency against both promastigotes and amastigotes, leading to more promising antileishmanial agents. Considering the unsubstituted benzothiazole, except for the 4-position that displayed only moderate activity, the introduction of this group led to high levels of antileishmanial activity against both forms of the parasite. Amongst these derivatives, 2-(benzothiazol-2-ylamino)-10H-acridin-9-one 39a emerged as the most promising antileishmanial agent, converging high activity and TI levels. In turn, introducing a 6-nitro group on the benzothiazole ring tends to decrease this class of compound’s efficiency against promastigotes, with IC50 values above 200 µM. However, 4-(6-nitrobenzothiazol-2-ylamino)-10H-acridin-9-one 39b displayed high antileishmanial activity against Leishmania amastigotes associated with an elevated SI. Finally, with only one exception, the presence of a 6-amino group on the benzothiazole led to a decrease in the antileishmanial activity against promastigotes. The exception contemplates 1-(6-aminobenzothiazol-2-ylamino)-10H-acridin-9-one 39c, a compound highly capable of inhibiting the amastigote form of the parasite and rendering an elevated SI value.
One year later, the same research group continued focusing their efforts on evaluating acridine’s antileishmanial potential by synthesizing and evaluating the antileishmanial activities of both 4-mono and 4,5-disubstituted acridines (
Figure 13) [
36]. The results of this study demonstrated that the mono-substituted acridines present stronger antileishmanial activities than their disubstituted homologs, with the latter group being weakly active against the promastigote form of the parasite. Furthermore, it became clear that compounds containing a nitrogen bond are also more active than those with an oxygen bond. Notably, in the case of the mono-substituted acridines, only one compound demonstrated considerable inhibitory activity against the promastigote form of the parasite, consisting of the 4-ethylaminoacridine
40a (IC
50 = 1.7 µM; TI = 2.9), while two displayed promising selective anti-amastigote activities, characterized by a 4-methoxybenzoyl
40b (IC
50 = 9.4 µM; TI = 42.8) and 4-(
N-dimethylamino)benzoyl
40c (IC
50 = 4.7 µM; TI = 30.4) groups, respectively. Regarding the 4,5-disubstituted acridines, some of these derivatives demonstrated considerable antileishmanial activity against both forms of the parasite with emphasis on 4,5-bis (hydroxymethyl)acridine
41 (IC
50 = 0.6 µM; TI > 200). Interestingly, several disubstituted acridines were characterized by an amastigote-specific activity, which might suggest that these compounds may act against the
Leishmania parasite through a different mechanism of action, unrelated to DNA intercalation. This assumption is supported by the fact that these acridine derivatives are capable of preventing macrophage infection through the reduction of parasite internalization, in addition to their selective antileishmanial activity against amastigotes.
In 2007, Di Giorgio et al. synthesized two more series of acridine derivatives, monofunctional 6-substituted acridine and bifunctional 3,6-disubstituted acridine derivatives, and evaluated their antileishmanial activities [
37]. The results of this research work demonstrated that some derivatives from this particular type of acridines present promising antileishmanial activities against both forms of the parasite. However, due to the high cytotoxicity shown by many of the derivatives, it becomes crucial to emphasize
N-[6-(acetylamino)acridin-3-yl]acetamide
42a and
N-[6-(benzoylamino)acridin-3-yl]benzamide
42b, which display the highest levels of selectivity (
Figure 14).
This study also allowed us to identify some molecular features that promote the specificity of the compounds for
Leishmania parasites instead of human cells, with variations depending on the parasite’s form. Against the promastigote form, the optimal substituent pattern consists of two benzoylamino groups in disubstituted derivatives. The introduction of 4′-chloro or 4′-fluoro atoms leads to a significant increase in antileishmanial activity. In turn, against the amastigote form of the parasite, both 3,6-acetylamino and 3,6-benzoylamino groups are the best features for this activity, suggesting the necessity for a symmetric conformation for a specific anti-amastigote activity. From a mechanism of action perspective, it became clear that, despite the proven ability of acridines to bind DNA [
38,
39,
40], their antileishmanial activity is not related to DNA binding. Instead, this might be associated with interferences on
Leishmania–macrophage interactions or even the interaction of acridine derivatives with several cellular structures.
Based on the current use of benznidazole for the treatment of Chagas disease, Papadopoulou et al. screened a series of forty-two 3-nitro-1
H-1,2,4-triazole-based and 2-nitroimidazole-based aromatic and aliphatic amines for their antiparasitic properties, with emphasis on 4 acridine derivatives (
Figure 15) [
41]. Even though these derivatives demonstrated considerable levels of antileishmanial activity against axenic amastigotes of
L. donovani, with IC
50 values from 9.09 to 35.37 µM, their cytotoxicity levels were even higher, with IC
50 values from 0.308 to 20.52 µM.
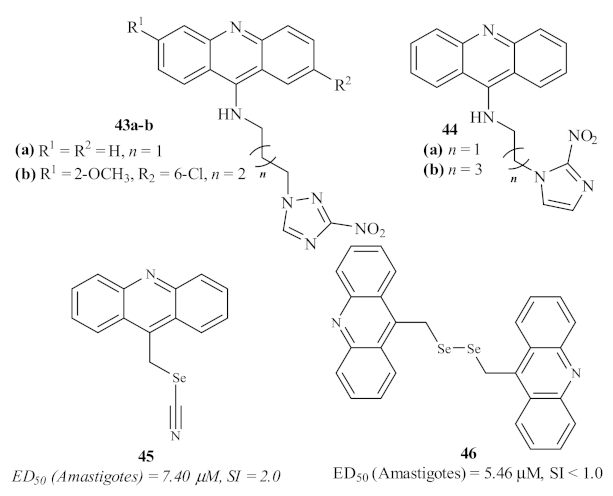
In 2016, Baquedano et al. evaluated a series of twenty-three heteroaryl selenocyanates and diselenides for their antileishmanial activity against the amastigote form of
L. infantum, including two acridines (
Figure 15) [
42]. Through the incorporation of these two functionalities bearing selenium atoms, this group intended to perform a rational design of potent and selective antileishmanial agents. These acridines, acridin-9-ylmethyl selenocyanate
45 and 9,9-(diselenodiyldimethanediyl)diacridine
46, demonstrated good levels of antileishmanial activity, with the diselenide-containing derivative being more active than the one containing the selenocyanate moiety. However, due to their high levels of cytotoxicity, these acridine derivatives were revealed not to be suitable for further development of novel antileishmanial agents.
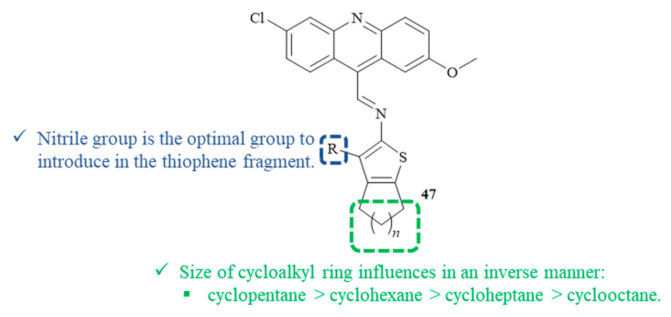
In 2018, a Brazilian research group synthesized and evaluated eight compounds containing both 2-aminocycloalkyl[
b]thiophene and acridine fragments for their antileishmanial activity against the promastigote form of
L. amazonensis (
Figure 16) [
43]. Concerning the cytotoxicity evaluation, it is essential to mention that none of the thiophene–acridine hybrids showed any cytotoxicity against human erythrocytes up to 1000 µM. Regarding their antileishmanial activity, the hybrids containing the 6,9-dichloro-2-methoxyacridine and the 2-aminothiophene fragments
47 demonstrated good antipromastigote activity with IC
50 levels from 9.60 µM to 69.11 µM. Structurally, the size of the cycloalkyl ring of the thiophene fragment clearly influences the compound’s antileishmanial activity through an inverse manner, following the order cyclopentane > cyclohexane > cycloheptane > cyclooctane. Furthermore, the substituent group present in the thiophene fragment also influences the antileishmanial activity, emphasizing the introduction of a nitrile radical. Based on these shreds of evidence, the most effective antileishmanial agent derived from this research work consist of the hybrid containing the nitrile group and a cyclopentane simultaneously, with an IC
50 of 9.60 µM. Through some molecular docking studies, this research group suggests that the antileishmanial activity might be related to the potential inhibition of the enzyme pyruvate kinase, which, in association with the ability of the acridine fragment to intercalate with DNA, indicates a dual mechanism of action.
In the same year, following the previous works from Giorgi et al. [
37], Mahajan et al. designed, synthesized, and evaluated a series of thirty-three 7-arylbenzo[
c]acridine-5,6-diones (
Figure 17) for their potential against both promastigote and amastigote form of
L. donovani [
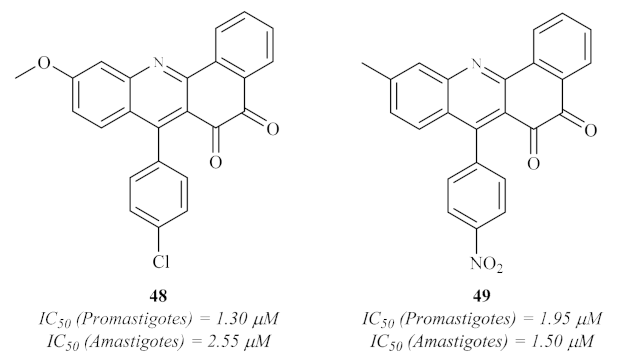
44]. The results demonstrated that several derivatives present significant antileishmanial activity against both promastigotes and axenic amastigotes. Interesting to mention is the fact that, against promastigotes, three compounds demonstrated activity levels similar to pentamidine (IC
50 = 1.89 µM); six compounds showed considerable antileishmanial properties (IC
50 from 2.00 µM to 2.56 µM); and ten were moderately active (IC
50 from 4.03 µM to 10.44 µM). In turn, against amastigotes, four derivatives were clearly more active than pentamidine (IC
50 = 9.57 µM), with IC
50 values of 1.50–1.95 µM, and fifteen others were demonstrating significant activities, with IC
50 values below 11.31 µM. In the case of the antipromastigote activity, the optimal substitution pattern consists of combining a 9-(4-chlorophenyl) group and a 2-methoxy group
48, while the optimal anti-amastigote activity is composed of a 9-(4-nitrophenyl) group and a 2-methyl group
49.
In 2020, based on previous evidence of the antiparasitic properties of artemisinin analogs [
45], Aucamp et al. synthesized a series of acridines and artemisinin–acridine hybrids (
Figure 18). They evaluated them for their in vitro antileishmanial activity against the promastigote form of three different strains of
Leishmania, two of
L. donovani and one of
L. major [
46]. The results of this research demonstrated that all the acridine derivatives presented significant levels of activity against all the
Leishmania strains, with IC
50 values in the micromolar range. However, with one exception, these derivatives also showed high levels of cytotoxicity, leading to undesirable SI (<9), which hinders the possibility of these compounds being used to develop novel antileishmanial agents. The exception consisted of a particular artemisinin–acridine hybrid
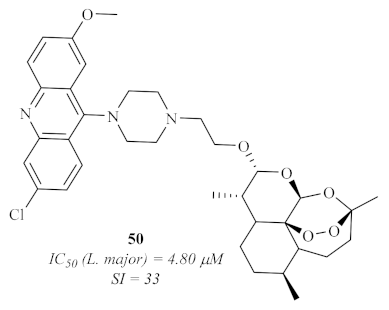
50 (
Figure 18) that demonstrated a promising SI (33), with low levels of cytotoxicity (>100 µM) and a considerable level of antileishmanial activity, especially against
L. major (4.80 µM). Important to mention is the fact that the hybridization of the acridine derivatives with artemisinin promotes an increase in the antileishmanial activity while decreasing the cytotoxicity levels of the derivatives, suggesting that acridine hybrids with other pharmacophores might be a solution to overcome acridine’s toxicity.
3. Xanthenes
The class of xanthene derivatives
51 constitutes an important class of oxygen-containing tricyclic compounds, characterized by a dibenzo[
b,e]pyran core, widely associated with a broad range of pharmaceutical and biological properties, such as antimicrobial, antiproliferative, antiviral, antioxidant, and anti-inflammatory activities, amongst others (
Figure 19) [
47,
48].
Even though numerous reports have described these compounds as constituents of natural extracts throughout the past decades, the majority of the known xanthenes are synthesized. The synthesis of this type of compound might be performed through the cyclization of specific building blocks or by modifying a most commonly found class of xanthene derivatives, namely xanthones. This versatile structural fragment allows the introduction of virtually any substituent group in its core. Depending on the substituent group desired, different synthetic approaches might be performed to obtain the intended xanthene derivative. Sen et al. described one of the first syntheses of the xanthene nucleus in 1925, in which 3-hydroxyxanthene was obtained through the condensation of saligenin and resorcinol [
49]. After that, numerous synthetic methods for the obtention of xanthenes with distinct substitution patterns have been described in literature, including the functionalization of particular core positions to synthesize broad spectra of xanthenes and the application of greener procedures [
50,
51,
52]. Thus, this has allowed the use of xanthene in a huge range of biological activities, leading to the increasing importance of xanthenes in medicinal chemistry [
48]. Besides its biological properties, this type of compound has been widely used for a broad range of applications, such as sensitizers in PDT, dyes in the food industry, industrial materials, and chemical probes for the visualization of biomolecules [
52,
53,
54,
55].
By the end of the 20th century, through bibliographic research, Chibale et al. understood that several aromatic hydrophobic tricyclic moieties had already been reported as competitive trypanothione reductase (TR) inhibitors [
56]. Thus, based on this knowledge, the research group decided to synthesize and evaluate a novel series of 9,9-dimethylxanthene tricyclics (
52,
53, and
54) against
L. donovani through the assessment of their trypanothione reductase (TR) inhibitory potential (
Figure 20). Their results demonstrated that, considering the inhibition of TR, compounds containing longer carbon chains (two
53 or three methylene groups
54) between the tricyclic moiety and the secondary amino group show stronger inhibition of TR in comparison with those containing only one
52b or no methylene spacer
52a. Interestingly, the efficiency of these compounds against TR was not replicated for the in vitro studies against the amastigote stage of
L. donovani, with compound
52c being the most effective (ED
50 = 0.55 µM), suggesting that TR is not the target of its activity. This lack of correlation between TR inhibition and in vitro sensitivity of
L. donovani amastigotes may be explained by the fact that TR inhibition is not entirely responsible for the effect of these compounds on the amastigotes. In turn, cell penetration and the parasite’s metabolism might also play a crucial role in the efficiency of these compounds against amastigotes.
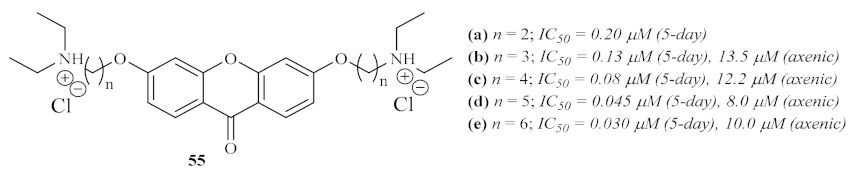
Three years later, in 2003, another research group focused their efforts on using a particular series of heme-binding xanthones, 6-bis-ω-diethylaminoalkoxyxanthones
55, against
L. mexicana (
Figure 21) [
57]. These compounds were selected since xanthones have already been described as potential antimalarial agents by both forming stable soluble complexes with heme and preventing their aggregation into insoluble hemozoin. Furthermore, it has been observed that these dibasic compounds accumulate in the parasite’s acidic vacuole and, considering the peculiar nutritional necessities of
Leishmania, it was expected that the parasites would be significantly sensitive to the effects of heme-binding xanthones. The results of this research work revealed that all the evaluated derivatives present strong effects against both intracellular amastigotes and axenic amastigotes of
L. mexicana, with IC
50 values in the nanomolar range. Regarding the 5-day assay, it was possible to verify the strong influence of chain length on the compounds’ antileishmanial activity, with the activity increasing with the number of methylene groups in the carbon chain. Another piece of evidence obtained through this work contemplates a strong correlation between the heme affinity of the compounds and their antileishmanial activity. Interestingly, despite the same order of efficiency, these compounds were considerably less effective against the axenic amastigotes. Finally, considering all the evidence, this group proposes that these compounds undergo a cationization and accumulation process within the acidic phagolysosomes of the host, being further capable of effectively complexing with any source of heme and, consequently, starving the intracellular parasites.
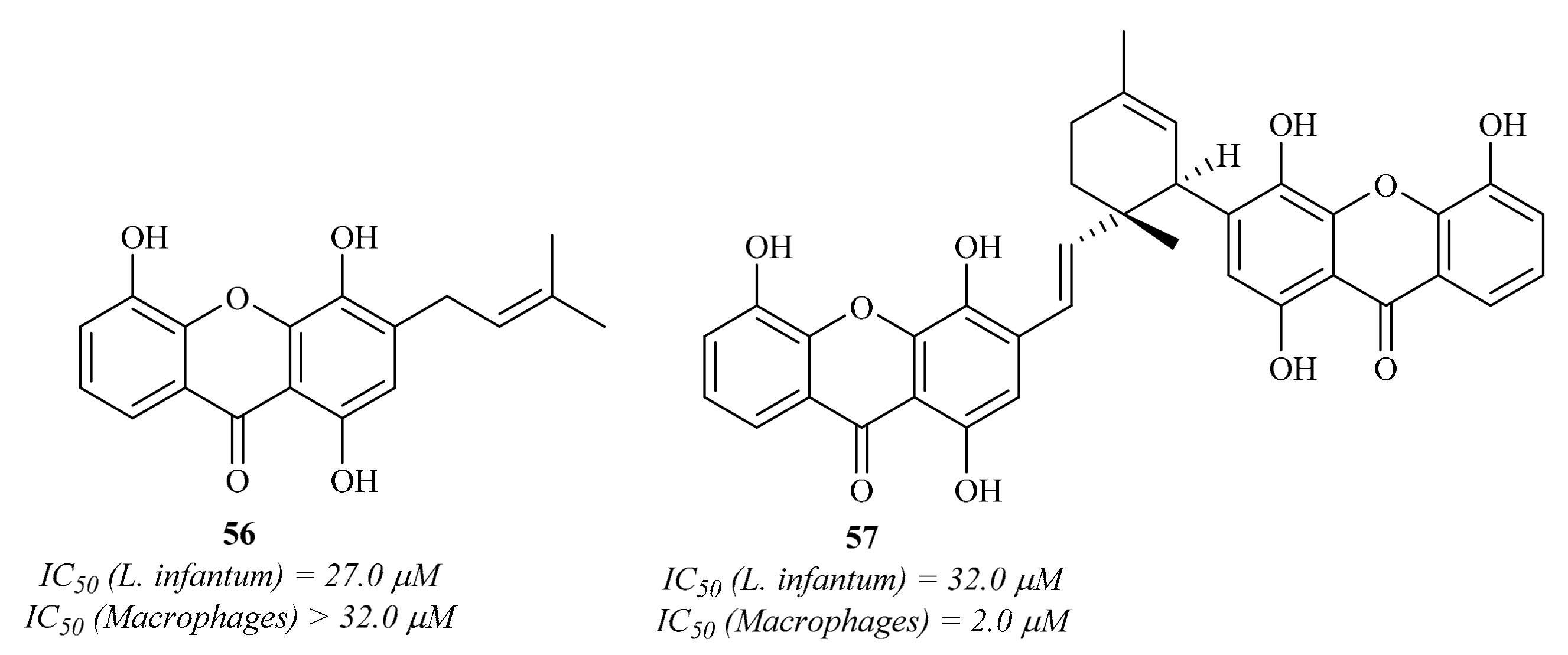
Two years later, Mbwambo et al. isolated from the root bark of
Garcinia livingstonei T. Anderson and evaluated a series of xanthones for their antiparasitic activities against the intracellular amastigotes of
L. infantum [
58]. The results of this work demonstrated that, from the five isolated xanthones, only two present considerable antileishmanial activity (
Figure 22), emphasizing 1,4,5-trihydroxy-3-(3-methylbut-2-enyl)-9
H-xanthen-9-one
56. However, these compounds also demonstrated high cytotoxicity values, with compound
57 being even more effective against macrophages than against the
Leishmania parasites.
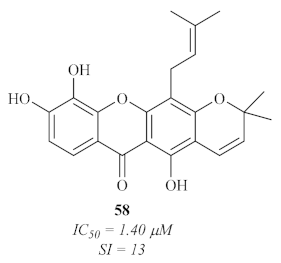
In 2007, an international research group decided to study the phenolic compounds from two Cameroonian medicinal plants,
Allanblackia monticola, and
Symphonia globulifera [
59]. Amongst the entire phenolic content of these plants, three xanthones were isolated and evaluated against the axenic amastigotes of
L. donovani, with only one of them
58 (
Figure 23) presenting both promising antileishmanial activity and cytotoxicity levels.
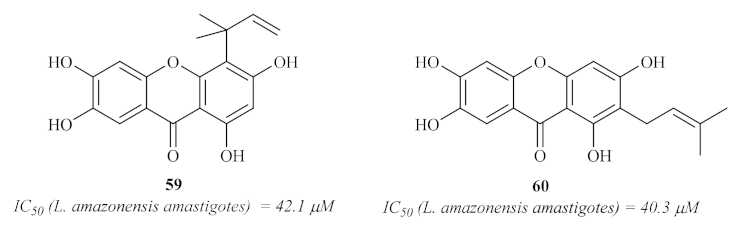
In 2008, Azebaze et al. isolated seven xanthones from the stem bark of
Allanblackia gabonensis and evaluated them against axenic amastigotes of
L. amazonensis [
60]. These preliminary studies showed that some of these xanthones present promising antileishmanial activities, emphasizing the tetraoxygenated prenylated xanthones activities when compared with the ones presented by dioxygenated simple xanthones. Additionally, the results suggest that their antileishmanial activity might be related to the presence of the 2- or 4-prenyl group. From the seven isolated xanthones, it is important to emphasize compounds
59 and
60 (
Figure 24) as the most effective against the
L. amazonensis axenic amastigotes.
This same year, another research group focused on characterizing the natural compounds from the marine-derived fungus
Chaetomium species. This led to the isolation of three new xanthones with unusual and rare structural features (
Figure 25) [
61]. Thus, these novel xanthone derivatives were evaluated for their antileishmanial potential against the amastigotes of
L. donovani. The results demonstrated that all the evaluated xanthones present considerable antileishmanial potential in the micromolar range.
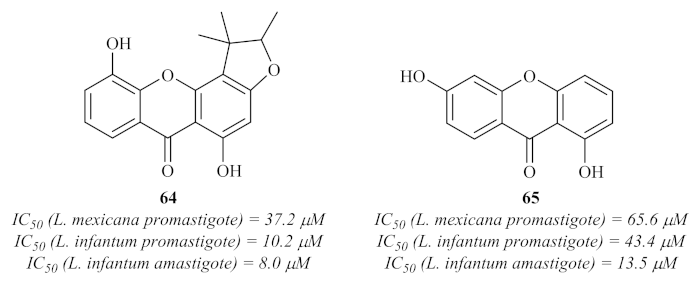
Nevertheless, in 2008, Hay et al. evaluated the polyphenolic content of an endemic plant from New Caledonia,
Garcinia vieillardii Pierre, against both
L. mexicana and
L. infantum [
62]. Five xanthones were further isolated from this phenolic content, characterized and evaluated for their antileishmanial properties, with only two of them (
64 and
65) showing considerable activity against the promastigote stage of both species of
Leishmania (
Figure 26). Regarding their potential against the amastigote stage of the parasite, these two compounds also presented significant activity levels, demonstrating that these two acridones are efficient against both stages of the parasite and capable of passing through the macrophage membranes.
The search for antileishmanial agents from natural sources continued throughout the decade, and a year later, in 2009, another research group isolated four xanthone derivatives from the roots of
Andrographis paniculate (Burm.f.) Nees [
63]. These xanthones were evaluated in vitro for their antiprotozoal activity, particularly against the amastigotes of
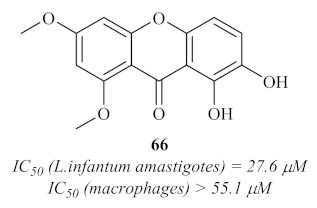
L. infantum, and the results demonstrated that, from the four isolated xanthones, only one (
66) showed considerable antileishmanial activity, with an IC
50 value of 27.6 µM (
Figure 27).
A few years later, in 2013, another research group focused their efforts on evaluating the in vitro antileishmanial of both dichloromethane and ethyl acetate extracts of
Garcinia mangostana Linn. against
L. infantum amastigotes [
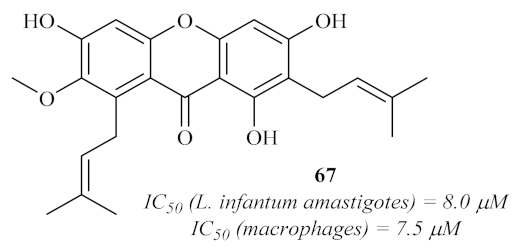
64]. Furthermore, this antileishmanial evaluation was also extended to the major constituent of the plant, namely α-mangostin
67 (
Figure 28). Considering the evaluation of this particular compound, the results demonstrated a promising antileishmanial potential, with an IC
50 of 8.0 µM. However, its cytotoxicity is even higher (IC
50 = 7.5 µM), which disqualifies its potential use against leishmaniasis. Interesting to verify is the fact that, as in the works of Azebaze et al. [
60], multiple hydroxy groups and a 2-prenyl group seem to be a key feature for the high antileishmanial activity of this type of compound.
In the same year, a Brazilian research group evaluated the antileishmanial potential of two xanthones from
Clusia pernambucensis G. Mariz. against
L. amazonensis amastigotes (
Figure 29) [
65]. Based on the results, these two xanthones only demonstrated moderate activity against
L. amazonensis, being still the most active compounds isolated from this plant. Thus, these results corroborate the works of Azebaze et al. [
60], Dua et al. [
63], and Kelly et al. [
57], in which both the importance of prenyl groups at 2- and 4-positions, as well as the hydroxy and methoxy groups, were previously described against other species of
Leishmania (
L. infantum and
L. mexicana). Despite their antileishmanial potential, these compounds also demonstrated high levels of cytotoxicity against murine macrophages, with IC
50 values similar to those observed against the parasite.
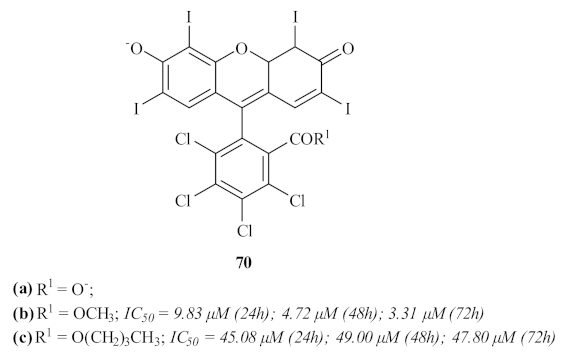
A few years later, another Brazilian research group intended to apply the widely studied technique of photodynamic therapy (PDT) to treat cutaneous leishmaniasis through the use of three promising xanthones [
66]. The selective derivatives, namely xanthene rose bengal B
70a and its correspondent methyl
70b and butyl esters
70c, presented favorable characteristics for their use in PTD, and their antileishmanial activity was assessed against
L. amazonensis promastigotes (
Figure 30). Through their use in PDT, the results showed that only the xanthene rose bengal B
70a did not present any activity against the promastigotes. In turn, the other two compounds (
70b and
70c) demonstrated considerable activity values in an illumination-dependent setting, with compound
70c as the most active of the two. Despite their proven antileishmanial potential, these compounds demonstrated high levels of cytotoxicity against macrophages, which not only makes their evaluation against the intracellular amastigotes of the parasite impossible but also impairs any further use of these compounds for the generalized treatment of leishmaniasis. Nevertheless, the authors do not rule out their potential application as photosensitizers in PDT to treat cutaneous leishmaniasis, the appropriate evaluation of their absorption and toxicity being crucial for this purpose.
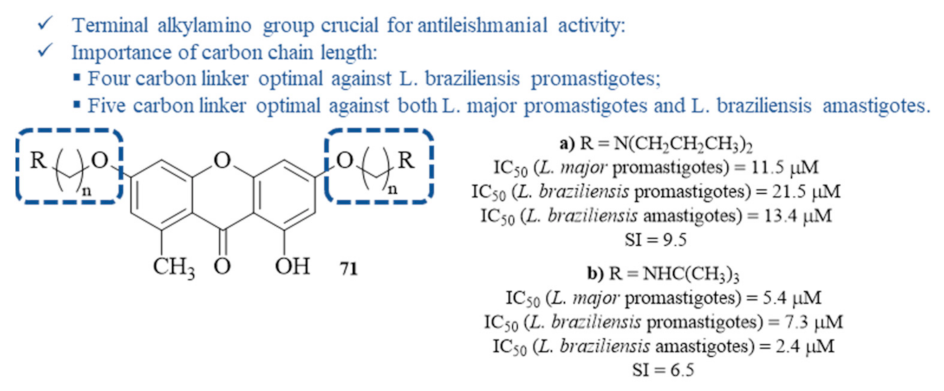
By the beginning of this decade, Micheletti et al. synthesized and evaluated a series of xanthones containing a diverse range of side chains at 3- and 6-positions for their antileishmanial activity against
L. major promastigotes and
L. braziliensis promastigotes and amastigotes (
Figure 31) [
67]. Considering the antileishmanial activity against the parasite’s promastigote stage, most of the derivatives containing terminal alkylamino groups demonstrated higher activity levels than the positive control, amphotericin B, being more active against
L. braziliensis. Thus, the most active compound against
L. braziliensis contains a four-carbon spacer between the amino group and the oxygen atom, with an IC
50 value of 0.1 µM, while the most active compound against
L. major bears a five-carbon spacer, with an IC
50 value of 5.4 µM. Against the
L. braziliensis amastigotes, the most active is also the compound bearing a five-carbon linker, with an IC
50 value of 2.4 µM. Furthermore, the wide variety of substituent groups enables an interpretation of the result to establish a structure–activity relationship study. This study, corroborating another work conducted against
L. Mexicana [
57], demonstrated that, in general, the xanthones bearing terminal alkylamino groups are highly effective against
Leishmania, with a clear correlation with the length of the carbon chain. The remaining compounds, lacking a side carbon chain with a terminal alkylamino group, only showed moderate to no activity against both the promastigotes and amastigotes of
Leishmania, confirming the relevance of this particular structural feature to the antileishmanial activity. Finally, all these compounds were evaluated for their cytotoxicity, demonstrating that some also present high activity levels against VERO cells. Determining the SI makes it important to highlight compounds
71a and
71b as the safest to use against Leishmania, with compound
71a presenting the highest SI despite its equally high IC
50 of 13.4 µM against
L. braziliensis amastigotes.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
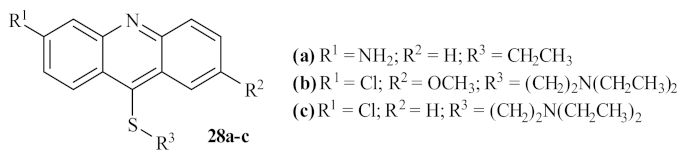{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
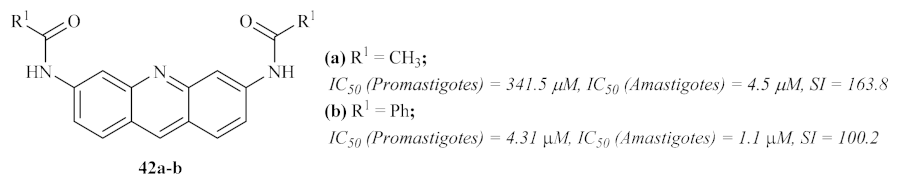{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






























