
Biased, Bitopic, Opioid–Adrenergic Tethered Compounds May Improve Specificity, Lower Dosage and Enhance Agonist or Antagonist Function with Reduced Risk of Tolerance and Addiction


Abstract
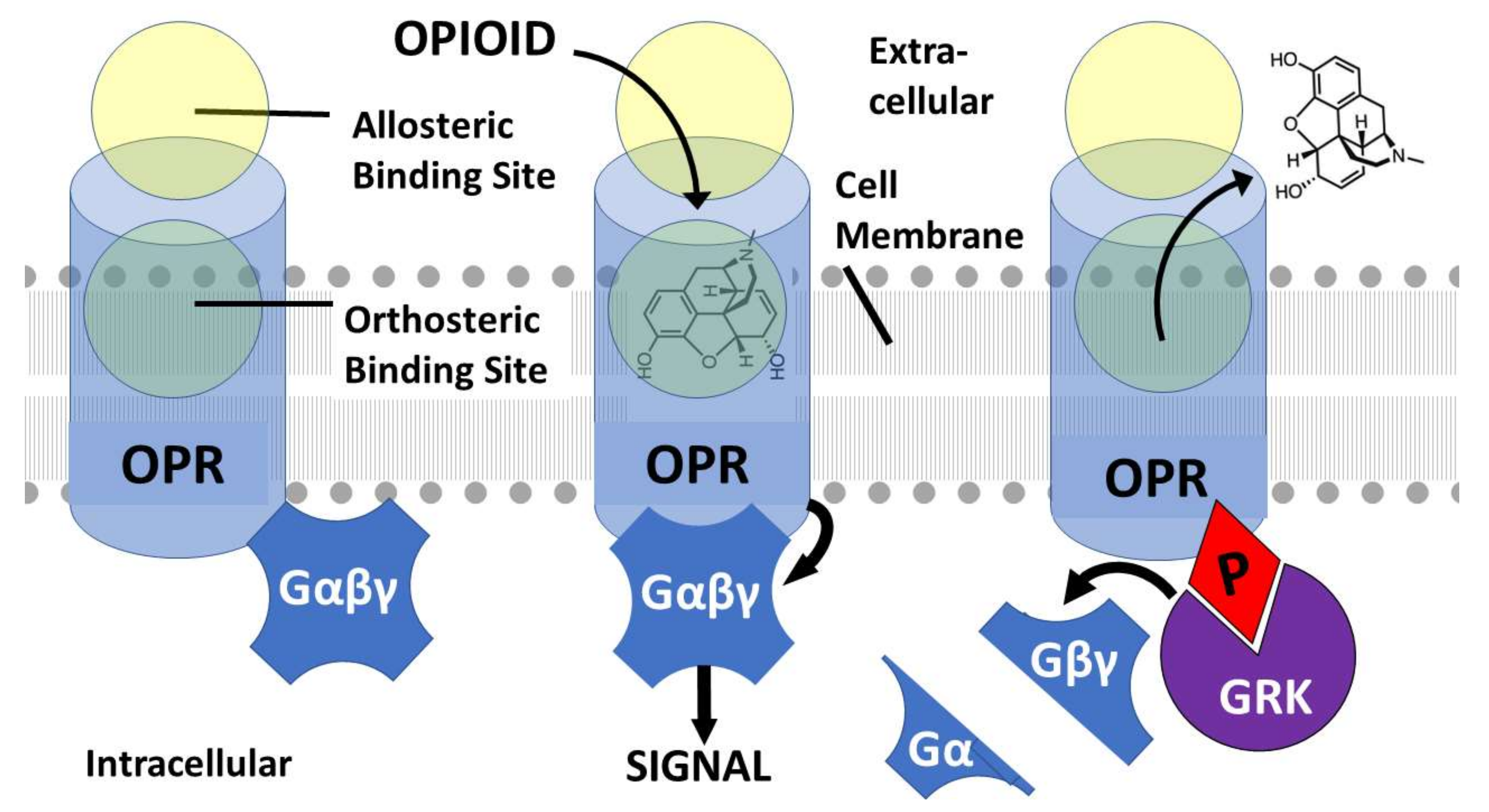
:1. Introduction
2. Mechanisms Underlying Opioid–Adrenergic Enhancement
3. Designing Heterobitopic Agonists, Antagonists, or Mixed Action Drugs to Optimize Opioid–Adrenergic Enhancement
“Bivalent ligands often display high affinity/avidity for and long residence time at their target. The [mechanism] responsible is the synergy that emanates from the simultaneous binding of their two pharmacophores to their respective target sites…. The first binding event prompts the still free pharmacophore to stay into ‘forced proximity’ of its target site, such lanes can be looked into by the equations that also apply to induced fit binding mechanisms. Interestingly, the simplest equations apply when bivalency goes along with a large gain in avidity. The overall bivalent ligand association and dissociation will be swifter than via each lane apart, but it is the lane that allows the fastest bidirectional ‘transit’ between the free and the fully bound target that is chiefly solicited. The bivalent ligand’s residence time is governed not only by the stability of the fully bound complex but also by the ability of freshly dissociated pharmacophores to successfully rebind.”
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Trescot, A.M.; Datta, S.; Lee, M.; Hansen, H. Opioid pharmacology. Pain Physician 2008, 11 (Suppl. S2), S133–S153. [Google Scholar] [CrossRef] [PubMed]
- Azzam, A.A.H.; McDonald, J.; Lambert, D.G. Hot topics in opioid pharmacology: Mixed and biased opioids. Br. J. Anaesth. 2019, 122, e136–e145. [Google Scholar] [CrossRef] [PubMed]
- Currow, D.C.; Quinn, S.; Greene, A.; Bull, J.; Johnson, M.J.; Abernethy, A.P. The longitudinal pattern of response when morphine is used to treat chronic refractory dyspnea. J. Palliat. Med. 2013, 16, 881–886. [Google Scholar] [CrossRef] [Green Version]
- Mahler, D.A. Opioids for refractory dyspnea. Expert Rev. Respir. Med. 2013, 7, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.; Strang, J. Medication Treatment of Opioid Use Disorder. Biol. Psychiatry 2020, 87, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Schwenk, E.; Grant, A.E.; Torjman, M.C.; McNulty, S.E.; Baratta, J.L.; Viscusi, E.R. The Efficacy of Peripheral Opioid Antagonists in Opioid-Induced Constipation and Postoperative Ileus: A systematic review of the literature. Reg. Anesth. Pain Med. 2017, 42, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.-Y.; Roh, D.-H.; Yeo, J.-H.; Woo, J.; Han, S.H.; Kim, K.-S. Analgesic Efficacy of α2 Adrenergic Receptor Agonists Depends on the Chronic State of Neuropathic Pain: Role of Regulator of G Protein Signaling 4. Neuroscience 2021, 455, 177–194. [Google Scholar] [CrossRef] [PubMed]
- Bahari, Z.; Meftahi, G.H. Spinal α 2-adrenoceptors and neuropathic pain modulation; therapeutic target. J. Cereb. Blood Flow Metab. 2019, 176, 2366–2381. [Google Scholar] [CrossRef] [PubMed]
- Boyd, R.E. Alpha2-adrenergic receptor agonists as analgesics. Curr. Top. Med. Chem. 2001, 1, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Albertson, T.E.; Chenoweth, J.; Ford, J.; Owen, K.; Sutter, M.E. Is It Prime Time for Alpha2-Adrenocepter Agonists in the Treatment of Withdrawal Syndromes? J. Med. Toxicol. 2014, 10, 369–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gowing, L.; Farrell, M.; Ali, R.; White, J. Alpha2-adrenergic agonists for the management of opioid withdrawal. Cochrane Database Syst. Rev. 2009, 2, CD002024. [Google Scholar] [CrossRef]
- Butterworth, J.F., IV; Mackey, D.C.; Wasnick, J.D. Chapter 14. Adrenergic Agonists & Antagonists. In Morgan & Mikhail’s Clinical Anesthesiology 5e; Butterworth, J.F., IV, Mackey, D.C., Wasnick, J.D., Eds.; McGraw Hill: New York, NY, USA, 2013; Available online: https://accessmedicine.mhmedical.com/content.aspx?bookid=564§ionid=42800545 (accessed on 17 December 2021).
- Farzam, K.; Kidron, A.; Lakhkar, A.D. Adrenergic Drugs. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK534230/ (accessed on 17 December 2021).
- Edinoff, A.N.; Houk, G.M.; Patil, S.; Siddaiah, H.B.; Kaye, A.J.; Iyengar, P.S.; Cornett, E.M.; Imani, F.; Mahmoudi, K.; Kaye, A.M.; et al. Adjuvant Drugs for Peripheral Nerve Blocks: The Role of Alpha-2 Agonists, Dexamethasone, Midazolam, and Non-steroidal Anti-inflammatory Drugs. Anesthesiol. Pain Med. 2021, 11, 117197. [Google Scholar] [CrossRef] [PubMed]
- Emelife, P.I.; Eng, M.R.; Menard, B.L.; Meyers, A.S.; Cornett, E.M.; Urman, R.D.; Kaye, A.D. Adjunct medications for peripheral and neuraxial anesthesia. Best Pr. Res. Clin. Anaesthesiol. 2018, 32, 83–99. [Google Scholar] [CrossRef] [PubMed]
- Helander, E.M.; Menard, B.L.; Harmon, C.M.; Homra, B.K.; Allain, A.V.; Bordelon, G.J.; Wyche, M.Q.; Padnos, I.W.; Lavrova, A.; Kaye, A.D. Multimodal Analgesia, Current Concepts, and Acute Pain Considerations. Curr. Pain Headache Rep. 2017, 21, 3. [Google Scholar] [CrossRef] [PubMed]
- Blaudszun, G.; Lysakowski, C.; Elia, N.; Tramèr, M.R. Effect of Perioperative Systemic α2 Agonists on Postoperative Morphine Consumption and Pain Intensity: Systematic review and meta-analysis of randomized controlled trials. Anesthesiology 2012, 116, 1312–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samoshkin, A.; Convertino, M.; Viet, C.T.; Wieskopf, J.S.; Kambur, O.; Marcovitz, J.; Patel, P.; Stone, L.S.; Kalso, E.; Mogil, J.S.; et al. Structural and functional interactions between six-transmembrane μ-opioid receptors and β2-adrenoreceptors modulate opioid signaling. Sci. Rep. 2015, 5, 18198. [Google Scholar] [CrossRef]
- Liang, D.-Y.; Shi, X.; Li, X.; Li, J.; Clark, J.D. The β2 adrenergic receptor regulates morphine tolerance and physical dependence. Behav. Brain Res. 2007, 181, 118–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, T.K.; Mishriky, B.M.; Klinger, R.Y.; Habib, A.S. The impact of neuraxial clonidine on postoperative analgesia and perioperative adverse effects in women having elective Caesarean section–a systematic review and meta-analysis. Br. J. Anaesth. 2018, 120, 228–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundblad, M.; Lönnqvist, P.-A. Adjunct analgesic drugs to local anaesthetics for neuroaxial blocks in children. Curr. Opin. Anaesthesiol. 2016, 29, 626–631. [Google Scholar] [CrossRef] [PubMed]
- Kundra, S.; Gupta, S.; Grewal, A.; Tewari, A.; Singh, R. Effect of clonidine and/or fentanyl in combination with intrathecal bupivacaine for lower limb surgery. J. Anaesthesiol. Clin. Pharmacol. 2015, 31, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Talwar, V.; Chopra, P. Low dose intrathecal clonidine and fentanyl added to hyperbaric bupivacaine prolongs analgesia in gynecological surgery. J. Anaesthesiol. Clin. Pharmacol. 2014, 30, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Engelman, E.; Marsala, C. Efficacy of adding clonidine to intrathecal morphine in acute postoperative pain: Meta-analysis. Br. J. Anaesth. 2013, 110, 21–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozdemir, N.; Kaya, F.N.; Gurbet, A.; Yilmazlar, A.; Demirag, B.; Mandiraci, B.O. Comparison of Intraarticular Bupivacaine and Levobupivacaine with Morphine and Epinephrine for Knee Arthroscopy. Eurasian J. Med. 2013, 45, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, A.V.; Berend, K.R.; Mallory, T.H.; Dodds, K.L.; Adams, J. Soft Tissue and Intra-articular Injection of Bupivacaine, Epinephrine, and Morphine Has a Beneficial Effect after Total Knee Arthroplasty. Clin. Orthop. Relat. Res. 2004, 428, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Massone, M.L.; Lampugnani, E.; Calevo, M.G.; Gandolfo, A.; Montobbio, G.; Fossa, S. Effetti di una dose di clonidina epidurale associata a morfina intratecale sull’analgesia postoperatoria [The effects of a dose of epidural clonidine combined with in-trathecal morphine for postoperative analgesia]. Minerva Anestesiol. 1998, 64, 289–296. [Google Scholar]
- Goyagi, T.; Nishikawa, T. The Addition of Epinephrine Enhances Postoperative Analgesia by Intrathecal Morphine. Anesth. Analg. 1995, 81, 508–513. [Google Scholar] [CrossRef]
- Abouleish, E.; Rawal, N.; Tobon-Randall, B.; Rivera-Weiss, M.; Meyer, B.; Wu, A.; Rashad, M.N. A Clinical and Laboratory Study to Compare the Addition of 0.2 mg of Morphine, 0.2 mg of Epinephrine, or Their Combination to Hyperbaric Bupivacaine for Spinal Anesthesia in Cesarean Section. Anesth. Analg. 1993, 77, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Andrieu, G.; Roth, B.; Ousmane, L.; Castaner, M.; Petillot, P.; Vallet, B.; Villers, A.; Lebuffe, G. The efficacy of intrathecal morphine with or without clonidine for postoperative analgesia after radical prostatectomy. Anesth. Analg. 2009, 108, 1954–1957. [Google Scholar] [CrossRef] [PubMed]
- Khandelwal, M.; Ahmed, F.; Sharma, A. A comparative study of the effect of clonidine, fentanyl, and the combination of both as adjuvant to intrathecal bupivacaine for postoperative analgesia in total abdominal hysterectomy. J. Anaesthesiol. Clin. Pharmacol. 2017, 33, 102–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goyagi, T.; Nishikawa, T. Oral Clonidine Premedication Enhances the Quality of Postoperative Analgesia by Intrathecal Morphine. Anesth. Analg. 1996, 82, 1192–1196. [Google Scholar] [CrossRef] [PubMed]
- Goyagi, T.; Tanaka, M.; Nishikawa, T. Oral Clonidine Premedication Enhances Postoperative Analgesia by Epidural Morphine. Anesth. Analg. 1999, 89, 1487–1491. [Google Scholar] [CrossRef] [PubMed]
- Fournier, R.; Van Gessel, E.; Weber, A.; Gamulin, Z. Epinephrine and clonidine do not improve intrathecal sufentanil analgesia after total hip replacement. Br. J. Anaesth. 2002, 89, 562–566. [Google Scholar] [CrossRef] [Green Version]
- Gursoy, S.; Ozdemir, E.; Bagcivan, I.; Altun, A.; Durmus, N. Effects of alpha 2-adrenoceptor agonists dexmedetomidine and guanfacine on morphine analgesia and tolerance in rats. Upsala J. Med. Sci. 2011, 116, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Honey, B.L.; Benefield, R.J.; Miller, J.L.; Johnson, P.N. α2-Receptor Agonists for Treatment and Prevention of Iatrogenic Opioid Abstinence Syndrome in Critically Ill Patients. Ann. Pharmacother. 2009, 43, 1506–1511. [Google Scholar] [CrossRef] [PubMed]
- Satarian, L.; Javan, M.; Fathollahi, Y. Epinephrine inhibits analgesic tolerance to intrathecal administrated morphine and increases the expression of calcium–calmodulin-dependent protein kinase IIα. Neurosci. Lett. 2008, 430, 213–217. [Google Scholar] [CrossRef]
- Bentley, G.; Newton, S.; Starr, J. Studies on the antinociceptive action of α-agonist drugs and their interactions with opioid mechanisms. J. Cereb. Blood Flow Metab. 1983, 79, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Tonner, P.H. Additives used to reduce perioperative opioid consumption 1: Alpha2-agonists. Best Pr. Res. Clin. Anaesthesiol. 2017, 31, 505–512. [Google Scholar] [CrossRef]
- Keating, G.M. Dexmedetomidine: A Review of Its Use for Sedation in the Intensive Care Setting. Drugs 2015, 75, 1119–1130. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Bu, J.; Wu, X.; Deng, L.; Chi, M.; Ma, C.; Shi, X.; Wang, G. Upregulation of μ-Opioid Receptor in the Rat Spinal Cord Contributes to the α2-Adrenoceptor Agonist Dexmedetomidine-Induced Attenuation of Chronic Morphine Tolerance in Cancer Pain. J. Pain Res. 2020, 13, 2617–2627. [Google Scholar] [CrossRef] [PubMed]
- Özdoǧan, U.K.; Lähdesmäki, J.; Hakala, K.; Scheinin, M. The involvement of α2A-adrenoceptors in morphine analgesia, tolerance and withdrawal in mice. Eur. J. Pharmacol. 2004, 497, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Özdoğan, U.K.; Lähdesmäki, J.; Scheinin, M. Influence of prazosin and clonidine on morphine analgesia, tolerance and withdrawal in mice. Eur. J. Pharmacol. 2003, 460, 127–134. [Google Scholar] [CrossRef]
- Del Bello, F.; Mattioli, L.; Ghelfi, F.; Giannella, M.; Piergentili, A.; Quaglia, W.; Cardinaletti, C.; Perfumi, M.; Thomas, R.J.; Zanelli, U.; et al. Fruitful Adrenergic α2C-Agonism/α2A-Antagonism Combination to Prevent and Contrast Morphine Tolerance and Dependence. J. Med. Chem. 2010, 53, 7825–7835. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Liu, G.; Zhou, Z.; Yang, J.; Su, C. Successful Treatment of Refractory Cancer Pain and Depression with Continuous Intrathecal Administration of Dexmedetomidine and Morphine: A Case Report. Pain Ther. 2020, 9, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Kapoor, S.; Durkin, B. Analgesic management of acute pain in the opioid-tolerant patient. Curr. Opin. Anaesthesiol. 2015, 28, 398–402. [Google Scholar] [CrossRef]
- Aldunate, J.; Yojay, L.; Mardones, J. Studies on the mechanism of the action of morphine on the peristalsis of guinea pig ileum in situ. Naunyn-Schmiedebergs Arch. Exp. Pathol. Pharmakol. 1975, 291, 395–403. [Google Scholar] [CrossRef]
- Heimanes, R.L. Catecholamines and the actions of morphine on the guinea-pig ileum. Arch. Int. Pharmacodyn. Ther. 1975, 216, 11–18. [Google Scholar] [PubMed]
- Valdes, A.M.; Abhishek, A.; Muir, K.; Zhang, W.; Maciewicz, R.A.; Doherty, M. Association of beta-blocker use with less prevalent joint pain and lower opioid requirement in people with osteoarthritis. Arthritis Care Res. 2016, 69, 1076–1081. [Google Scholar] [CrossRef] [Green Version]
- Greenwell, T.N.; Walker, B.M.; Cottone, P.; Zorrilla, E.P.; Koob, G.F. The α1 adrenergic receptor antagonist prazosin reduces heroin self-administration in rats with extended access to heroin administration. Pharmacol. Biochem. Behav. 2009, 91, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Milne, B.; Jhamandas, K.; Sutak, M.; Grenier, P.; Cahill, C.M. Analgesia, enhancement of spinal morphine antinociception, and inhibition of tolerance by ultra-low dose of the α2A-adrenoceptor selective antagonist BRL44408. Eur. J. Pharmacol. 2014, 743, 89–97. [Google Scholar] [CrossRef]
- Takahashi, M.; Okada, T.; Kaneto, H. Differential Roles of the Adrenal Gland in the Suppression of Morphine Antinociceptive Tolerance Development by.ALPHA.- and.BETA.-Adrenergic Blockers. Jpn. J. Pharmacol. 1991, 55, 555–558. [Google Scholar] [CrossRef]
- Yu, S.K.H.; Tait, G.; Karkouti, K.; Wijeysundera, D.; McCluskey, S.; Beattie, W.S. The Safety of Perioperative Esmolol: A systematic review and meta-analysis of randomized controlled trials. Anesth. Analg. 2011, 112, 267–281. [Google Scholar] [CrossRef] [Green Version]
- Harless, M.; Depp, C.; Collins, S.; Hewer, I. Role of Esmolol in Perioperative Analgesia and Anesthesia: A Literature Review. AANA J. 2015, 83, 167–177. [Google Scholar] [PubMed]
- Härkänen, L.; Halonen, J.; Selander, T.; Kokki, H. Beta-adrenergic antagonists during general anesthesia reduced postoperative pain: A systematic review and a meta-analysis of randomized controlled trials. J. Anesth. 2015, 29, 934–943. [Google Scholar] [CrossRef] [PubMed]
- Gelineau, A.M.; King, M.R.; Ladha, K.S.; Burns, S.M.; Houle, T.; Anderson, T.A. Intraoperative Esmolol as an Adjunct for Perioperative Opioid and Postoperative Pain Reduction: A systematic review, meta-analysis, and meta-regression. Anesth. Analg. 2018, 126, 1035–1049. [Google Scholar] [CrossRef] [PubMed]
- Bahr, M.P.; Williams, B.A. Esmolol, Antinociception, and Its Potential Opioid-Sparing Role in Routine Anesthesia Care. Reg. Anesth. Pain Med. 2018, 43, 815–818. [Google Scholar] [CrossRef] [PubMed]
- Boronat, M.A.; Olmos, G.; García-Sevilla, J.A. Attenuation of tolerance to opioid-induced antinociception and protection against morphine-induced decrease of neurofilament proteins by idazoxan and other I2-imidazoline ligands. J. Cereb. Blood Flow Metab. 1998, 125, 175–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chabot-Doré, A.-J.; Schuster, D.J.; Stone, L.S.; Wilcox, G.L. Analgesic synergy between opioid and α2-adrenoceptors. J. Cereb. Blood Flow Metab. 2014, 172, 388–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiruvenkatarajan, V.; Wood, R.; Watts, R.; Currie, J.; Wahba, M.; Van Wijk, R. The intraoperative use of non-opioid adjuvant analgesic agents: A survey of anaesthetists in Australia and New Zealand. BMC Anesthesiol. 2019, 19, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, A.F.; Kalso, E.A.; McQuay, H.J.; Dickenson, A.H. Evidence for the involvement of the μ but not δ opioid receptor subtype in the synergistic interaction between opioid and α2 adrenergic antinociception in the rat spinal cord. Neurosci. Lett. 1992, 139, 65–68. [Google Scholar] [CrossRef]
- Aira, Z.; Barrenetxea, T.; Buesa, I.; Azkue, J.J. Plasticity of α2-adrenergic spinal antinociception following nerve injury: Selective, bidirectional interaction with the delta opioid receptor. Brain Res. 2015, 1594, 190–203. [Google Scholar] [CrossRef]
- Nguyen, V.T.; Wu, Y.; Guillory, A.N.; McConnell, B.K.; Fujise, K.; Huang, M.-H. Delta-opioid augments cardiac contraction through β-adrenergic and CGRP-receptor co-signaling. Peptides 2012, 33, 77–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, C.E.; Habash, T.; Dykstra, L.A.; Picker, M.J. Discriminative-stimulus effects of morphine in combination with α- and β-noradrenergic agonists and antagonists in rats. Pharmacol. Biochem. Behav. 1996, 53, 979–986. [Google Scholar] [CrossRef]
- Zarrindast, M.-R.; Ramezani-Tehrani, B.; Ghadimi, M. Effects of adrenoceptor agonists and antagonists on morphine-induced Straub tail in mice. Pharmacol. Biochem. Behav. 2002, 72, 203–207. [Google Scholar] [CrossRef]
- Haenecour, A.S.; Seto, W.; Urbain, C.M.; Stephens, D.; Laussen, P.C.; Balit, C.R. Prolonged dexmedetomidine infusion and drug withdrawal in critically ill children. J. Pediatr. Pharmacol. Ther. 2017, 22, 453–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.M.; Caylor, K.; Gockenbach, N. Evaluating the transition from dexmedetomidine to clonidine for the prevention of withdrawal in critically ill pediatric patients. J. Pediatr. Pharmacol. Ther. 2020, 25, 104–110. [Google Scholar] [CrossRef]
- Bhatt, K.; Quan, A.T.; Baumgartner, L.; Jia, S.; Croci, R.; Puntillo, K.; Ramsay, J.; Bouajram, R.H. Effects of a Clonidine Taper on Dexmedetomidine Use and Withdrawal in Adult Critically Ill Patients—A Pilot Study. Crit. Care Explor. 2020, 2, e0245. [Google Scholar] [CrossRef] [PubMed]
- Syrovatkina, V.; Alegre, K.O.; Dey, R.; Huang, X.-Y. Regulation, Signaling, and Physiological Functions of G-Proteins. J. Mol. Biol. 2016, 428, 3850–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weis, W.I.; Kobilka, B.K. The Molecular Basis of G Protein–Coupled Receptor Activation. Annu. Rev. Biochem. 2018, 87, 897–919. [Google Scholar] [CrossRef]
- Chevillard, F.; Kelemen, Á.; Baker, J.G.; Aranyodi, V.A.; Balzer, F.; Kolb, P.; Keserű, G.M. Fragment evolution for GPCRs: The role of secondary binding sites in optimization. Chem. Commun. 2021, 57, 10516–10519. [Google Scholar] [CrossRef] [PubMed]
- Egyed, A.; Kelemen, Á.A.; Vass, M.; Visegrády, A.; Thee, S.A.; Wang, Z.; de Graaf, C.; Brea, J.; Loza, M.I.; Leurs, R.; et al. Controlling the selectivity of aminergic GPCR ligands from the extracellular vestibule. Bioorganic Chem. 2021, 111, 104832. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, Y. Recent Advances in the Drug Discovery and Development of Dualsteric/Bitopic Activators of G Protein-Coupled Receptors. Curr. Top. Med. Chem. 2019, 19, 2378–2392. [Google Scholar] [CrossRef]
- Fronik, P.; Gaiser, B.I.; Sejer Pedersen, D. Bitopic Ligands and Metastable Binding Sites: Opportunities for G Protein-Coupled Receptor (GPCR) Medicinal Chemistry. J. Med. Chem. 2017, 60, 4126–4134. [Google Scholar] [CrossRef] [PubMed]
- Munro, T.A.; Xu, W.; Ho, D.M.; Liu-Chen, L.-Y.; Cohen, B.M. Studies toward bivalent κ opioids derived from salvinorin A: Heteromethylation of the furan ring reduces affinity. Beilstein, J. Org. Chem. 2013, 9, 2916–2924. [Google Scholar] [CrossRef] [PubMed]
- Valant, C.; Sexton, P.M.; Christopoulos, A. Orthosteric/Allosteric Bitopic Ligands: Going Hybrid at GPCRs. Mol. Interv. 2009, 9, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Pepe, S.; van den Brink, O.W.; Lakatta, E.; Xiao, R.-P. Cross-talk of opioid peptide receptor and β-adrenergic receptor signalling in the heart. Cardiovasc. Res. 2004, 63, 414–422. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.-C.; Wang, H.-X.; Zhang, W.-M.; Wong, T.M. Cross-talk between Cardiac kappa-Opioid and β-Adrenergic Receptors in Developing Hypertensive Rats. J. Mol. Cell. Cardiol. 1999, 31, 597–605. [Google Scholar] [CrossRef]
- Cussac, D.; Rauly-Lestienne, I.; Heusler, P.; Finana, F.; Cathala, C.; Bernois, S.; De Vries, L. μ-opioid and 5-HT1A receptors heterodimerize and show signalling crosstalk via G protein and MAP-kinase pathways. Cell. Signal. 2012, 24, 1648–1657. [Google Scholar] [CrossRef] [PubMed]
- Ballesta, J.J.; Orts, A. Interaction of opioid drugs with human platelet α2-adrenoceptors. Eur. J. Pharmacol. Mol. Pharmacol. 1992, 227, 349–351. [Google Scholar] [CrossRef]
- Ide, S.; Minami, M.; Ishihara, K.; Uhl, G.R.; Sora, I.; Ikeda, K. Mu opioid receptor-dependent and independent components in effects of tramadol. Neuropharmacology 2006, 51, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Höcker, J.; Böhm, R.; Meybohm, P.; Gruenewald, M.; Renner, J.; Ohnesorge, H.; Scholz, J.; Bein, B. Interaction of morphine but not fentanyl with cerebral α2-adrenoceptors in α2-adrenoceptor knockout mice. J. Pharm. Pharmacol. 2009, 61, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Höcker, J.; Weber, B.; Tonner, P.H.; Scholz, J.; Brand, P.-A.; Ohnesorge, H.; Bein, B. Meperidine, remifentanil and tramadol but not sufentanil interact with α2-adrenoceptors in α2A-, α2B- and α2C-adrenoceptor knock out mice brain. Eur. J. Pharmacol. 2008, 582, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R.; Churchill, B.; Turke, M.; Subhramanyam, U.K.T.; Labahn, J.; Bernstein, R. Mutual Enhancement of Opioid and Adrenergic Receptors by Combinations of Opioids and Adrenergic Ligands Is Reflected in Molecular Complementarity of Ligands: Drug Development Possibilities. Int. J. Mol. Sci. 2019, 20, 4137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Root-Bernstein, R.; Churchill, B.; Turke, M. Glutathione and Glutathione-Like Sequences of Opioid and Aminergic Receptors Bind Ascorbic Acid, Adrenergic and Opioid Drugs Mediating Antioxidant Function: Relevance for Anesthesia and Abuse. Int. J. Mol. Sci. 2020, 21, 6230. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R.; Turke, M.; Subhramanyam, U.K.T.; Churchill, B.; Labahn, J. Adrenergic Agonists Bind to Adrenergic-Receptor-Like Regions of the Mu Opioid Receptor, Enhancing Morphine and Methionine-Enkephalin Binding: A New Approach to “Biased Opioids”? Int. J. Mol. Sci. 2018, 19, 272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Root-Bernstein, R. Adrenergic agonists and the mu opioid receptor. In The Neurobiology, Physiology, and Behavior of Pain; Rajendram, R., Patel, V.B., Preedy, V.R., Eds.; Elsevier Science: Amsterdam, The Netherland, 2022; pp. 1–22. [Google Scholar]
- Root-Bernstein, R.S.; Dillon, P.F. Fostering adventure research. A case study of the discovery that ascorbic acid enhances adrenergic drug activity. Drug Develop. Res. 2002, 57, 58–74. [Google Scholar] [CrossRef]
- Root-Bernstein, R.; Churchill, B. Co-Evolution of Opioid and Adrenergic Ligands and Receptors: Shared, Complementary Modules Explain Evolution of Functional Interactions and Suggest Novel Engineering Possibilities. Life 2021, 11, 1217. [Google Scholar] [CrossRef] [PubMed]
- Onogi, T.; Minami, M.; Katao, Y.; Nakagawa, T.; Aoki, Y.; Toya, T.; Katsumata, S.; Satoh, M. DAMGO, a mu-opioid receptor selective agonist, distinguishes between mu- and delta-opioid receptors around their first extracellular loops. FEBS Lett. 1995, 357, 93–97. [Google Scholar] [CrossRef] [Green Version]
- Metzger, T.G.; Ferguson, D.M. On the role of extracellular loops of opioid receptors in conferring ligand selectivity. FEBS Lett. 1995, 375, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Paterlini, M.G. The function of the extracellular regions in opioid receptor binding: Insights from computational biology. Curr. Top. Med. Chem. 2005, 5, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Strasser, A.; Wittmann, H.-J.; Seifert, R. Binding Kinetics and Pathways of Ligands to GPCRs. Trends Pharmacol. Sci. 2017, 38, 717–732. [Google Scholar] [CrossRef] [PubMed]
- Gaiser, B.I.; Danielsen, M.; Marcher-Rørsted, E.; Jørgensen, K.R.; Wróbel, T.M.; Frykman, M.; Johansson, H.; Bräuner-Osborne, H.; Gloriam, D.E.; Mathiesen, J.M.; et al. Probing the Existence of a Metastable Binding Site at the β2-Adrenergic Receptor with Homobivalent Bitopic Ligands. J. Med. Chem. 2019, 62, 7806–7839. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Kaindl, J.; Clark, M.J.; Hübner, H.; Hirata, K.; Sunahara, R.K.; Gmeiner, P.; Kobilka, B.K.; Liu, X. Binding pathway determines norepinephrine selectivity for the human β1AR over β2AR. Cell Res. 2021, 31, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Malcolm, D.S.; Zaloga, G.P.; Willey, S.C.; Amir, S.; Holaday, J.W. Naloxone potentiates epinephrine’s pressor actions in endotoxemic rats. Circ. Shock. 1988, 25, 259–265. [Google Scholar] [PubMed]
- Lechner, R.B. Naloxone potentiates the inotropic effects of isoproterenol in vitro by a nonopiate receptor mechanism. Circ. Shock 1992, 38, 157–164. [Google Scholar] [PubMed]
- Lechner, R.B. Naloxone potentiates inotropic but not chronotropic effects of isoproterenol in vitro. Circ. Shock 1993, 39, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Barron, A.B.; Gaugl, J.F.; Caffrey, J.L. (+) Naloxone potentiates the inotropic effect of epinephrine in the isolated dog heart. Circ. Shock 1993, 40, 206–211. [Google Scholar]
- Gu, H.; Gaugl, J.F.; Barron, B.A.; Caffrey, J.L. Naloxone enhances cardiac contractile responses to epinephrine without altering epinephrine uptake from plasma. Circ. Shock 1990, 32, 257–271. [Google Scholar]
- Caffrey, J.L.; Hathorne, L.F.; Carter, G.C.; Sinclair, R.J. Naloxone potentiates contractile responses to epinephrine in isolated canine arteries. Circ. Shock 1990, 31, 317–332. [Google Scholar] [PubMed]
- Caffrey, J.L.; Stoll, S.T.; Sinclair, R.J.; Barron, B.A. (+) naloxone enhances vascular contractile responses to added epinephrine. Prog. Clin. Biol. Res. 1990, 328, 375–378. [Google Scholar]
- Grenier, P.; Wiercigroch, D.; Olmstead, M.C.; Cahill, C.M. Dissociation between morphine-induced spinal gliosis and analgesic tolerance by ultra-low-dose α2-adrenergic and cannabinoid CB1-receptor antagonists. Behav. Pharmacol. 2018, 29, 241–254. [Google Scholar] [CrossRef]
- Livingston, E.K.; Traynor, J.R. Allostery at opioid receptors: Modulation with small molecule ligands. J. Cereb. Blood Flow Metab. 2018, 175, 2846–2856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livingston, K.E.; Stanczyk, M.A.; Burford, N.T.; Alt, A.; Canals, M.; Traynor, J.R. Pharmacologic Evidence for a Putative Conserved Allosteric Site on Opioid Receptors. Mol. Pharmacol. 2017, 93, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Burford, N.T.; Clark, M.J.; Wehrman, T.S.; Gerritz, S.; Banks, M.; O’Connell, J.; Traynor, J.R.; Alt, A. Discovery of positive allosteric modulators and silent allosteric modulators of the μ-opioid receptor. Proc. Natl. Acad. Sci. USA 2013, 110, 10830–10835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandasamy, R.; Hillhouse, T.M.; Livingston, K.E.; Kochan, K.E.; Meurice, C.; Eans, S.O.; Li, M.-H.; White, A.D.; Roques, B.P.; McLaughlin, J.P.; et al. Positive allosteric modulation of the mu-opioid receptor produces analgesia with reduced side effects. Proc. Natl. Acad. Sci. USA 2021, 118, 2000017118. [Google Scholar] [CrossRef] [PubMed]
- Filizola, M. Insights from molecular dynamics simulations to exploit new trends for the development of improved opioid drugs. Neurosci. Lett. 2019, 700, 50–55. [Google Scholar] [CrossRef]
- Shang, Y.; Yeatman, H.R.; Provasi, D.; Alt, A.; Christopoulos, A.; Canals, M.; Filizola, M. Proposed Mode of Binding and Action of Positive Allosteric Modulators at Opioid Receptors. ACS Chem. Biol. 2016, 11, 1220–1229. [Google Scholar] [CrossRef] [PubMed]
- Bisignano, P.; Burford, N.T.; Shang, Y.; Marlow, B.; Livingston, K.E.; Fenton, A.M.; Rockwell, K.; Budenholzer, L.; Traynor, J.R.; Gerritz, S.W.; et al. Ligand-Based Discovery of a New Scaffold for Allosteric Modulation of the μ-Opioid Receptor. J. Chem. Inf. Model. 2015, 55, 1836–1843. [Google Scholar] [CrossRef] [Green Version]
- Bartuzi, D.; Kędzierska, E.; Kaczor, A.A.; Schmidhammer, H.; Matosiuk, D. Novel Positive Allosteric Modulators of µ Opioid Receptor—Insight from In Silico and In Vivo Studies. Int. J. Mol. Sci. 2020, 21, 8463. [Google Scholar] [CrossRef]
- Bartuzi, D.; Kaczor, A.A.; Matosiuk, D. Activation and Allosteric Modulation of Human μ Opioid Receptor in Molecular Dynamics. J. Chem. Inf. Model. 2015, 55, 2421–2434. [Google Scholar] [CrossRef]
- Uprety, R.; Che, T.; Zaidi, S.A.; Grinnell, S.G.; Varga, B.R.; Faouzi, A.; Slocum, S.T.; Allaoa, A.; Varadi, A.; Nelson, M.; et al. Controlling opioid receptor functional selectivity by targeting distinct subpockets of the orthosteric site. eLife 2021, 10. [Google Scholar] [CrossRef]
- Root-Bernstein, R.; Dillon, P.F.; Hollingsworth, R. A tethered ascorbate-norepinephrine compound, 4-UT, displays long-acting adrenergic activity on rabbit aortic smooth muscle. Drug Dev. Res. 2008, 69, 242–250. [Google Scholar] [CrossRef]
- Jordan, B.A.; Trapaidze, N.; Gomes, I.; Nivarthi, R.; Devi, L.A. Oligomerization of opioid receptors with beta 2-adrenergic receptors: A role in trafficking and mitogen-activated protein kinase activation. Proc. Natl. Acad. Sci. USA 2001, 98, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Vilardaga, J.-P.; Nikolaev, V.O.; Lorenz, K.; Ferrandon, S.; Zhuang, Z.; Lohse, M.J. Conformational cross-talk between α2A-adrenergic and μ-opioid receptors controls cell signaling. Nat. Chem. Biol. 2008, 4, 126–131. [Google Scholar] [CrossRef]
- Fujita, W.; Gomes, I.A.; Devi, L. Revolution in GPCR signalling: Opioid receptor heteromers as novel therapeutic targets: IUPHAR Review 10. J. Cereb. Blood Flow Metab. 2014, 171, 4155–4176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andurkar, S.V.; Gendler, L.; Gulati, A. Tramadol antinociception is potentiated by clonidine through α2-adrenergic and I2-imidazoline but not by endothelin ETA receptors in mice. Eur. J. Pharmacol. 2012, 683, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Gulati, A.; Bhalla, S.; Matwyshyn, G.; Zhang, Z.; Andurkar, S.V. Determination of Adrenergic and Imidazoline Receptor Involvement in Augmentation of Morphine and Oxycodone Analgesia by Clonidine and BMS182874. Pharmacology 2009, 83, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Kremer, M.; Megat, S.; Bohren, Y.; Wurtz, X.; Nexon, L.; Ceredig, R.A.; Doridot, S.; Massotte, D.; Salvat, E.; Yalcin, I.; et al. Delta opioid receptors are essential to the antiallodynic action of Β2-mimetics in a model of neuropathic pain. Mol. Pain 2020, 16, 1744806920912931. [Google Scholar] [CrossRef] [Green Version]
- Vulliemoz, Y.; Virag, L.; Whittington, R.A. Interaction of the α-2 adrenergic- and opioid receptor with the cGMP system in the mouse cerebellum. Brain Res. 1998, 813, 26–31. [Google Scholar] [CrossRef]
- Roerig, S.C.; Lei, S.; Kitto, K.; Hylden, J.K.; Wilcox, G.L. Spinal interactions between opioid and noradrenergic agonists in mice: Multiplicativity involves delta and α-2 receptors. J. Pharmacol. Exp. Ther. 1992, 262, 365–374. [Google Scholar]
- Park, W.K.; Chang, C.H.; Chae, J.E.; Kim, M.H.; Cho, Y.L.; Ahn, D.S. Phosphodiesterase inhibition by naloxone augments the inotropic actions of β-adrenergic stimulation. Acta Anaesthesiol. Scand. 2009, 53, 1043–1051. [Google Scholar] [CrossRef]
- Newman, A.H.; Battiti, F.O.; Bonifazi, A.; Philip, S. Portoghese Medicinal Chemistry Lectureship: Designing Bivalent or Bitopic Molecules for G-Protein Coupled Receptors. The Whole Is Greater Than the Sum of Its Parts. J. Med. Chem. 2020, 63, 1779–1797. [Google Scholar] [CrossRef] [PubMed]
- Lalchandani, S.G.; Lei, L.; Zheng, W.; Suni, M.M.; Moore, B.M.; Liggett, S.B.; Miller, D.D.; Feller, D.R. Yohimbine Dimers Exhibiting Selectivity for the Human α2c-Adrenoceptor Subtype. J. Pharmacol. Exp. Ther. 2002, 303, 979–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bavadekar, S.A.; Hong, S.-S.; Lee, S.-I.; Miller, D.D.; Feller, D.R. Bioisosteric phentolamine analogs as selective human α2- versus α1-adrenoceptor ligands. Eur. J. Pharmacol. 2008, 590, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Knapp, B.I.; Bidlack, J.M.; Neumeyer, J.L. Pharmacological properties of bivalent ligands containing butorphan linked to nalbuphine, naltrexone, and naloxone at mu, delta, and kappa opioid receptors. J. Med. Chem. 2007, 50, 2254–2258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decker, M.; Fulton, B.S.; Zhang, B.; Knapp, B.I.; Bidlack, J.M.; Neumeyer, J.L. Univalent and Bivalent Ligands of Butorphan: Characteristics of the Linking Chain Determine the Affinity and Potency of Such Opioid Ligands. J. Med. Chem. 2009, 52, 7389–7396. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Zhang, T.; Sromek, A.W.; Scrimale, T.; Bidlack, J.M.; Neumeyer, J.L. Synthesis and binding affinity of novel mono- and bivalent morphinan ligands for κ, μ, and δ opioid receptors. Bioorganic Med. Chem. 2011, 19, 2808–2816. [Google Scholar] [CrossRef] [Green Version]
- Balboni, G.; Salvadori, S.; Marczak, E.D.; Knapp, B.I.; Bidlack, J.M.; Lazarus, L.H.; Peng, X.; Si, Y.G.; Neumeyer, J.L. Opioid bifunctional ligands from morphine and the opioid pharmacophore Dmt-Tic. Eur. J. Med. Chem. 2011, 46, 799–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Cao, D.; Gillespie, J.C.; Mendez, R.E.; Selley, D.E.; Liu-Chen, L.-Y.; Zhang, Y. Exploring the putative mechanism of allosteric modulations by mixed-action kappa/mu opioid receptor bitopic modulators. Futur. Med. Chem. 2021, 13, 551–573. [Google Scholar] [CrossRef] [PubMed]
- Le Naour, M.; Lunzer, M.M.; Powers, M.D.; Kalyuzhny, A.E.; Benneyworth, M.A.; Thomas, M.J.; Portoghese, P.S. Putative kappa opioid heteromers as targets for developing analgesics free of adverse effects. J. Med. Chem. 2014, 57, 6383–6392. [Google Scholar] [CrossRef]
- Obeng, S.; Wang, H.; Jali, A.; Stevens, D.L.; Akbarali, H.I.; Dewey, W.L.; Selley, D.E.; Zhang, Y. Structure–Activity Relationship Studies of 6α- and 6β-Indolylacetamidonaltrexamine Derivatives as Bitopic Mu Opioid Receptor Modulators and Elaboration of the “Message-Address Concept” to Comprehend Their Functional Conversion. ACS Chem. Neurosci. 2018, 10, 1075–1090. [Google Scholar] [CrossRef] [PubMed]
- Campbell, A.; Wakelin, L.P.G.; Denny, W.A.; Finch, A.M. Homobivalent Conjugation Increases the Allosteric Effect of 9-aminoacridine at the α1-Adrenergic Receptors. Mol. Pharmacol. 2016, 91, 135–144. [Google Scholar] [CrossRef]
- Valant, C.; Lane, J.R.; Sexton, P.M.; Christopoulos, A. The Best of Both Worlds? Bitopic Orthosteric/Allosteric Ligands of G Protein–Coupled Receptors. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 153–178. [Google Scholar] [CrossRef] [PubMed]
- Neumeyer, J.L.; Zhang, A.; Xiong, W.; Gu, X.-H.; Hilbert, J.E.; Knapp, B.I.; Negus, S.S.; Mello, A.N.K.; Bidlack, J.M. Design and Synthesis of Novel Dimeric Morphinan Ligands for kappa and micro Opioid Receptors. J. Med. Chem. 2003, 46, 5162–5170. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Gao, M.; Zhang, Q.; Zhang, C.; Yang, X.; Huang, Z.; An, J. Design, synthesis, and biological characterization of novel PEG-linked dimeric modulators for CXCR4. Bioorganic Med. Chem. 2016, 24, 5393–5399. [Google Scholar] [CrossRef] [Green Version]
- Nomura, W.; Aikawa, H.; Taketomi, S.; Tanabe, M.; Mizuguchi, T.; Tamamura, H. Exploration of labeling by near infrared dyes of the polyproline linker for bivalent-type CXCR4 ligands. Bioorganic Med. Chem. 2015, 23, 6967–6973. [Google Scholar] [CrossRef]
- Fulton, B.S.; Knapp, B.L.; Bidlack, J.M.; Neumeyer, J.L. Effect of linker substitution on the binding of butorphan univalent and bivalent ligands to opioid receptors. Bioorganic Med. Chem. Lett. 2010, 20, 1507–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobrovnik, S.A. The influence of rigid or flexible linkage between two ligands on the effective affinity and avidity for reversible interactions with bivalent receptors. J. Mol. Recognit. 2007, 20, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Olson, K.M.; Traynor, J.R.; Alt, A. Allosteric Modulator Leads Hiding in Plain Site: Developing Peptide and Peptidomimetics as GPCR Allosteric Modulators. Front. Chem. 2021, 9, 671483. [Google Scholar] [CrossRef]
- Erez, M.; Takemori, A.E.; Portoghese, P.S. Narcotic antagonistic potency of bivalent ligands which contain β-naltrexamine. Evidence for bridging between proximal recognition sites. J. Med. Chem. 1982, 25, 847–849. [Google Scholar] [CrossRef] [PubMed]
- Vo, L.; Drummond, P.D. Effect of combined opioid receptor and α2-adrenoceptor blockade on anxiety and electrically evoked startle responses. J. Psychopharmacol. 2017, 31, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Vo, L.; Hood, S.; Drummond, P.D. Involvement of Opioid Receptors and α2-Adrenoceptors in Inhibitory Pain Modulation Processes: A Double-Blind Placebo-Controlled Crossover Study. J. Pain 2016, 17, 1164–1173. [Google Scholar] [CrossRef] [PubMed]
- Vauquelin, G. Simplified models for heterobivalent ligand binding: When are they applicable and which are the factors that affect their target residence time. Naunyn-Schmiedebergs Arch. Exp. Pathol. Pharmakol. 2013, 386, 949–962. [Google Scholar] [CrossRef] [PubMed]
- Vauquelin, G.; Charlton, S. Exploring avidity: Understanding the potential gains in functional affinity and target residence time of bivalent and heterobivalent ligands. J. Cereb. Blood Flow Metab. 2013, 168, 1771–1785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lezoualc’h, F.; Jockers, R. Bivalent Ligands as Specific Pharmacological Tools for G Protein-Coupled Receptor Dimers. Curr. Drug Discov. Technol. 2008, 5, 312–318. [Google Scholar] [CrossRef]
- Huang, B.; Onge, C.M.S.; Ma, H.; Zhang, Y. Design of bivalent ligands targeting putative GPCR dimers. Drug Discov. Today 2021, 26, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Arnatt, C.K.; Li, G.; Haney, K.M.; Ding, D.; Jacob, J.C.; Selley, D.E.; Zhang, Y. Design and synthesis of a bivalent ligand to explore the putative heterodimerization of the mu opioid receptor and the chemokine receptor CCR5. Org. Biomol. Chem. 2012, 10, 2633–2646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldhoer, M.; Fong, J.; Jones, R.M.; Lunzer, M.M.; Sharma, S.K.; Kostenis, E.; Portoghese, P.S.; Whistler, J.L. A heterodimer-selective agonist shows in vivo relevance of G protein-coupled receptor dimers. Proc. Natl. Acad. Sci. USA 2005, 102, 9050–9055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Bhushan, R.G.; Daniels, D.J.; Portoghese, P.S. Interaction of bivalent ligand KDN21 with heterodimeric δ-κopioid re-ceptors in human embryonic kidney 293 cells. Mol. Pharmacol. 2005, 68, 1079–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhushan, R.G.; Sharma, S.K.; Xie, Z.; Daniels, D.J.; Portoghese, P.S. A bivalent ligand (KDN-21) reveals spinal delta and kappa opioid receptors are organized as heterodimers that give rise to delta (1) and kappa (2) phenotypes. Selective targeting of del-ta-kappa heterodimers. J. Med. Chem. 2004, 47, 2969–2972. [Google Scholar] [CrossRef] [PubMed]
- Mahler, D.A.; Murray, J.A.; Waterman, L.A.; Ward, J.; Kraemer, W.J.; Zhang, X.; Baird, J.C. Endogenous opioids modify dyspnoea during treadmill exercise in patients with COPD. Eur. Respir. J. 2009, 33, 771–777. [Google Scholar] [CrossRef] [Green Version]
- Tataris, K.L.; Weber, J.M.; Stein-Spencer, L.; Aks, S.E. The effect of prehospital nebulized naloxone on suspected heroin-induced bronchospasm. Am. J. Emerg. Med. 2013, 31, 717–718. [Google Scholar] [CrossRef] [PubMed]
- Dillon, P.F.; Root-Bernstein, R.S.; Lieder, C.M. Antioxidant-independent ascorbate enhancement of catecholamine-induced contractions of vascular smooth muscle. Am. J. Physiol. Circ. Physiol. 2004, 286, H2353–H2360. [Google Scholar] [CrossRef] [PubMed]
- Dillon, P.F.; Root-Bernstein, R.; Robinson, N.E.; Abraham, W.M.; Berney, C. Receptor-Mediated Enhancement of Beta Adrenergic Drug Activity by Ascorbate In Vitro and In Vivo. PLoS ONE 2010, 5, e15130. [Google Scholar] [CrossRef] [PubMed] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Adrenergic Drug | Opioid Drug | Improved Anesthesia or Analgesia | Increases Anesthesia Duration | Opioid Sparing | Prevents Opioid Tolerance | Reverses Opioid Tolerance | No Benefits Reported |
|---|---|---|---|---|---|---|---|
| Epinephrine (α and β agonist) | Morphine (MOR) | [20,21,25,26,27,28,29] | [20,21,25,26,27,28,30,31] | [17,20,25,29, 36,39,40] | [36,37,38] | ||
| Epinephrine (α and β agonist) | Sufentanil (MOR) | [34] | |||||
| Noradrenaline (α1 and 2 agonist) | Morphine (MOR) | [14,58] | [14,58] | ||||
| Clonidine (α2 agonist) | Morphine (MOR) | [14,20,21,27,30,32,33,58] | [14,20,21,22,27,30,32,33,58] | [17,36,39,40, 58] | [36,38,42,43,58] | [42,43] | |
| Clonidine (α2 agonist) | Fentanyl (MOR) | [20,21,22,31,58] | [20,31,58] | ||||
| Clonidine (α2 agonist) | Meperidine (MOR, KOR) | [58] | [58] | ||||
| Clonidine (α2 agonist) | DAMGO (MOR) | [58] | [58] | ||||
| Clonidine (α2 agonist) | Sufentanil (MOR) | [34] | |||||
| Guanfacine (α2A agonist) | Morphine (MOR) | [35,36,37,38] | [35] | [61] | |||
| Guanfacine (α2A agonist) | DAMGO (MOR) | [61 | |||||
| Guanfacine (α2A agonist) | Deltorphin (DOR) | [61] | |||||
| Dexmedetomidine (α2 agonist) | Morphine (MOR) | [40,61] | [61] | [35,36,37,38,41,42,43] | [35,41,45] | ||
| Prazosin (α1 antagonist) | Morphine (MOR) | [50] | [42,43,50] | [43] | |||
| Phentolamine (α1 antagonist) | Morphine (MOR) | [52] | [52] | ||||
| BRL44408 (α2 antagonist) | Morphine (MOR) | [51] | [51] | [51] | |||
| Idazoxan (α2 antagonist) | Morphine (MOR) | [39] | [39,58] | [58] | |||
| Esmolol (β1 agonist) | Morphine (MOR) | [55,56] | [53,54,55,56,57] | [53,54,55,56,57] | |||
| Propranolol (β1 and 2 antagonist) | Morphine (MOR) | [52] | [52] |
| Opioid Ligand | Adrenergic | Effect on OPR | Effect on ADR | Effect on OPR-ADR Dimerization | Effect on OPR-OPR Dimerization | Effect on ADR-ADR Dimerization |
|---|---|---|---|---|---|---|
| Ligand | ||||||
| AGONIST | AGONIST | ENHANCED AGONIST | ENHANCED AGONIST | ENHANCED Dimerization | ENHANCED Dimerization | ENHANCED Dimerization |
| AGONIST | Antagonist | ENHANCED AGONIST | ENHANCED Antagonist | Blocked Dimerization? | ENHANCED Dimerization | Blocked Dimerization |
| Antagonist | AGONIST | ENHANCED Antagonist | ENHANCED AGONIST | Blocked Dimerization? | Blocked Dimerization | ENHANCED Dimerization |
| Antagonist | Antagonist | ENHANCED Antagonist | ENHANCED Antagonist | Blocked Dimerization | Blocked Dimerization | Blocked Dimerization |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Root-Bernstein, R. Biased, Bitopic, Opioid–Adrenergic Tethered Compounds May Improve Specificity, Lower Dosage and Enhance Agonist or Antagonist Function with Reduced Risk of Tolerance and Addiction. Pharmaceuticals 2022, 15, 214. https://doi.org/10.3390/ph15020214
Root-Bernstein R. Biased, Bitopic, Opioid–Adrenergic Tethered Compounds May Improve Specificity, Lower Dosage and Enhance Agonist or Antagonist Function with Reduced Risk of Tolerance and Addiction. Pharmaceuticals. 2022; 15(2):214. https://doi.org/10.3390/ph15020214
Chicago/Turabian StyleRoot-Bernstein, Robert. 2022. "Biased, Bitopic, Opioid–Adrenergic Tethered Compounds May Improve Specificity, Lower Dosage and Enhance Agonist or Antagonist Function with Reduced Risk of Tolerance and Addiction" Pharmaceuticals 15, no. 2: 214. https://doi.org/10.3390/ph15020214
APA StyleRoot-Bernstein, R. (2022). Biased, Bitopic, Opioid–Adrenergic Tethered Compounds May Improve Specificity, Lower Dosage and Enhance Agonist or Antagonist Function with Reduced Risk of Tolerance and Addiction. Pharmaceuticals, 15(2), 214. https://doi.org/10.3390/ph15020214