
The Transthyretin/Oleuropein Aglycone Complex: A New Tool against TTR Amyloidosis




Abstract
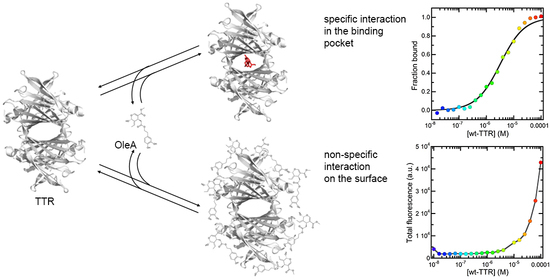
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Molecular Docking of Polyphenols to Native TTR
2.2. Excitation−Emission Matrices
2.3. Investigation of the Interaction between OleA and wt-TTR
2.4. Investigation of the Interaction between OleA and TTR Mutants
2.5. Biophysical Characterization of the Emission by OleA
2.6. Effects of Oleuropein Aglycone on TTR under Physiological Conditions
3. Discussion
4. Materials and Methods
4.1. Oleuropein Deglycosylation
4.2. Preparation of TTR Samples
4.3. Docking Experiments
4.4. Interaction between OleA and wt-TTR
4.5. Biophysical Characterization of OleA Emission
4.6. Intrinsic Fluorescence Measurements
4.7. Fluorimetric Binding Assays
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gião, T.; Saavedra, J.; Cotrina, E.; Quintana, J.; Llop, J.; Arsequell, G.; Cardoso, I. Undiscovered Roles for Transthyretin: From a Transporter Protein to a New Therapeutic Target for Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 2075. [Google Scholar] [CrossRef] [Green Version]
- Wojtczak, A.; Cody, V.; Luft, J.R.; Pangborn, W. Structures of human transthyretin complexed with thyroxine at 2.0 angstrom resolution and 3′,5′-dinitro-N-acetyl-l-thyronine at 2.2 angstrom resolution. Acta Crystallogr. D Biol. Crystallogr. 1996, 52, 758–765. [Google Scholar] [CrossRef] [PubMed]
- Cendron, L.; Trovato, A.; Seno, F.; Folli, C.; Alfieri, B.; Zanotti, G.; Berni, R. Amyloidogenic potential of transthyretin variants insights from structural and computational analyses. J. Biol. Chem. 2009, 284, 25832–25841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCutchen, S.L.; Lai, Z.; Miroy, G.J.; Kelly, J.W.; Colón, W. Comparison of lethal and nonlethal transthyretin variants and their relationship to amyloid disease. Biochemistry 1995, 34, 13527–13536. [Google Scholar] [CrossRef] [PubMed]
- Blevins, G.; Macaulay, R.; Harder, S.; Fladeland, D.; Yamashita, T.; Yazaki, M.; Hamidi, A.K.; Benson, M.D.; Donat, J.R. Oculoleptomeningeal amyloidosis in a large kindred with a new transthyretin variant Tyr69His. Neurology 2003, 60, 1625–1630. [Google Scholar] [CrossRef] [PubMed]
- Connors, L.H.; Lim, A.; Prokaeva, T.; Roskens, V.A.; Costello, C.E. Tabulation of human transthyretin (TTR) variants, 2003. Amyloid 2003, 10, 160–184. [Google Scholar] [CrossRef]
- Hagiwara, K.; Ochi, H.; Suzuki, S.; Shimizu, Y.; Tokuda, T.; Murai, H.; Shigeto, H.; Ohyagi, Y.; Iwata, M.; Iwaki, T.; et al. Highly selective leptomeningeal amyloidosis with transthyretin variant Ala25Thr. Neurology 2009, 72, 1358–1360. [Google Scholar] [CrossRef]
- Uemichi, T.; Uitti, R.J.; Koeppen, A.H.; Donat, J.R.; Benson, M.D. Oculoleptomeningeal amyloidosis associated with a new transthyretin variant Ser64. Arch. Neurol. 1999, 56, 1152–1155. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, I.; Almeida, M.R.; Ferreira, N.; Arsequell, G.; Valencia, G.; Saraiva, M.J. Comparative in vitro and ex vivo activities of selected inhibitors of transthyretin aggregation: Relevance in drug design. Biochem. J. 2007, 408, 131–138. [Google Scholar] [CrossRef] [Green Version]
- Quintas, A.; Vaz, D.C.; Cardoso, I.; Saraiva, M.J.; Brito, R.M. Tetramer dissociation and monomer partial unfolding precedes protofibril formation in amyloidogenic transthyretin variants. J. Biol. Chem. 2001, 276, 27207–27213. [Google Scholar] [CrossRef] [Green Version]
- Shnyrov, V.L.; Villar, E.; Zhadan, G.G.; Sanchez-Ruiz, J.M.; Quintas, A.; Saraiva, M.J.; Brito, R.M. Comparative calorimetric study of non-amyloidogenic and amyloidogenic variants of the homotetrameric protein transthyretin. Biophys. Chem. 2000, 88, 61–67. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Smith, C.S.; Petrassi, H.M.; Hammarstrom, P.; White, J.T.; Sacchettini, J.C.; Kelly, J.W. An engineered transthyretin monomer that is non amyloidogenic, unless it is partially denatured. Biochemistry 2001, 40, 11442–11452. [Google Scholar] [CrossRef] [PubMed]
- Westermark, P.; Johansson, B.; Natvig, J.B. Senile cardiac amyloidosis: Evidence of two different amyloid substances in the ageing heart. Scand. J. Immunol. 1979, 10, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Cornwell, G.G., 3rd; Murdoch, W.L.; Kyle, R.A.; Westermark, P.; Pitkänen, P. Frequency and distribution of senile cardiovascular amyloid. A clinicopathologic correlation. Am. J. Med. 1983, 75, 618–623. [Google Scholar] [CrossRef]
- Goren, H.; Steinberg, M.C.; Farboody, G.H. Familial oculoleptomeningeal amyloidosis. Brain 1980, 103, 473–495. [Google Scholar] [CrossRef]
- Keetch, C.A.; Bromley, E.H.; McCammon, M.G.; Wang, N.; Christodoulou, J.; Robinson, C.V. L55P transthyretin accelerates subunit exchange and leads to rapid formation of hybrid tetramers. J. Biol. Chem. 2005, 280, 41667–41674. [Google Scholar] [CrossRef] [Green Version]
- McCutchen, S.L.; Colon, W.; Kelly, J.W. Transthyretin mutation Leu-55-Pro significantly alters tetramer stability and increases amyloidogenicity. Biochemistry 1993, 32, 12119–12127. [Google Scholar] [CrossRef]
- Sebastião, M.P.; Saraiva, M.J.; Damas, A.M. The crystal structure of amyloidogenic Leu55 --> Pro transthyretin variant reveals a possible pathway for transthyretin polymerization into amyloid fibrils. J. Biol. Chem. 1998, 273, 24715–24722. [Google Scholar] [CrossRef] [Green Version]
- Obici, L.; Merlini, G. An overview of drugs currently under investigation for the treatment of transthyretin-related hereditary amyloidosis. Expert. Opin. Investig. Drugs 2014, 23, 1239–1251. [Google Scholar] [CrossRef]
- Berk, J.L.; Suhr, O.B.; Obici, L.; Sekijima, Y.; Zeldenrust, S.R.; Yamashit, T.; Heneghan, M.A.; Gorevic, P.D.; Litchy, W.J.; Wiesman, J.F.; et al. Repurposing diflunisal for familial amyloid polyneuropathy. A randomized clinical trial. JAMA 2013, 310, 2658–2667. [Google Scholar] [CrossRef] [Green Version]
- Coelho, T.; Maia, L.F.; da Silva, A.M.; Cruz, M.W.; Planté-Bordeneuve, V.; Suhr, O.B.; Conceiçao, I.; Schmidt, H.H.; Trigo, P.; Kelly, J.W.; et al. Long-term effects of tafamidis for the treatment of transthyretin familial amyloid polyneuropathy. J. Neurol. 2013, 260, 2802–2814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Judge, D.P.; Heitner, S.B.; Falk, R.H.; Maurer, M.S.; Shah, S.J.; Witteles, R.M.; Grogan, M.; Selby, V.N.; Jacoby, D.; Hanna, M.; et al. Transthyretin stabilization by AG10 in symptomatic transthyretin amyloid cardiomyopathy. J. Am. Coll. Cardiol. 2019, 74, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, J.P.; Hanna, M. Cardiac amyloidosis: An update on diagnosis and treatment. Cleve Clin. J. Med. 2017, 84, 12–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. New Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Trivella, D.B.; dos Reis, C.V.; Lima, L.M.; Foguel, D.; Polikarpov, I. Flavonoid interactions with human transthyretin: Combined structural and thermodynamic analysis. J. Struct. Biol. 2012, 180, 143–153. [Google Scholar] [CrossRef]
- Bourgault, S.; Choi, S.; Buxbaum, J.N.; Kelly, J.W.; Price, J.L.; Reixach, N. Mechanisms of transthyretin cardiomyocyte toxicity inhibition by resveratrol analogs. Biochem. Biophys. Res. Commun. 2011, 410, 707–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, N.; Goncalves, N.P.; Saraiva, M.J.; Almeida, M.R. Curcumin: A multi-target disease-modifying agent for late-stage transthyretin amyloidosis. Sci. Rep. 2016, 6, 26623. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, N.; Cardoso, I.; Domingues, M.R.; Vitorino, R.; Bastos, M.; Bai, G.; Saraiva, M.J.; Almeida, M.R. Binding of epigallocatechin-3-gallate to transthyretin modulates its amyloidogenicity. FEBS Lett. 2009, 583, 3569–3576. [Google Scholar] [CrossRef] [Green Version]
- Leri, M.; Nosi, D.; Natalello, A.; Porcari, R.; Ramazzotti, M.; Chiti, F.; Bellotti, V.; Doglia, S.M.; Stefani, M.; Bucciantini, M. The polyphenol Oleuropein aglycone hinders the growth of toxic transthyretin amyloid assemblies. J. Nutr. Biochem. 2016, 30, 153–166. [Google Scholar] [CrossRef]
- Ferreira, N.; Saraiva, M.J.; Almeida, M.R. Natural polyphenols inhibit different steps of the process of transthyretin (TTR) amyloid fibril formationn. FEBS Lett. 2011, 585, 2424–2430. [Google Scholar] [CrossRef] [Green Version]
- Corazza, A.; Verona, G.; Waudby, C.A.; Mangione, P.P.; Bingham, R.; Uings, I.; Canetti, D.; Nocerino, P.; Taylor, G.W.; Pepys, M.B.; et al. Binding of Monovalent and Bivalent Ligands by Transthyretin Causes Different Short- and Long-Distance Conformational Changes. J. Med. Chem. 2019, 62, 8274–8283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinho, E.; Grootveld, M.; Soares, G.; Henriques, M. Cyclodextrin-based hydrogels toward improved wound dressings. Crit. Rev. Biotechnol. 2014, 34, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Santoro, M.M.; Bolen, D.W. Unfolding free energy changes determined by the linear extrapolation method. 1. Unfolding of phenylmethanesulfonyl alpha-chymotrypsin using different denaturants. Biochemistry 1988, 27, 8063–8068. [Google Scholar] [CrossRef] [PubMed]
- Pawar, A.P.; Dubay, K.F.; Zurdo, J.; Chiti, F.; Vendruscolo, M.; Dobson, C.M. Prediction of "aggregation-prone" and "aggregation-susceptible" regions in proteins associated with neurodegenerative diseases. J. Mol. Biol. 2005, 350, 379–392. [Google Scholar] [CrossRef]
- Gazit, E. A possible role for pi-stacking in the self-assembly of amyloid fibrils. FASEB J. 2002, 16, 77–83. [Google Scholar] [CrossRef]
- Porat, Y.; Mazor, Y.; Efrat, S.; Gazit, E. Inhibition of islet amyloid polypeptide fibril formation: A potential role for heteroaromatic interactions. Biochemistry 2004, 43, 14454–14462. [Google Scholar] [CrossRef]
- Sekijima, Y.; Kelly, J.W.; Ikeda, S. Pathogenesis of and therapeutic strategies to ameliorate the transthyretin amyloidoses. Curr. Pharm. Des. 2008, 14, 3219–3230. [Google Scholar] [CrossRef]
- Ge, J.F.; Qiao, J.P.; Qi, C.C.; Wang, C.W.; Zhou, J.N. The binding of resveratrol to monomer and fibril amyloid beta. Neurochem. Int. 2012, 61, 1192–1201. [Google Scholar] [CrossRef]
- Yang, F.S.; Lim, G.P.; Begum, A.N.; Ubeda, O.J.; Simmons, M.R.; Ambegaokar, S.S.; Chen, P.P.; Kayed, R.; Glabe, C.G.; Frautschy, S.A.; et al. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J. Biol. Chem. 2005, 280, 5892–5901. [Google Scholar] [CrossRef] [Green Version]
- Florio, P.; Folli, C.; Cianci, M.; Del Rio, D.; Zanotti, G.; Berni, R. Transthyretin Binding Heterogeneity and Anti-amyloidogenic Activity of Natural Polyphenols and Their Metabolites. J. Biol. Chem. 2015, 290, 29769–29780. [Google Scholar] [CrossRef] [Green Version]
- Bezerra, F.; Saraiva, M.J.; Almeida, M.R. Modulation of the Mechanisms Driving Transthyretin Amyloidosis. Front Mol. Neurosci. 2020, 13, 592644. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, N.; Saraiva, M.J.; Almeida, M.R. Uncovering the Neuroprotective Mechanisms of Curcumin on Transthyretin Amyloidosis. Int. J. Mol. Sci. 2019, 20, 1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pullakhandam, R.; Srinivas, P.N.B.S.; Nair, M.K.; Reddy, G.B. Binding and stabilization of transthyretin by curcumin. Arch. Biochem. Biophys. 2009, 485, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.; Pal, A.; Albusairi, W.; Joo, H.; Pappas, B.; Haque Tuhin, M.T.; Liang, D.; Jampala, R.; Liu, F.; Khan, J.; et al. Enthalpy-Driven Stabilization of Transthyretin by AG10 Mimics a Naturally Occurring Genetic Variant That Protects from Transthyretin Amyloidosis. J. Med. Chem. 2018, 61, 7862–7876. [Google Scholar] [CrossRef]
- Rigacci, S.; Guidotti, V.; Bucciantini, M.; Parri, M.; Nediani, C.; Cerbai, E.; Stefani, M.; Berti, A. Oleuropein aglycon prevents cytotoxic amyloid aggregation of human amylin. J. Nutr. Biochem 2010, 21, 726–735. [Google Scholar] [CrossRef]
- Mangione, P.P.; Porcari, R.; Gillmore, J.D.; Pucci, P.; Monti, M.; Porcari, M.; Giorgetti, S.; Marchese, L.; Raimondi, S.; Serpell, L.C.; et al. Proteolytic cleavage of Ser52Pro variant transthyretin triggers its amyloid fibrillogenesis. Proc. Natl. Acad. Sci. USA 2014, 111, 1539–1544. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Morais-de-Sá, E.; Neto-Silva, R.M.; Pereira, P.J.; Saraiva, M.J.; Damas, A.M. The binding of 2,4-dinitrophenol to wild-type and amyloidogenic transthyretin. Acta Crystallogr. D Biol. Crystallogr. 2006, 62, 512–519. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15--Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Kumar, V.; Sharma, V.K.; Kalonia, D.S. Second derivative tryptophan fluorescence spectroscopy as a tool to characterize partially unfolded intermediates of proteins. Int. J. Pharm. 2005, 294, 193–199. [Google Scholar] [CrossRef]
- Frazier, R.A.; Papadopoulou, A.; Mueller-Harvey, I.; Kissoon, D.; Green, R.J. Probing protein-tannin interactions by isothermal titration microcalorimetry. J. Agric. Food Chem. 2003, 51, 5189–5195. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bemporad, F.; Leri, M.; Ramazzotti, M.; Stefani, M.; Bucciantini, M. The Transthyretin/Oleuropein Aglycone Complex: A New Tool against TTR Amyloidosis. Pharmaceuticals 2022, 15, 277. https://doi.org/10.3390/ph15030277
Bemporad F, Leri M, Ramazzotti M, Stefani M, Bucciantini M. The Transthyretin/Oleuropein Aglycone Complex: A New Tool against TTR Amyloidosis. Pharmaceuticals. 2022; 15(3):277. https://doi.org/10.3390/ph15030277
Chicago/Turabian StyleBemporad, Francesco, Manuela Leri, Matteo Ramazzotti, Massimo Stefani, and Monica Bucciantini. 2022. "The Transthyretin/Oleuropein Aglycone Complex: A New Tool against TTR Amyloidosis" Pharmaceuticals 15, no. 3: 277. https://doi.org/10.3390/ph15030277
APA StyleBemporad, F., Leri, M., Ramazzotti, M., Stefani, M., & Bucciantini, M. (2022). The Transthyretin/Oleuropein Aglycone Complex: A New Tool against TTR Amyloidosis. Pharmaceuticals, 15(3), 277. https://doi.org/10.3390/ph15030277