
Design and Synthesis of a Novel 4-aryl-N-(2-alkoxythieno [2,3-b]pyrazine-3-yl)-4-arylpiperazine-1-carboxamide DGG200064 Showed Therapeutic Effect on Colon Cancer through G2/M Arrest

 , , ,
, , ,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Cell-Based SRB Assay: Cytotoxicity and SAR Analysis
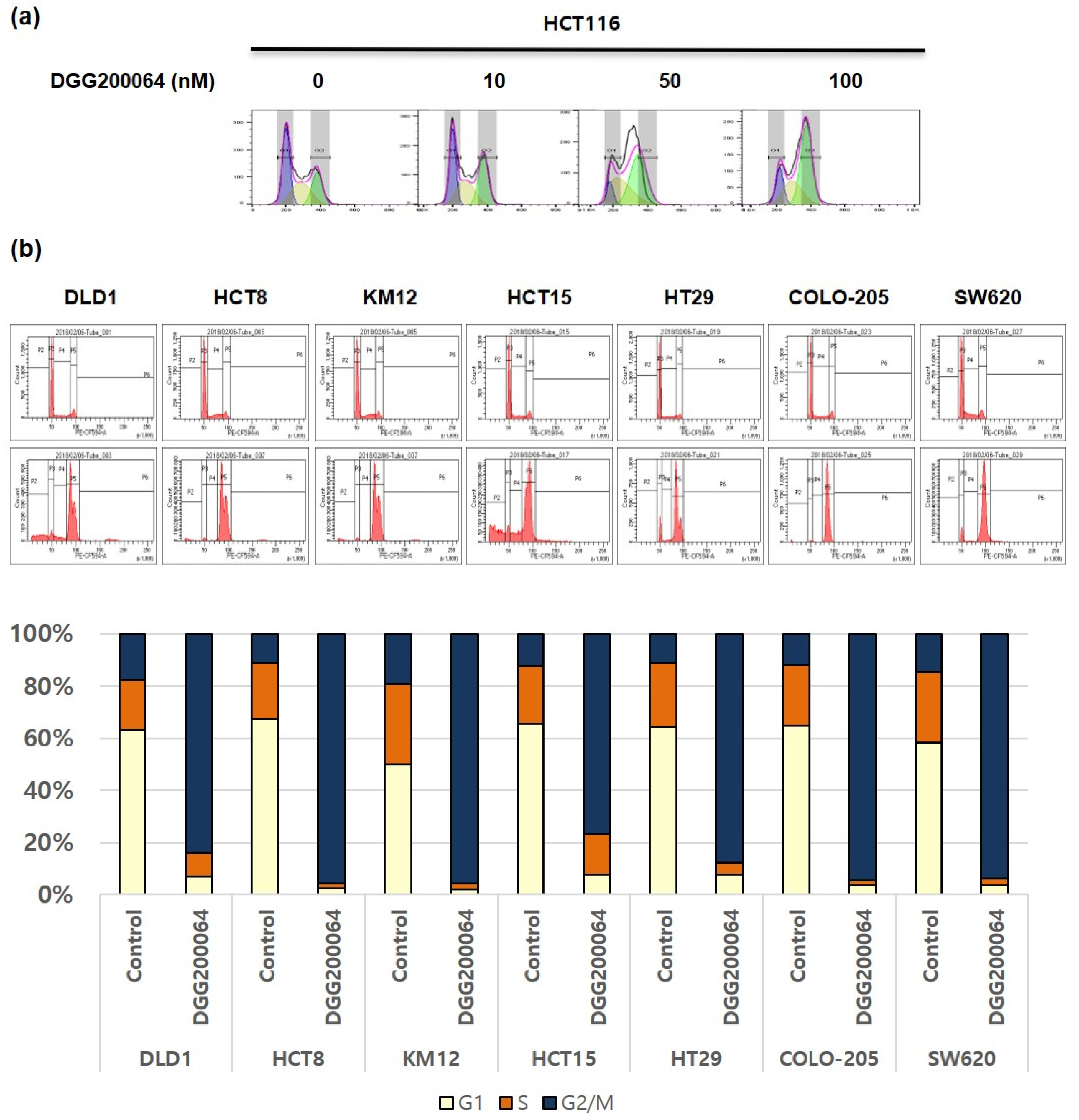
2.2. Induced G2/M Cell Cycle Arrest by DGG200064
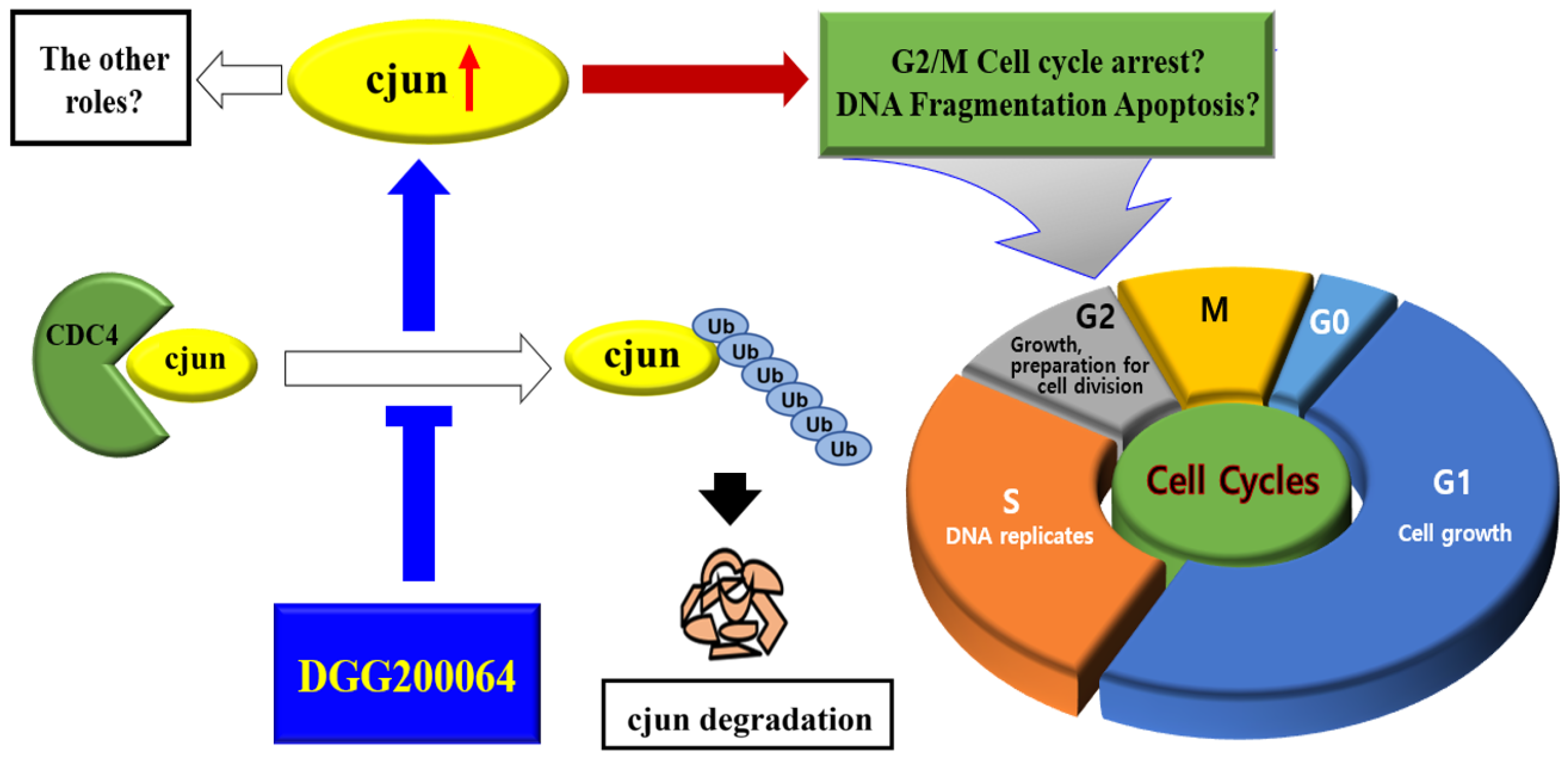
2.3. c-Jun Stabilization by DGG200064
2.4. Selective Inhibition of c-Jun Ubiquitination through Interruption of FBXW7/c-Jun Interaction
2.5. Identification of the Interaction Inhibition Site between FBXW7 and c-Jun by Docking Study
2.6. DGG200064 Treatment Abrogated CRC in Xenograft Models with an Increase in the c-Jun Level
2.7. Pharmacokinetic Analysis and Study
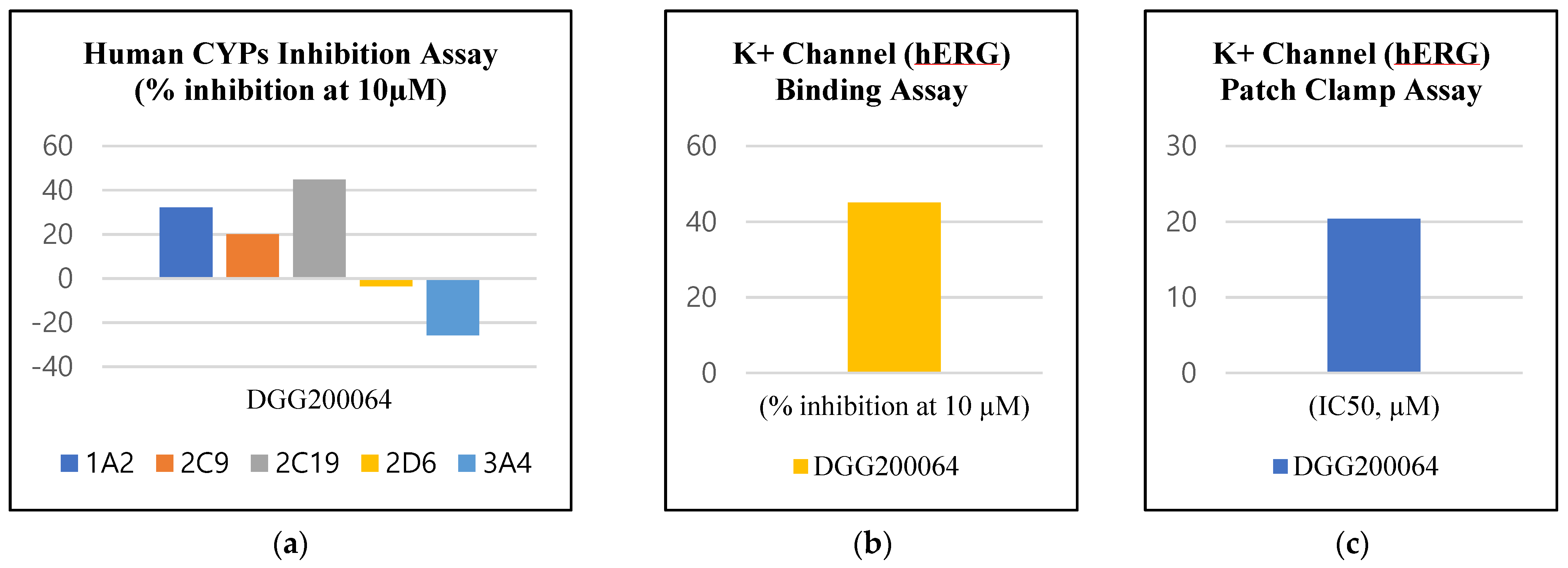
2.8. Evaluation of Physicochemical Properties
3. Materials and Methods
3.1. General Methods
3.2. Synthesis of 6-chloro-5-((trimethylsilyl)ethynyl)pyrazin-2-amine 2
3.3. Synthesis of thieno [2,3-b]pyrazin-3-amine 3
3.4. Synthesis of 2,6-dichlorothieno [2,3-b]pyrazin-3-amine 4
3.5. Synthesis of the Key Intermediates, 6-chloro-2-alkoxyoxythieno [2,3-b]pyrazin-3-amines 5a, 5b
3.6. Synthesis of 2-alkoxyoxythieno [2,3-b]pyrazin-3-amines 5c, 5d
3.7. Synthesis of diphenyl(6-substituted-2-alkoxythieno [3,2-b]pyrazin-3-yl)imino-dicarbonates 6a–6c
3.8. Synthesis of the Final Products, N-(6-substituted-2-alkoxythieno [2,3-b]pyrazin-3-yl)-4-(3,5-dimethoxyphenyl)piperazine-1-carboxamide 7a–7i
3.9. General Antibodies and Reagents
3.10. Cell Culture and siRNA
3.11. Sulforhodamine B (SRB) Assay
3.12. Western Blotting
3.13. Immunoprecipitation
3.14. Preclinical Xenograft Tumor Models
3.15. Immunohistochemistry (IHC)
3.16. Cell Cycle Distribution
3.17. Immunofluorescence Analysis
3.18. Statistical Analysis
3.19. Pharmacokinetic and Physicochemical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bester, A.C.; Roniger, M.; Oren, Y.S.; Im, M.M.; Sarni, D.; Chaoat, M.; Bensimon, A.; Zamir, G.; Shewach, D.S.; Kerem, B. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 2011, 145, 435–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, M.E.; Corsino, P.E.; Narayan, S.; Law, B.K. Cyclin-Dependent Kinase Inhibitors as Anticancer Therapeutics. Mol. Pharmacol. 2015, 88, 846–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobbelstein, M.; Sorensen, C.S. Exploiting replicative stress to treat cancer. Nat. Rev. Drug Discov. 2015, 14, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parry, D.; Guzi, T.; Shanahan, F.; Davis, N.; Prabhavalkar, D.; Wiswell, D.; Seghezzi, W.; Paruch, K.; Dwyer, M.P.; Doll, R.; et al. Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol. Cancer Ther. 2010, 9, 2344–2353. [Google Scholar] [CrossRef] [Green Version]
- Feldmann, G.; Mishra, A.; Bisht, S.; Karikari, C.; Garrido-Laguna, I.; Rasheed, Z.; Ottenhof, N.A.; Dadon, T.; Alvarez, H.; Fendrich, V.; et al. Cyclin-dependent kinase inhibitor Dinaciclib (SCH727965) inhibits pancreatic cancer growth and progression in murine xenograft models. Cancer Biol. Ther. 2014, 12, 598–609. [Google Scholar] [CrossRef] [Green Version]
- Lacrima, K.; Valentini, A.; Lambertini, C.; Taborelli, M.; Rinaldi, A.; Zucca, E.; Catapano, C.; Cavalli, F.; Gianella-Borradori, A.; Maccallum, D.E.; et al. In vitro activity of cyclin-dependent kinase inhibitor CYC202 (Seliciclib, R-roscovitine) in mantle cell lymphomas. Ann. Oncol. 2005, 16, 1169–1176. [Google Scholar] [CrossRef]
- Arguello, F.; Alexander, M.; Sterry, J.A.; Tudor, G.; Smith, E.M.; Kalavar, N.T.; Greene, J.F., Jr.; Koss, W.; Morgan, C.D.; Stinson, S.F.; et al. Flavopiridol induces apoptosis of normal lymphoid cells, causes immunosuppression, and has potent antitumor activity In vivo against human leukemia and lymphoma xenografts. Blood 1998, 91, 2482–2490. [Google Scholar]
- Gopalan, P.K.; Villegas, A.G.; Cao, C.; Pinder-Schenck, M.; Chiappori, A.; Hou, W.; Zajac-Kaye, M.; Ivey, A.M.; Kaye, F.J. CDK4/6 inhibition stabilizes disease in patients with p16-null non-small cell lung cancer and is synergistic with mTOR inhibition. Oncotarget 2018, 9, 37352–37366. [Google Scholar] [CrossRef] [Green Version]
- Vaughn, D.J.; Hwang, W.T.; Lal, P.; Rosen, M.A.; Gallagher, M.; O’Dwyer, P.J. Phase 2 trial of the cyclin-dependent kinase 4/6 inhibitor palbociclib in patients with retinoblastoma protein-expressing germ cell tumors. Cancer 2015, 121, 1463–1468. [Google Scholar] [CrossRef] [Green Version]
- Hart, L.S.; Rader, J.; Raman, P.; Batra, V.; Russell, M.R.; Tsang, M.; Gagliardi, M.; Chen, L.; Martinez, D.; Li, Y.; et al. Preclinical Therapeutic Synergy of MEK1/2 and CDK4/6 Inhibition in Neuroblastoma. Clin. Cancer Res. 2017, 23, 1785–1796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, C.; Diaz, H.B.; McNeely, S.; Barnard, D.; Dempsey, J.; Blosser, W.; Beckmann, R.; Barda, D.; Marshall, M.S. LY2606368 Causes Replication Catastrophe and Antitumor Effects through CHK1-Dependent Mechanisms. Mol. Cancer Ther. 2015, 14, 2004–2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leijen, S.; van Geel, R.M.; Sonke, G.S.; de Jong, D.; Rosenberg, E.H.; Marchetti, S.; Pluim, D.; van Werkhoven, E.; Rose, S.; Lee, M.A.; et al. Phase II Study of WEE1 Inhibitor AZD1775 Plus Carboplatin in Patients with TP53-Mutated Ovarian Cancer Refractory or Resistant to First-Line Therapy Within 3 Months. J. Clin. Oncol. 2016, 34, 4354–4361. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Zhang, Y.; Chen, S.; Kmieciak, M.; Leng, Y.; Lin, H.; Rizzo, K.A.; Dumur, C.I.; Ferreira-Gonzalez, A.; Dai, Y.; et al. A regimen combining the Wee1 inhibitor AZD1775 with HDAC inhibitors targets human acute myeloid leukemia cells harboring various genetic mutations. Leukemia 2015, 29, 807–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudolph, D.; Impagnatiello, M.A.; Blaukopf, C.; Sommer, C.; Gerlich, D.W.; Roth, M.; Tontsch-Grunt, U.; Wernitznig, A.; Savarese, F.; Hofmann, M.H.; et al. Efficacy and mechanism of action of volasertib, a potent and selective inhibitor of Polo-like kinases, in preclinical models of acute myeloid leukemia. J. Pharmacol. Exp. Ther. 2015, 352, 579–589. [Google Scholar] [CrossRef] [Green Version]
- Gorlick, R.; Kolb, E.A.; Keir, S.T.; Maris, J.M.; Reynolds, C.P.; Kang, M.H.; Carol, H.; Lock, R.; Billups, C.A.; Kurmasheva, R.T.; et al. Initial testing (stage 1) of the Polo-like kinase inhibitor volasertib (BI 6727), by the Pediatric Preclinical Testing Program. Pediatr. Blood Cancer 2014, 61, 158–164. [Google Scholar] [CrossRef] [Green Version]
- Manfredi, M.G.; Ecsedy, J.A.; Chakravarty, A.; Silverman, L.; Zhang, M.; Hoar, K.M.; Stroud, S.G.; Chen, W.; Shinde, V.; Huck, J.J.; et al. Characterization of Alisertib (MLN8237), an investigational small-molecule inhibitor of aurora A kinase using novel in vivo pharmacodynamic assays. Clin. Cancer Res. 2011, 17, 7614–7624. [Google Scholar] [CrossRef] [Green Version]
- Kelly, K.R.; Nawrocki, S.T.; Espitia, C.M.; Zhang, M.; Yang, J.J.; Padmanabhan, S.; Ecsedy, J.; Giles, F.J.; Carew, J.S. Targeting Aurora A kinase activity with the investigational agent alisertib increases the efficacy of cytarabine through a FOXO-dependent mechanism. Int. J. Cancer 2012, 131, 2693–2703. [Google Scholar] [CrossRef] [Green Version]
- DiPaola, R.S. To Arrest or Not to G2-M Cell-Cycle Arrest. Clin. Cancer Res. 2002, 8, 3311–3314. [Google Scholar]
- Tyagi, A.K.; Singh, R.P.; Agarwal, C.; Chan, D.C.; Agarwal, R. Silibinin strongly synergizes human prostate cancer DU145 cells to doxorubicin-induced growth inhibition, G2-M arrest, and apoptosis. Clin. Cancer Res. 2002, 8, 3512–3519. [Google Scholar]
- Cabrera, M.; Gomez, N.; Remes Lenicov, F.; Echeverría, E.; Shayo, C.; Moglioni, A.; Fernández, N.; Davio, C. G2/M Cell Cycle Arrest and Tumor Selective Apoptosis of Acute Leukemia Cells by a Promising Benzophenone Thiosemicarbazone Compound. PLoS ONE 2015, 10, e0136878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Zhang, B.; Gao, F.; Shi, R. Modulation of G2/M cell cycle arrest and apoptosis by luteolin in human colon cancer cells and xenografts. Oncol. Lett. 2018, 15, 1559–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Saini, N.; Parris, A.B.; Zhao, M.; Yang, X. Ganetespib induces G2/M cell cycle arrest and apoptosis in gastric cancer cells through targeting of receptor tyrosine kinase signaling. Int. J. Oncol. 2017, 51, 967–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, J.Y.; Choi, H.-S.; Seo, J.-S.; La, H.-J.; Yoo, S.-E.; Gong, Y.-D. Method for the Solid-Phase Parallel Synthesis of a 6-Alkylamino-2-(functionalized-aminomethyl)-2H-1-benzopyran Library. J. Comb. Chem. 2006, 8, 897–906. [Google Scholar] [CrossRef]
- Hwang, J.Y.; Gong, Y.-D. Solid-Phase Synthesis of the 2-Aminobenzoxazole Library Using Thioether Linkage as the Safety-Catch Linker. J. Comb. Chem. 2006, 8, 297–303. [Google Scholar] [CrossRef]
- Hwang, J.Y.; Choi, H.S.; Lee, D.H.; Yoo, S.-E.; Gong, Y.-D. Solid-Phase Synthesis of 5-Amino-1-(Substituted Thiocarbamoyl)pyrazole and 1,2,4-Triazole Derivatives via Dithiocarbazate Linker. J. Comb. Chem. 2005, 7, 136–141. [Google Scholar] [CrossRef]
- Hwang, J.Y.; Choi, H.-S.; Lee, D.H.; Gong, Y.-D. Solid-Phase Synthesis of 1,3,4-Oxadiazole and 1,3,4-Thiadiazole Derivatives via Selective, Reagent-Based Cyclization of Acyldithiocarbazate Resins. J. Comb. Chem. 2005, 7, 816–819. [Google Scholar] [CrossRef]
- Abdilidinova, A.; Yang, S.-J.; Gong, Y.-D. Solid-phase parallel synthesis of 1,3,4-oxadiazole based peptidomimetic library as a potential modulator of protein-protein interactions. Tetrahedron 2018, 74, 684–691. [Google Scholar] [CrossRef]
- Yang, S.-J.; Lee, S.-H.; Kwak, H.-J.; Gong, Y.-D. Regioselective Synthesis of 2-Amino-Substituted 1,3,4-Oxadiazole and 1,3,4-Thiadiazole Derivatives via Reagent-Based Cyclization of Thiosemicarbazide Intermediate. J. Org. Chem. 2013, 78, 438–444. [Google Scholar] [CrossRef]
- Yang, S.-J.; Choe, J.-H.; Gong, Y.-D. Solid-Phase Synthesis of 1,3,4-Thiadiazole Derivatives via Desulfurative Cyclization of Thiosemicarbazide Intermediate Resin. ACS Comb. Sci. 2016, 18, 499–506. [Google Scholar] [CrossRef]
- Ha, J.-E.; Yang, S.-J.; Gong, Y.-D. Construction of 1,3,4-Oxadiazole and 1,3,4-Thiadiazole Library with a High Level of Skeletal Diversity Based on Branching Diversity-Oriented Synthesis on Solid-Phase Supports. ACS Comb. Sci. 2018, 20, 82–97. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.; Park, J.-H.; Lee, D.-H.; Gong, Y.-D. Traceless Solid-Phase Synthesis of 2,4,6-Trisubstituted Thiazolo[4,5-d]pyrimidine-5,7-dione Derivatives. J. Comb. Chem. 2009, 11, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.; Park, J.-H.; Jeon, M.-K.; Gong, Y.-D. Solid-Phase Synthesis of 1,3,6-Trisubstituted-1H-thiazolo[4,5-c][1,2]thiazin-4(3H)one-2,2-dioxide Derivatives using Traceless Linker. J. Comb. Chem. 2009, 11, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.-D.; Lee, T.-H. Combinatorial Syntheses of Five-Membered Ring Heterocycles Using Carbon Disulfide and a Solid Support. J. Comb. Chem. 2010, 12, 393–409. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-J.; Kwon, H.-J.; Han, S.-Y.; Gong, Y.-D. Synthesis of 2-Amino-5-Carboxamide Thiazole Derivatives via Dehydrative Cyclization of Thiourea Intermediate Resin on Solid Phase. ACS Comb. Sci. 2019, 21, 380–388. [Google Scholar] [CrossRef]
- Kwon, H.-J.; Kim, Y.-J.; Han, S.-Y.; Gong, Y.-D. Design and Solid-Phase Parallel Synthesis of 2,4,5-Trisubstituted Thiazole Derivatives via Cyclization Reaction with a Carbamimidothioate Linker. ACS Comb. Sci. 2019, 21, 482–488. [Google Scholar]
- Lee, I.Y.; Kim, S.Y.; Lee, J.Y.; Yu, C.M.; Lee, D.H.; Gong, Y.-D. Solution-phase parallel synthesis of new 2H-pyrimido-[4,5-e][1,2,4]triazin-3-ylidenecyanamides. Tetrahedron Lett. 2004, 45, 9319–9322. [Google Scholar] [CrossRef]
- Yoon, H.-J.; Yang, S.-J.; Gong, Y.-D. Synthesis of 2-Alkoxy/Thioalkoxy Benzo[d]imidazoles and 2-Thione Benzo[d]imidazoles via a Phase-Based, Chemoselective Reaction. ACS Comb. Sci. 2017, 19, 738–747. [Google Scholar] [CrossRef]
- Ryu, H.-J.; Yang, S.-J.; Lee, J.-H.; Gong, Y.-D. Construction of Druglike 2-Amido Benzo[d]imidazole Analogues via Desulfurative Cyclization of Thiourea Intermediate Resin on Solid-Phase. ACS Comb. Sci. 2018, 20, 282–291. [Google Scholar] [CrossRef]
- Gong, Y.D.; Kwak, S.H.; Lee, E.S. Novel 4-(aryl)-N-(2-alkoxythieno[3,2-b]pyrazin-3-yl)-piperazine-1-carboxamide Derivative, and Antiproliferative Effect Thereof. U.S. Patent 15/533,076, 9 November 2017. [Google Scholar]
- Wisdom, R.; Johnson, R.S.; Moore, C. c-Jun regulates cell cycle progression and apoptosis by distinct mechanisms. EMBO J. 1999, 18, 188–197. [Google Scholar] [CrossRef] [Green Version]
- Hershko, A. Ubiquitin: Roles in protein modification and breakdown. Cell 1983, 34, 11–12. [Google Scholar] [CrossRef]
- Nakayama, K.I.; Nakayama, K. Ubiquitin ligases: Cell-cycle control and cancer. Nat. Rev. Cancer 2006, 6, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Hao, B.; Oehlmann, S.; Sowa, M.E.; Harper, J.W.; Pavletich, N.P. Structure of a Fbw7-Skp1-cyclin E complex: Multisite-phosphorylated substrate recognition by SCF ubiquitin ligases. Mol. Cell 2007, 26, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Takagi, T.; Ramachandran, C.; Bermejo, M.; Yamashita, S.; Yu, L.X.; Amidon, G.L. A Provisional Biopharmaceutical Classification of the Top 200 Oral Drug Products in the United States, Great Britain, Spain, and Japan. Mol. Pharm. 2006, 3, 631–643. [Google Scholar] [CrossRef]
- Hann, M.M.; Oprea, T.I. Pursuing the leadlikeness concept in pharmaceutical research. Curr. Opin. Chem. Biol. 2004, 8, 255–263. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| No | X | R1 | R3 | Yields (%) |
| 7a | Cl | Me | 3,5-dimethoxy | 66 |
| 7b | Cl | Me | 3,5-dimethyl | 63 |
| 7c | H | Me | 3,5-dimethoxy | 78 |
| 7d | H | Me | 3,5-dimethyl | 87 |
| 7e | H | Me | 3,5-fluoro | 95 |
| 7f | H | Me | 3-methoxy-5-methyl | 92 |
| 7g | H | Me | 3-fluoro-5-methoxy | 90 |
| 7h | H | Me | 3-fluoro-5-methyl | 95 |
| 7i | H | Et | 3,5-dimethoxy | 78 |
| No | HCT116 | HCT15 | Colo205 | KM12 | SW620 | HCT8 | HT29 |
|---|---|---|---|---|---|---|---|
| 7a | 344 | 484 | 173 | 273 | 427 | 761 | 408 |
| 7b | 244 | 437 | 900 | 244 | 273 | 900 | 342 |
| 7c | 12 | 26 | 24 | 8 | 27 | 54 | 17 |
| 7d | 33 | 38 | 40 | 32 | 38 | 59 | 26 |
| 7e | 307 | 507 | 287 | 426 | 288 | 269 | 672 |
| 7f | 1.9 | 36 | 28 | 21 | 34 | 37 | 21 |
| 7g | 45 | 30 | 14 | 22 | 29 | 39 | 57 |
| 7h | 32 | 42 | 28 | 38 | 40 | 51 | 29 |
| 7i | 71 | 68 | 35 | 74 | 53 | 58 | 35 |
| Species | SD Rats | ICR Mice | ||
|---|---|---|---|---|
| Dosing Route | IV | PO | IV | PO |
| Dose (mg/kg) | 1 | 5 | 5 | 5 |
| Tmax (h) | 0.083 | 4.05 | - | 0.667 |
| Cmax (μg/mL) | 0.551 | 0.076 | - | 0.798 |
| T1/2 (h) | 1.11 | 7.496 | 2.98 | 5.2 |
| AUClast (μg·h/m) | 0.703 | 0.707 | 16.4 | 2.26 |
| AUCinf (μg·h/m) | 0.708 | 0.964 | 16.6 | 2.34 |
| CL (mL/h/k) | 1,512 | - | 305 | - |
| Vss (mL/kg) | 2,308 | - | 1.310 | - |
| F (%) | - | 20.11 | - | 13.8 |
| Properties | 7c | |||
|---|---|---|---|---|
| pKa | 2.39, 11.60 | |||
| logP | 2.63 | |||
| Permeability | Pe (10−6 cm/s) | 29.49 | ||
| logPe | −4.53 | |||
| Class | High | |||
| Solubility (logS) | pH | pH2.0 | pH7.4 | pH9.0 |
| Equilibrium | −2.68 | −3.22 | −3.21 | |
| Kinetic | −3.13 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, E.-S.; Kim, N.; Kang, J.H.; Abdildinova, A.; Lee, S.-H.; Lee, M.H.; Kang, N.S.; Koo, T.-S.; Kim, S.-Y.; Gong, Y.-D. Design and Synthesis of a Novel 4-aryl-N-(2-alkoxythieno [2,3-b]pyrazine-3-yl)-4-arylpiperazine-1-carboxamide DGG200064 Showed Therapeutic Effect on Colon Cancer through G2/M Arrest. Pharmaceuticals 2022, 15, 502. https://doi.org/10.3390/ph15050502
Lee E-S, Kim N, Kang JH, Abdildinova A, Lee S-H, Lee MH, Kang NS, Koo T-S, Kim S-Y, Gong Y-D. Design and Synthesis of a Novel 4-aryl-N-(2-alkoxythieno [2,3-b]pyrazine-3-yl)-4-arylpiperazine-1-carboxamide DGG200064 Showed Therapeutic Effect on Colon Cancer through G2/M Arrest. Pharmaceuticals. 2022; 15(5):502. https://doi.org/10.3390/ph15050502
Chicago/Turabian StyleLee, Eun-Sil, Nayeon Kim, Joon Hee Kang, Aizhan Abdildinova, Seon-Hyeong Lee, Myung Hwi Lee, Nam Sook Kang, Tae-Sung Koo, Soo-Youl Kim, and Young-Dae Gong. 2022. "Design and Synthesis of a Novel 4-aryl-N-(2-alkoxythieno [2,3-b]pyrazine-3-yl)-4-arylpiperazine-1-carboxamide DGG200064 Showed Therapeutic Effect on Colon Cancer through G2/M Arrest" Pharmaceuticals 15, no. 5: 502. https://doi.org/10.3390/ph15050502
APA StyleLee, E. -S., Kim, N., Kang, J. H., Abdildinova, A., Lee, S. -H., Lee, M. H., Kang, N. S., Koo, T. -S., Kim, S. -Y., & Gong, Y. -D. (2022). Design and Synthesis of a Novel 4-aryl-N-(2-alkoxythieno [2,3-b]pyrazine-3-yl)-4-arylpiperazine-1-carboxamide DGG200064 Showed Therapeutic Effect on Colon Cancer through G2/M Arrest. Pharmaceuticals, 15(5), 502. https://doi.org/10.3390/ph15050502