
A Mini Review on Isatin, an Anticancer Scaffold with Potential Activities against Neglected Tropical Diseases (NTDs)


Abstract
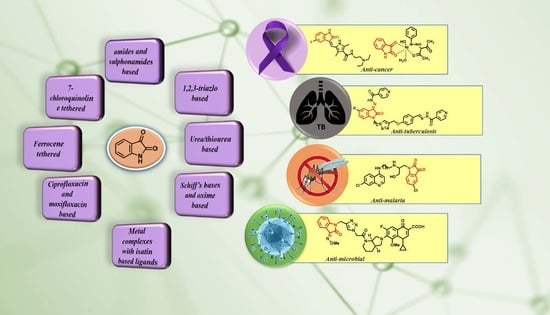
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Anticancer Activities of Functionalized Isatins
2.1. Isatin-Based Amides and Sulphonamides
2.2. 1H-1,2,3-Triazole-Tethered Isatin Hybrids
2.3. Spiro Compounds Based on Isatins
2.4. Urea/Thiourea-Based Isatin-Derivatives
2.5. Isatin-Based Schiff’s Bases and Oximes
2.6. Metal Complexes of Isatin Containing Ligands
2.7. Miscellaneous Compounds Containing Isatin Core
3. Antimycobacterial/Tubercular Activities of Isatin-Based Scaffolds
3.1. Ciprofloxacin-Isatin and Moxifloxacin-Isatin Hybrids
3.2. Spiro-Isatin Derivatives
3.3. Schiff’s Bases and Oximes of Functionalized Isatins
3.4. Ferrocene-Tethered Isatin Hybrids
3.5. Miscellaneous Isatin Derivatives with Antimycobacterial Potential
4. Antiplasmodial/Malarial Activities of Isatin-Based Scaffolds
4.1. Isatin-7-Chloroquinoline Hybrids
4.2. Isatin-Based Schiff’s Bases
4.3. Miscellaneous Antiplasmodial Isatin Derivatives
5. Antimicrobial Activities of Isatin-Based Scaffolds
5.1. Isatin-Schiff’s Bases/Oximes
5.2. Isatin-Ciprofloxacin/Isatin-Moxifloxacin Hybrids
5.3. Miscellaneous Isatin Scaffolds with Antimicrobial Activities
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Hotez, P.J.; Aksoy, S.; Brindley, P.J. What constitutes a neglected tropical disease? PLoS Negl. Trop. Dis. 2010, 14, e0008001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization (WHO). Neglected Tropical Diseases. Available online: https://www.who.int/news-room/q-a-detail/neglected-tropical-diseases (accessed on 24 February 2021).
- DNDi, Drugs for Neglected Disease Initiative, Drugs Neglected Dis. Initiat. 2021. Available online: https://dndi.org/ (accessed on 24 February 2021).
- World Health Organization. Working to Overcome the Global Impact of Neglected Tropical Diseases: First WHO Report on Neglected Tropical Diseases; World Health Organization: Geneva, Switzerland, 2010; Volume 1. [Google Scholar]
- Esmaeelian, B.; Abbott, C.A.; Le Leu, R.K.; Benkendorff, K. 6-Bromoisatin found in muricid mollusc extracts inhibits colon cancer cell proliferation and induces apoptosis, preventing early-stage tumor formation in a colorectal cancer rodent model. Mar. Drugs. 2014, 12, 17–35. [Google Scholar] [CrossRef] [PubMed]
- Varuna, S.R.K. Isatin and its derivatives: A survey of recent syntheses, reactions, and applications. Med. Chem. Commun. 2019, 10, 351–368. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.; Singh, N.P.; Kumar, K. Recent Advancement of Synthesis of Isatins as a Versatile Pharmacophore: A review. Drug Res. 2021, 71, 115–121. [Google Scholar]
- Kaur, M.; Singh, M.; Chadha, N.; Silakari, O. Oxindole: A chemical prism carrying plethora of therapeutic benefits. Eur. J. Med. Chem. 2016, 123, 858–894. [Google Scholar] [CrossRef]
- Zhang, Y.Z.; Du, H.Z.; Liu, H.L.; He, Q.S.; Xu, Z. Isatin dimers and their biological activities. Arch. Pharm. 2020, 353, 1900299. [Google Scholar] [CrossRef]
- International Agency for Research on, All Cancers Factsheet. World Health Organ. 2020, 419, 199–200. Available online: https://gco.iarc.fr/today/data/factsheets/cancers/39-All-cancers-fact-sheet.pdf (accessed on 24 February 2022).
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries, CA. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Enoque, R.; De Paiva, F.; Vieira, E.G.; Rodrigues, D. Anticancer Compounds Based on Isatin-Derivatives: Strategies to Ameliorate Selectivity and Efficiency. Front. Mol. Biosci. 2021, 7, 627272. [Google Scholar]
- Gao, S.; Zang, J.; Gao, Q.; Liang, X.; Ding, Q.; Li, X.; Xu, W.; Chou, C.J.; Zhang, Y. Design, synthesis and anti-tumor activity study of novel histone deacetylase inhibitors containing isatin-based caps and o-phenylenediamine-based zinc binding groups. Bioorg. Med. Chem. 2017, 25, 2981–2994. [Google Scholar] [CrossRef]
- Abo-Ashour, M.F.; Eldehna, W.M.; Nocentini, A.; Ibrahim, H.S.; Bua, S.; Abou-Seri, S.M.; Supuran, C.T. Novel hydrazido benzenesulfonamides-isatin conjugates: Synthesis, carbonic anhydrase inhibitory activity and molecular modeling studies. Eur. J. Med. Chem. 2018, 157, 28–36. [Google Scholar] [CrossRef]
- Abo-Ashour, M.F.; Eldehna, W.M.; Nocentini, A.; Bonardi, A.; Bua, S.; Ibrahim, H.S.; Elaasser, M.M.; Kryštof, V.; Jorda, R.; Gratteri, P.; et al. 3-Hydrazinoisatin-based benzenesulfonamides as novel carbonic anhydrase inhibitors endowed with anticancer activity: Synthesis, in vitro biological evaluation and in silico insights. Eur. J. Med. Chem. 2019, 184, 111768. [Google Scholar] [CrossRef]
- Eldehna, W.M.; Fares, M.; Ceruso, M.; Ghabbour, H.A.; Abou-Seri, S.M.; Abdel-Aziz, H.A.; Abou El Ella, D.A.; Supuran, C.T. Amido/ureidosubstituted benzenesulfonamides-isatin conjugates as low nanomolar/subnanomolar inhibitors of the tumor-associated carbonic anhydrase isoform XII. Eur. J. Med. Chem. 2016, 110, 259–266. [Google Scholar] [CrossRef]
- Eldehna, W.M.; Al-Ansary, G.H.; Bua, S.; Nocentini, A.; Gratteri, P.; Altoukhy, A.; Ghabbour, H.; Ahmed, H.Y.; Supuran, C.T. Novel indolin-2-one-based sulfonamides as carbonic anhydrase inhibitors: Synthesis, in vitro biological evaluation against carbonic anhydrases isoforms I, II, IV and VII and molecular docking studies. Eur. J. Med. Chem. 2017, 127, 521–530. [Google Scholar] [CrossRef]
- Eldehna, W.M.; Nocentini, A.; Al-Rashood, S.T.; Hassan, G.S.; Alkahtani, H.M.; Almehizia, A.A.; Reda, A.M.; Abdel-Aziz, H.A.; Supuran, C.T. Tumor-associated carbonic anhydrase isoform IX and XII inhibitory properties of certain isatin-bearing sulfonamides endowed with in vitro antitumor activity towards colon cancer. Bioorg. Chem. 2018, 81, 425–432. [Google Scholar] [CrossRef]
- Eldehna, W.M.; Abo-Ashour, M.F.; Nocentini, A.; El-Haggar, R.S.; Bua, S.; Bonardi, A.; Al-Rashood, S.T.; Hassan, G.S.; Gratteri, P.; Abdel-Aziz, H.A.; et al. Enhancement of the tail hydrophobic interactions within the carbonic anhydrase IX active site via structural extension: Design and synthesis of novel N-substituted isatins-SLC-0111 hybrids as carbonic anhydrase inhibitors and antitumor agents. Eur. J. Med. Chem. 2019, 162, 147–160. [Google Scholar] [CrossRef]
- Wang, J.; Yun, D.; Yao, J.; Fu, W.; Huang, F.; Chen, L.; Wei, T.; Yu, C.; Xu, H.; Zhou, X.; et al. Design, synthesis and QSAR study of novel isatin analogues inspired Michael acceptor as potential anticancer compounds. Eur. J. Med. Chem. 2018, 144, 493–503. [Google Scholar] [CrossRef]
- Yu, S.; Liu, Y.; Zhang, Z.; Zhang, J.; Zhao, G. Design, synthesis and biological evaluation of novel 2,3-indolinedione derivatives against mantle cell lymphoma. Bioorg. Med. Chem. 2019, 27, 3319–3327. [Google Scholar] [CrossRef]
- George, R.F.; Bua, S.; Supuran, C.T.; Awadallah, F.M. Synthesis of some N-aroyl-2-oxindole benzenesulfonamide conjugates with carbonic anhydrase inhibitory activity. Bioorg. Chem. 2020, 96, 103635. [Google Scholar] [CrossRef]
- Panga, S.; Podila, N.K.; Ciddi, V. Design, Synthesis, Characterization, and In Vitro Evaluation of Isatin-Pomalidomide Hybrids for Cytotoxicity against Multiple Myeloma Cell Lines. J. Heterocycl. Chem. 2018, 55, 2919–2928. [Google Scholar] [CrossRef]
- Eldehna, W.M.; El Hassab, M.A.; Abo-Ashour, M.F.; Al-Warhi, T.; Elaasser, M.M.; Safwat, N.A.; Suliman, H.; Ahmed, M.F.; Al-Rashood, S.T.; Abdel-Aziz, H.A.; et al. Development of isatin-thiazolo[3,2-a]benzimidazole hybrids as novel CDK2 inhibitors with potent in vitro apoptotic anti-proliferative activity: Synthesis, biological and molecular dynamics investigations. Bioorg. Chem. 2021, 110, 104748. [Google Scholar] [CrossRef]
- Yu, B.; Wang, S.Q.; Qi, P.P.; Yang, D.X.; Tang, K.; Liu, H.M. Design and synthesis of isatin/triazole conjugates that induce apoptosis and inhibit migration of MGC-803 cells. Eur. J. Med. Chem. 2016, 124, 350–360. [Google Scholar] [CrossRef]
- Nagarsenkar, A.; Guntuku, L.; Guggilapu, S.D.; Danthi Bai, K.; Gannoju, S.; Naidu, V.G.M.; Bathini, N.B. Synthesis and apoptosis inducing studies of triazole linked 3-benzylidene isatin derivatives. Eur. J. Med. Chem. 2016, 124, 782–793. [Google Scholar] [CrossRef]
- Kumar, S.; Gu, L.; Palma, G.; Kaur, M.; Singh-Pillay, A.; Singh, P.; Kumar, V. Design, synthesis, anti-proliferative evaluation and docking studies of 1: H-1,2,3-triazole tethered ospemifene-isatin conjugates as selective estrogen receptor modulators. New J. Chem. 2018, 42, 3703–3713. [Google Scholar] [CrossRef]
- Aneja, B.; Khan, N.S.; Khan, P.; Queen, A.; Hussain, A.; Rehman, M.T.; Alajmi, M.F.; El-Seedi, H.R.; Ali, S.; Hassan, M.I.; et al. Design and development of Isatin-triazole hydrazones as potential inhibitors of microtubule affinity-regulating kinase 4 for the therapeutic management of cell proliferation and metastasis. Eur. J. Med. Chem. 2019, 163, 840–852. [Google Scholar] [CrossRef]
- Yu, B.; Qi, P.P.; Shi, X.J.; Huang, R.; Guo, H.; Zheng, Y.C.; Yu, D.Q.; Liu, H.M. Efficient synthesis of new antiproliferative steroidal hybrids using the molecular hybridization approach. Eur. J. Med. Chem. 2016, 117, 241–255. [Google Scholar] [CrossRef]
- Singh, P.; Sharma, P.; Anand, A.; Bedi, P.M.S.; Kaur, T.; Saxena, A.K.; Kumar, V. Azide-alkyne cycloaddition en route to novel 1H-1,2,3-triazole tethered isatin conjugates with in vitro cytotoxic evaluation. Eur. J. Med. Chem. 2012, 55, 455–461. [Google Scholar] [CrossRef]
- Sharma, M.; Sharma, S.; Buddhiraja, A.; Saxena, A.K.; Nepali, K.; Bedi, P.M.S. Synthesis and cytotoxicity studies of 3,5-diaryl N-acetyl pyrazoline-Isatin hybrids. Med. Chem. Res. 2014, 23, 4337–4344. [Google Scholar] [CrossRef]
- Sharma, S.; Gupta, M.K.; Saxena, A.K.; Bedi, P.M.S. Triazole linked mono carbonyl curcumin-isatin bifunctional hybrids as novel anti tubulin agents: Design, synthesis, biological evaluation and molecular modeling studies. Bioorg. Med. Chem. 2015, 23, 7165–7180. [Google Scholar] [CrossRef]
- Kamal, A.; Mahesh, R.; Nayak, V.L.; Babu, K.S.; Kumar, G.B.; Shaik, A.B.; Kapure, J.S.; Alarifi, A. Discovery of pyrrolospirooxindole derivatives as novel cyclin dependent kinase 4 (CDK4) inhibitors by catalyst-free, green approach. Eur. J. Med. Chem. 2016, 108, 476–485. [Google Scholar] [CrossRef]
- Islam, M.S.; Ghawas, H.M.; El-Senduny, F.F.; Al-Majid, A.M.; Elshaier, Y.A.M.M.; Badria, F.A.; Barakat, A. Synthesis of new thiazolo-pyrrolidine–(spirooxindole) tethered to 3-acylindole as anticancer agents. Bioorg. Chem. 2019, 82, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Lotfy, G.; Said, M.M.; El Ashry, E.S.H.; El Tamany, E.S.H.; Al-Dhfyan, A.; Abdel Aziz, Y.M.; Barakat, A. Synthesis of new spirooxindole-pyrrolothiazole derivatives: Anti-cancer activity and molecular docking. Bioorg. Med. Chem. 2017, 25, 1514–1523. [Google Scholar] [CrossRef] [PubMed]
- Nunes, R.C.; Ribeiro, C.J.A.; Monteiro, Â.; Rodrigues, C.M.P.; Amaral, J.D.; Santos, M.M.M. In vitro targeting of colon cancer cells using spiropyrazoline oxindoles. Eur. J. Med. Chem. 2017, 139, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Senwar, K.R.; Sharma, P.; Reddy, T.S.; Jeengar, M.K.; Nayak, V.L.; Naidu, V.G.M.; Kamal, A.; Shankaraiah, N. Spirooxindole-derived morpholine-fused-1,2,3-triazoles: Design, synthesis, cytotoxicity and apoptosis inducing studies. Eur. J. Med. Chem. 2015, 102, 413–424. [Google Scholar] [CrossRef]
- Eldehna, W.M.; El Kerdawy, A.M.; Al-Ansary, G.H.; Al-Rashood, S.T.; Ali, M.M.; Mahmoud, A.E. Type IIA-Type IIB protein tyrosine kinase inhibitors hybridization as an efficient approach for potent multikinase inhibitor development: Design, synthesis, anti-proliferative activity, multikinase inhibitory activity and molecular modeling of novel indolinone-based ureides and amides. Eur. J. Med. Chem. 2019, 163, 37–53. [Google Scholar]
- Eldehna, W.M.; Fares, M.; Ibrahim, H.S.; Alsherbiny, M.A.; Aly, M.H.; Ghabbour, H.A.; Abdel-Aziz, H.A. Synthesis and cytotoxic activity of biphenylurea derivatives containing indolin-2-one moieties. Molecules 2016, 21, 762. [Google Scholar] [CrossRef] [Green Version]
- Gabr, M.T.; El-Gohary, N.S.; El-Bendary, E.R.; El-Kerdawy, M.M.; Ni, N. Isatin-β-thiocarbohydrazones: Microwave-assisted synthesis, antitumor activity and structure-activity relationship. Eur. J. Med. Chem. 2017, 128, 36–44. [Google Scholar] [CrossRef]
- Althagafi, I.I.; Abouzied, A.S.; Farghaly, T.A.; Al-Qurashi, N.T.; Alfaifi, M.Y.; Shaaban, M.R.; Abdel Aziz, M.R. Novel Nano-sized bis-indoline Derivatives as Antitumor Agents. J. Heterocycl. Chem. 2019, 56, 391–399. [Google Scholar] [CrossRef]
- Jeong, P.; Moon, Y.; Lee, J.H.; Lee, S.D.; Park, J.; Lee, J.; Kim, J.; Lee, H.J.; Kim, N.Y.; Choi, J.; et al. Discovery of orally active indirubin-3′-oxime derivatives as potent type 1 FLT3 inhibitors for acute myeloid leukemia. Eur. J. Med. Chem. 2020, 195, 112205. [Google Scholar] [CrossRef]
- Zayed, M.F.; Ahmed, S.; Ihmaid, S.; Ahmed, H.E.A.; Rateb, H.S.; Ibrahim, S.R.M. Design, synthesis, cytotoxic evaluation and molecular docking of new fluoroquinazolinones as potent anticancer agents with dual EGFR kinase and tubulin polymerization inhibitory effects. Int. J. Mol. Sci. 2018, 19, 1731. [Google Scholar] [CrossRef] [Green Version]
- Nam, N.H.; Huong, T.L.; Mai Dung, D.T.; Phuong Dung, P.T.; Kim, D.T.; Oanh Quyen, D.; Thao, L.T.; Park, S.H.; Kim, K.R.; Han, B.W.; et al. Novel isatin-based hydroxamic acids as histone deacetylase inhibitors and antitumor agents. Eur. J. Med. Chem. 2013, 70, 477–486. [Google Scholar] [CrossRef]
- Dweedar, H.E.; Mahrous, H.; Ibrahim, H.S.; Abdel-Aziz, H.A. Analogue-based design, synthesis and biological evaluation of 3-substituted-(methylenehydrazono)indolin-2-ones as anticancer agents. Eur. J. Med. Chem. 2014, 78, 275–280. [Google Scholar] [CrossRef]
- Liang, C.; Xia, J.; Lei, D.; Li, X.; Yao, Q.; Gao, J. Synthesis, in vitro and in vivo antitumor activity of symmetrical bis-Schiff base derivatives of isatin. Eur. J. Med. Chem. 2014, 74, 742–750. [Google Scholar] [CrossRef]
- Ali, A.Q.; Teoh, S.G.; Eltayeb, N.E.; Khadeer Ahamed, M.B.; Abdul Majid, A.M.S. Synthesis of copper(II) complexes of isatin thiosemicarbazone derivatives: In vitro anti-cancer, DNA binding, and cleavage activities. Polyhedron 2014, 74, 6–15. [Google Scholar] [CrossRef]
- Aneesrahman, K.N.; Ramaiah, K.; Rohini, G.; Stefy, G.P.; Bhuvanesh, N.S.P.; Sreekanth, A. Synthesis and characterisations of copper (II) complexes of 5-methoxyisatin thiosemicarbazones: Effect of N-terminal substitution on DNA/protein binding and biological activities. Inorg. Chim. Acta. 2019, 492, 131–141. [Google Scholar] [CrossRef]
- Balachandran, C.; Haribabu, J.; Jeyalakshmi, K. Nickel (II) bis (isatin thiosemicarbazone) complexes induced apoptosis through mitochondrial signaling pathway and G0/G1 cell cycle arrest in. J. Inorg. Biochem. 2018, 182, 208–221. [Google Scholar] [CrossRef]
- Hunoor, R.S.; Patil, B.R.; Badiger, D.S.; Chandrashekhar, V.M.; Muchchandi, I.S.; Gudasi, K.B. Co (II), Ni (II), Cu (II) and Zn (II) complexes of isatinyl-2-aminobenzoylhydrazone: Synthesis, characterization and anticancer activity. Appl. Organomet. Chem. 2014, 29, 101–108. [Google Scholar] [CrossRef]
- Haribabu, J.; Alajrawy, O.I.; Jeyalakshmi, K.; Balachandran, C. N-substitution in isatin thiosemicarbazones decides nuclearity of Cu (II) complexes–Spectroscopic, molecular docking and cytotoxic studies. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2021, 246, 118963. [Google Scholar] [CrossRef]
- Haribabu, J.; Jeyalakshmi, K.; Arun, Y.; Bhuvanesh, N.S.P.; Perumal, P.T.; Karvembu, R. Synthesis, DNA/protein binding, molecular docking, DNA cleavage and in vitro anticancer activity of nickel(II) bis(thiosemicarbazone) complexes. RSC Adv. 2015, 5, 46031–46049. [Google Scholar] [CrossRef]
- Kandile, N.G.; Mohamed, M.I.; Ismaeel, H.M.; Kandile, N.G.; Mohamed, M.I.; Ismaeel, H.M. Antiproliferative effects of metal complexes of new isatin hydrazones against HCT116, MCF7 and HELA tumour cell lines. J. Enzym. Inhib. Med. Chem. 2012, 27, 330–338. [Google Scholar] [CrossRef]
- Youssef, H.M.; Abdulhamed, Y.K.; El-Reash, G.A.; Yousef, T.A. Cr (III) and Ni (II) complexes of isatin-hydrazone ligand: Preparation, characterization, DFT studies, biological activity, and ion-flotation separation of Ni (II). Inorg. Chem. Commun. 2022, 138, 109278. [Google Scholar] [CrossRef]
- Osman, S.A.; Mousa, H.A.; Yosef, H.A.A. Synthesis, characterization and cytotoxicity of mixed ligand Mn (II), Co (II) and Ni (II) complexes. J. Serban Chem. Soc. 2014, 79, 953–964. [Google Scholar] [CrossRef] [Green Version]
- Yekke-ghasemi, Z.; Ramezani, M.; Mague, T. Synthesis, characterization and bioactivity studies of new dithiocarbazate complexes. New J. Chem. 2020, 44, 8878–8889. [Google Scholar] [CrossRef]
- Evdokimov, N.M.; Magedov, I.V.; McBrayer, D.; Kornienko, A. Isatin derivatives with activity against apoptosis-resistant cancer cells. Bioorg. Med. Chem. Lett. 2016, 26, 1558–1560. [Google Scholar] [CrossRef] [Green Version]
- Senwar, K.R.; Reddy, T.S.; Thummuri, D.; Sharma, P.; Naidu, V.G.M.; Srinivasulu, G.; Shankaraiah, N. Design, synthesis and apoptosis inducing effect of novel (Z)-3-(3′-methoxy-4′-(2-amino-2-oxoethoxy)-benzylidene)indolin-2-ones as potential antitumour agents. Eur. J. Med. Chem. 2016, 118, 34–46. [Google Scholar] [CrossRef]
- Teng, Y.O.; Zhao, H.Y.; Wang, J.; Liu, H.; Le Gao, M.; Zhou, Y.; Han, K.L.; Fan, Z.C.; Zhang, Y.M.; Sun, H.; et al. Synthesis and anti-cancer activity evaluation of 5-(2-carboxyethenyl)-isatin derivatives. Eur. J. Med. Chem. 2016, 112, 145–156. [Google Scholar] [CrossRef]
- Sharma, P.; Thummuri, D.; Reddy, T.S.; Senwar, K.R.; Naidu, V.G.M.; Srinivasulu, G.; Bharghava, S.K.; Shankaraiah, N. New (E)-1-alkyl-1H-benzo[d]imidazol-2-yl)methylene)indolin-2-ones: Synthesis, in vitro cytotoxicity evaluation and apoptosis inducing studies. Eur. J. Med. Chem. 2016, 122, 584–600. [Google Scholar] [CrossRef]
- Song, Z.; Chen, C.P.; Liu, J.; Wen, X.; Sun, H.; Yuan, H. Design, synthesis, and biological evaluation of (2E)-(2-oxo-1, 2-dihydro-3H-indol-3-ylidene)acetate derivatives as anti-proliferative agents through ROS-induced cell apoptosis. Eur. J. Med. Chem. 2016, 124, 809–819. [Google Scholar] [CrossRef]
- Lozinskaya, N.A.; Babkov, D.A.; Zaryanova, E.V.; Bezsonova, E.N.; Efremov, A.M.; Tsymlyakov, M.D.; Anikina, L.V.; Zakharyascheva, O.Y.; Borisov, A.V.; Perfilova, V.N.; et al. Synthesis and biological evaluation of 3-substituted 2-oxindole derivatives as new glycogen synthase kinase 3β inhibitors. Bioorg. Med. Chem. 2019, 27, 1804–1817. [Google Scholar] [CrossRef]
- Zhang, Q.; Teng, Y.; Yuan, Y.; Ruan, T.; Wang, Q.; Gao, X.; Zhou, Y.; Han, K.; Yu, P.; Lu, K. Synthesis and cytotoxic studies of novel 5-phenylisatin derivatives and their anti-migration and anti-angiogenic evaluation. Eur. J. Med. Chem. 2018, 156, 800–814. [Google Scholar] [CrossRef]
- Dinavahi, S.S.; Gowda, R.; Bazewicz, C.G.; Battu, M.B.; Lin, J.M.; Chitren, R.J.; Pandey, M.K.; Amin, S.; Robertson, G.P.; Gowda, K. Design, synthesis characterization and biological evaluation of novel multi-isoform ALDH inhibitors as potential anticancer agents. Eur. J. Med. Chem. 2020, 187, 111962. [Google Scholar] [CrossRef] [PubMed]
- Rana, S.; Blowers, E.C.; Tebbe, C.; Contreras, J.I.; Radhakrishnan, P.; Kizhake, S.; Zhou, T.; Rajule, R.N.; Arnst, J.L.; Munkarah, A.R.; et al. Isatin Derived Spirocyclic Analogues with α-Methylene-γ-butyrolactone as Anticancer Agents: A Structure-Activity Relationship Study. J. Med. Chem. 2016, 59, 5121–5127. [Google Scholar] [CrossRef] [PubMed]
- Meleddu, R.; Petrikaite, V.; Distinto, S.; Arridu, A.; Angius, R.; Serusi, L.; Škarnulytė, L.; Endriulaitytė, U.; Paškevičiūtė, M.; Cottiglia, F.; et al. Investigating the Anticancer Activity of Isatin/Dihydropyrazole Hybrids. ACS Med. Chem. Lett. 2019, 10, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Karthikeyan, C.; Solomon, V.R.; Lee, H.; Trivedi, P. Design, synthesis and biological evaluation of some isatin-linked chalcones as novel anti-breast cancer agents: A molecular hybridization approach. Biomed. Prev. Nutr. 2013, 3, 325–330. [Google Scholar] [CrossRef]
- Sai Prathima, P.; Rajesh, P.; Venkateswara Rao, J.; Sai Kailash, U.; Sridhar, B.; Mohan Rao, M. “On water” expedient synthesis of 3-indolyl-3-hydroxy oxindole derivatives and their anticancer activity in vitro. Eur. J. Med. Chem. 2014, 84, 155–159. [Google Scholar] [CrossRef]
- Han, K.; Zhou, Y.; Liu, F.; Guo, Q.; Wang, P.; Yang, Y.; Song, B.; Liu, W.; Yao, Q.; Teng, Y.; et al. Design, synthesis and in vitro cytotoxicity evaluation of 5-(2-carboxyethenyl)isatin derivatives as anticancer agents. Bioorg. Med. Chem. Lett. 2014, 24, 591–594. [Google Scholar] [CrossRef]
- Schwartz, N.G.; Sandy, F.P.; Robert, H.P.; Adam, J.L. Tuberculosis—United States, 2019. Morb. Mortal. Wkly Rep. 2020, 69, 286. [Google Scholar] [CrossRef] [Green Version]
- Chakaya, J.; Khan, M.; Ntoumi, F.; Aklillu, E.; Fatima, R.; Mwaba, P.; Kapata, N.; Mfinanga, S.; Hasnain, S.E.; Katoto, P.D.M.C.; et al. Global Tuberculosis Report 2020–Reflections on the Global TB burden, treatment and prevention efforts. Int. J. Infect. Dis. 2021, 113, S7–S12. [Google Scholar] [CrossRef]
- Huszár, S.; Chibale, K.; Singh, V. The quest for the holy grail: New antitubercular chemical entities, targets and strategies. Drug Discov. Today 2020, 25, 772–780. [Google Scholar] [CrossRef]
- Yang, L.; Hu, X.; Chai, X.; Ye, Q.; Pang, J.; Li, D. Opportunities for overcoming tuberculosis: Emerging targets and their inhibitors. Drug Discov. Today 2022, 27, 326–336. [Google Scholar] [CrossRef]
- Perveen, S.; Kumari, D.; Singh, K.; Sharma, R. Tuberculosis drug discovery: Progression and future interventions in the wake of emerging resistance. Eur. J. Med. Chem. 2022, 229, 114066. [Google Scholar] [CrossRef]
- Xu, Z.; Song, X.; Hu, Y.; Qiang, M.; Lv, Z. Design, Synthesis and In Vitro Anti-mycobacterial Activities of 8-OMe Ciprofloxacin-1H-1,2,3-triazole-isatin-(thio) Semicarbazide/Oxime Hybrids. J. Heterocycl. Chem. 2018, 55, 192–198. [Google Scholar] [CrossRef]
- Xu, Z.; Song, X.F.; Fan, J.; Lv, Z.S. Design, Synthesis, and in vitro Anti-mycobacterial Evaluation of Propylene-1H-1,2,3-triazole-4-methylene-tethered (Thio)semicarbazone-isatin-moxifloxacin Hybrids. J. Heterocycl. Chem. 2018, 55, 77–82. [Google Scholar] [CrossRef]
- Yan, X.; Lv, Z.; Wen, J.; Zhao, S.; Xu, Z. Synthesis and in vitro evaluation of novel substituted isatin-propylene-1H-1,2,3-triazole-4-methylene-moxifloxacin hybrids for their anti-mycobacterial activities. Eur. J. Med. Chem. 2018, 143, 899–904. [Google Scholar] [CrossRef]
- Chen, R.; Zhang, H.; Ma, T.; Xue, H.; Miao, Z.; Chen, L.; Shi, X. Ciprofloxacin-1,2,3-triazole-isatin hybrids tethered via amide: Design, synthesis, and in vitro anti-mycobacterial activity evaluation. Bioorg. Med. Chem. Lett. 2019, 29, 2635–2637. [Google Scholar] [CrossRef]
- Rouatbi, F.; Askri, M.; Nana, F.; Kirsch, G.; Sriram, D.; Yogeeswari, P. Synthesis of new spirooxindole derivatives through 1,3-dipolar cycloaddition of azomethine ylides and their antitubercular activity. Tetrahedron Lett. 2016, 57, 163–167. [Google Scholar] [CrossRef]
- Borad, M.A.; Jethava, D.J.; Bhoi, M.N.; Patel, C.N.; Pandya, H.A.; Patel, H.D. Novel isoniazid-spirooxindole derivatives: Design, synthesis, biological evaluation, in silico ADMET prediction and computational studies. J. Mol. Struct. 2020, 1222, 128881. [Google Scholar] [CrossRef]
- Mhiri, C.; Boudriga, S.; Askri, M.; Knorr, M.; Sriram, D.; Yogeeswari, P.; Nana, F.; Golz, C.; Strohmann, C. Design of novel dispirooxindolopyrrolidine and dispirooxindolopyrrolothiazole derivatives as potential antitubercular agents. Bioorg. Med. Chem. Lett. 2015, 25, 4308–4313. [Google Scholar] [CrossRef]
- Chavan, P.V.; Pandit, K.S.; Desai, U.V.; Wadgaonkar, P.P.; Nawale, L.; Bhansali, S.; Sarkar, D. Click-chemistry-based multicomponent condensation approach for design and synthesis of spirochromene-tethered 1,2,3-triazoles as potential antitubercular agents. Res. Chem. Intermed. 2017, 43, 5675–5690. [Google Scholar] [CrossRef]
- Pogaku, V.; Krishna, V.S.; Sriram, D.; Rangan, K.; Basavoju, S. Ultrasonication-ionic liquid synergy for the synthesis of new potent anti-tuberculosis 1,2,4-triazol-1-yl-pyrazole based spirooxindolopyrrolizidines. Bioorg. Med. Chem. Lett. 2019, 29, 1682–1687. [Google Scholar] [CrossRef]
- Abdu-Allah, H.H.M.; Youssif, B.G.M.; Abdelrahman, M.H.; Abdel-Hamid, M.K.; Reshma, R.S.; Yogeeswari, P.; Aboul-Fadl, T.; Sriram, D. Synthesis and anti-mycobacterial activity of 4-(4-phenyl-1H-1,2,3-triazol-1-yl)salicylhydrazones: Revitalizing an old drug. Arch. Pharm. Res. 2017, 40, 168–179. [Google Scholar] [CrossRef]
- Yurttaş, L.; Ertaş, M.; Cankılıç Yılmaz, M.; Demirayak, Ş. Synthesis and Antimycobacterial Activity Evaluation of Isatin-derived 3-[(4-aryl-2-thiazolyl])hydrazone]-1H-indol-2,3-diones. Acta Pharm. Sci. 2017, 55, 51–58. [Google Scholar]
- Gao, F.; Yang, H.; Lu, T.; Chen, Z.; Ma, L.; Xu, Z.; Schaffer, P.; Lu, G. Design, synthesis and anti-mycobacterial activity evaluation of benzofuran-isatin hybrids. Eur. J. Med. Chem. 2018, 159, 277–281. [Google Scholar] [CrossRef]
- Gao, T.; Zeng, Z.; Wang, G.; Sun, S.; Liu, Y. Synthesis of Ethylene Tethered Isatin-Coumarin Hybrids and Evaluation of Their in vitro Antimycobacterial Activities. J. Heterocycl. Chem. 2018, 55, 1484–1488. [Google Scholar] [CrossRef]
- Hua, X.; Zhang, G.; Zhang, D.; Wu, Y. Design, Synthesis, and in vitro Anti-mycobacterial Activities of Propylene-tethered Heteronuclear Bis-isatin Derivatives. J. Heterocycl. Chem. 2018, 55, 1504–1508. [Google Scholar] [CrossRef]
- Xu, Y.; Guan, J.; Xu, Z.; Zhao, S. Design, synthesis and in vitro anti-mycobacterial activities of homonuclear and heteronuclear bis-isatin derivatives. Fitoterapia 2018, 127, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wang, G.Q.; Peng, Y.H.; Tang, X.Q.; Hu, G.W. Design, synthesis, and in vitro antimycobacterial activities of butylene tethered 7-fluoroisatin-isatin scaffolds. J. Heterocycl. Chem. 2019, 56, 3423–3428. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, R.; Zhang, T.; Yan, W.; Chen, Y.; Zhang, Y.; Zhou, M. Benzofuran-isatin-hydroxylimine/thiosemicarbazide hybrids: Design, synthesis and in vitro anti-mycobacterial activity evaluation. Chin. Chem. Lett. 2019, 30, 653–655. [Google Scholar] [CrossRef]
- Gao, F.; Wang, T.; Gao, M.; Zhang, X.; Liu, Z.; Zhao, S.J.; Lv, Z.S.; Xiao, J. Benzofuran-isatin-imine hybrids tethered via different length alkyl linkers: Design, synthesis and in vitro evaluation of anti-tubercular and anti-bacterial activities as well as cytotoxicity. Eur. J. Med. Chem. 2019, 165, 323–331. [Google Scholar] [CrossRef]
- Eldehna, W.M.; Fares, M.; Abdel-Aziz, M.M.; Abdel-Aziz, H.A. Design, synthesis and antitubercular activity of certain nicotinic acid hydrazides. Molecules 2015, 20, 8800–8815. [Google Scholar] [CrossRef] [Green Version]
- Karunanidhi, S.; Chandrasekaran, B.; Karpoormath, R.; Patel, H.M.; Kayamba, F.; Merugu, S.R.; Kumar, V.; Dhawan, S.; Kushwaha, B.; Mahlalela, M.C. Novel thiomorpholine tethered isatin hydrazones as potential inhibitors of resistant Mycobacterium tuberculosis. Bioorg. Chem. 2021, 115, 105133. [Google Scholar] [CrossRef]
- Elsayed, Z.M.; Eldehna, W.M.; Abdel-Aziz, M.M.; El Hassab, M.A.; Elkaeed, E.B.; Al-Warhi, T.; Abdel-Aziz, H.A.; Abou-Seri, S.M.; Mohammed, E.R. Development of novel isatin–nicotinohydrazide hybrids with potent activity against susceptible/resistant Mycobacterium tuberculosis and bronchitis causing–bacteria. J. Enzym. Inhib. Med. Chem. 2021, 36, 384–393. [Google Scholar] [CrossRef]
- Johansen, M.D.; Shalini; Kumar, S.; Raynaud, C.; Quan, D.H.; Britton, W.J.; Hansbro, P.M.; Kumar, V.; Kremer, L. Biological and biochemical evaluation of isatin-isoniazid hybrids as bactericidal candidates against mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2021, 65, e00011-21. [Google Scholar] [CrossRef]
- Kumar, K.; Carrère-Kremer, S.; Kremer, L.; Guérardel, Y.; Biot, C.; Kumar, V. 1H-1,2,3-Triazole-tethered isatin-ferrocene and isatin-ferrocenylchalcone conjugates: Synthesis and in vitro antitubercular evaluation. Organometallics 2013, 32, 5713–5719. [Google Scholar] [CrossRef]
- Kumar, K.; Biot, C.; Carrère-Kremer, S.; Kremer, L.; Guérardel, Y.; Roussel, P.; Kumar, V. Base-promoted expedient access to spiroisatins: Synthesis and antitubercular evaluation of 1H-1,2,3-triazole-tethered spiroisatin-ferrocene and isatin-ferrocene conjugates. Organometallics 2013, 32, 7386–7398. [Google Scholar] [CrossRef]
- Mubarak, H.S. Synthesis of Novel Triazole-incorporated Isatin Derivatives as Antifungal, Antitubercular, and Antioxidant Agents and Molecular Docking Study. J. Heterocycl. Chem. 2016, 54, 413–421. [Google Scholar]
- Jeankumar, V.U.; Alokam, R.; Sridevi, J.P.; Suryadevara, P.; Matikonda, S.S.; Peddi, S.; Sahithi, S.; Alvala, M.; Yogeeswari, P.; Sriram, D. Discovery and structure optimization of a series of isatin derivatives as Mycobacterium tuberculosis chorismate mutase inhibitors. Chem. Biol. Drug Des. 2014, 83, 498–506. [Google Scholar] [CrossRef]
- Kumar, S.B.; Ravinder, M.; Kishore, G.; Jayathirtha Rao, V.; Yogeeswari, P.; Sriram, D. Synthesis, antitubercular and anticancer activity of new Baylis-Hillman adduct-derived N-cinnamyl-substituted isatin derivatives. Med. Chem. Res. 2014, 23, 1934–1940. [Google Scholar] [CrossRef]
- Kumar, N.; Singh, R.; Rawat, D.S. Tetraoxanes: Synthetic and Medicinal Chemistry Perspective. Med. Res. Rev. 2012, 32, 581–610. [Google Scholar] [CrossRef]
- World Malaria Report. 2021. Available online: https://www.who.int/teams/global-malaria-programme/reports/world-malaria-report-2021 (accessed on 24 February 2022).
- Chauhan, M.; Saxena, A.; Saha, B. An insight in anti-malarial potential of indole scaffold: A review. Eur. J. Med. Chem. 2021, 218, 113400. [Google Scholar] [CrossRef]
- Zhou, W.; Wang, H.; Yang, Y.; Chen, Z.; Zou, C.; Zhang, J. Chloroquine against malaria, cancers and viral diseases. Drug Discov. Today 2020, 25, 2012–2022. [Google Scholar] [CrossRef]
- Gut, J.; Rosenthal, P.J.; Kumar, V. β-amino-alcohol tethered 4-aminoquinoline-isatin conjugates: Synthesis and antimalarial evaluation. Eur. J. Med. Chem. 2014, 84, 566–573. [Google Scholar]
- Raj, R.; Singh, P.; Singh, P.; Gut, J.; Rosenthal, P.J.; Kumar, V. Azide-alkyne cycloaddition en route to 1H-1,2,3-triazole-tethered 7-chloroquinoline-isatin chimeras: Synthesis and antimalarial evaluation. Eur. J. Med. Chem. 2013, 62, 590–596. [Google Scholar] [CrossRef] [Green Version]
- Raj, R.; Gut, J.; Rosenthal, P.J.; Kumar, V. 1H-1,2,3-Triazole-tethered isatin-7-chloroquinoline and 3-hydroxy-indole-7-chloroquinoline conjugates: Synthesis and antimalarial evaluation. Bioorg. Med. Chem. Lett. 2014, 24, 756–759. [Google Scholar] [CrossRef]
- Raj, R.; Biot, C.; Carrère-Kremer, S.; Kremer, L.; Guérardel, Y.; Gut, J.; Rosenthal, P.J.; Forge, D.; Kumar, V. 7-chloroquinoline-isatin conjugates: Antimalarial, antitubercular, and cytotoxic evaluatio. Chem. Biol. Drug Des. 2014, 83, 622–629. [Google Scholar] [CrossRef]
- Kumar, S.; Saini, A.; Legac, J.; Rosenthal, P.J.; Raj, R.; Kumar, V. Amalgamating Isatin/Indole/Nitroimidazole with 7-chloroquinolines via azide-alkyne cycloaddition: Synthesis, anti-plasmodial, and cytotoxic evaluation. Chem. Biol. Drug Des. 2020, 96, 1355–1651. [Google Scholar] [CrossRef]
- Akhaja, T.N.; Raval, J.P. Design, synthesis and in vitro evaluation of tetrahydropyrimidine-isatin hybrids as potential antitubercular and antimalarial agents. Chin. Chem. Lett. 2012, 23, 785–788. [Google Scholar] [CrossRef]
- Thakur, R.K.; Joshi, P.; Baranwal, P.; Sharma, G.; Shukla, S.K.; Tripathi, R.; Tripathi, R.P. Synthesis and antiplasmodial activity of glyco-conjugate hybrids of phenylhydrazono-indolinones and glycosylated 1,2,3-triazolyl-methyl-indoline-2,3-diones. Eur. J. Med. Chem. 2018, 155, 764–771. [Google Scholar] [CrossRef]
- Thakur, R.K.; Joshi, P.; Upadhyaya, K.; Singh, K.; Sharma, G.; Shukla, S.K.; Tripathi, R.; Tripathi, R.P. Synthesis of isatin based N1-alkylated 3-β-C-glycoconjugated-oxopropylidene oxindoles as potent antiplasmodial agents. Eur. J. Med. Chem. 2019, 162, 448–454. [Google Scholar] [CrossRef]
- Hans, R.H.; Wiid, I.J.F.; van Helden, P.D.; Wan, B.; Franzblau, S.G.; Gut, J.; Rosenthal, P.J.; Chibale, K. Novel thiolactone-isatin hybrids as potential antimalarial and antitubercular agents. Bioorg. Med. Chem. Lett. 2011, 21, 2055–2058. [Google Scholar] [CrossRef]
- Kumar, K.; Pradines, B.; Madamet, M.; Amalvict, R.; Benoit, N.; Kumar, V. 1H-1,2,3-triazole tethered isatin-ferrocene conjugates: Synthesis and in vitro antimalarial evaluation. Eur. J. Med. Chem. 2014, 87, 801–804. [Google Scholar] [CrossRef] [PubMed]
- Gaurav, G.L.; Manish, P.P. New approach for the synthesis of spiro indolinone incorporated 1,2,4-triazolo [1,5-a]quinoline derivatives and their pharmacological screening. Heterocycl. Lett. 2016, 6, 393–405. [Google Scholar]
- Bin Zaman, S.; Hussain, M.A.; Nye, R.; Mehta, V.; Taib, K. A Review on Antibiotic Resistance: Alarm Bells are Ringing Origin of antibiotic resistance. Cureus 2017, 9. [Google Scholar]
- Thanh, N.D.; Giang, N.T.K.; Quyen, T.H.; Huong, D.T.; Toan, V.N. Synthesis and evaluation of in vivo antioxidant, in vitro antibacterial, MRSA and antifungal activity of novel substituted isatin N-(2,3,4,6-tetra-O-acetyl-β-D-glucopyranosyl)thiosemicarbazones. Eur. J. Med. Chem. 2016, 123, 532–543. [Google Scholar] [CrossRef]
- Lian, Z.M.; Sun, J.; Zhu, H.L. Design, synthesis and antibacterial activity of isatin derivatives as FtsZ inhibitors. J. Mol. Struct. 2016, 1117, 8–16. [Google Scholar] [CrossRef]
- Abo-Ashour, M.F.; Eldehna, W.M.; George, R.F.; Abdel-Aziz, M.M.; Elaasser, M.M.; Abdel Gawad, N.M.; Gupta, A.; Bhakta, S.; Abou-Seri, S.M. Novel indole-thiazolidinone conjugates: Design, synthesis and whole-cell phenotypic evaluation as a novel class of antimicrobial agents. Eur. J. Med. Chem. 2018, 160, 49–60. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.S.; Su, M.M.; Xu, J.F.; Liu, Q.X.; Bai, L.F.; Hu, X.W.; Zhu, H.L. Discovery of novel oxoindolin derivatives as atypical dual inhibitors for DNA Gyrase and FabH. Bioorg. Chem. 2019, 93, 103309. [Google Scholar] [CrossRef]
- Salem, M.A.; Ragab, A.; El-Khalafawy, A.; Makhlouf, A.H.; Askar, A.A.; Ammar, Y.A. Design, synthesis, in vitro antimicrobial evaluation and molecular docking studies of indol-2-one tagged with morpholinosulfonyl moiety as DNA gyrase inhibitors. Bioorg. Chem. 2020, 96, 103619. [Google Scholar] [CrossRef]
- Song, G.Q.; Wang, W.M.; Li, Z.S.; Wang, Y.; Wang, J.G. First identification of isatin-β-thiosemicarbazones as novel inhibitors of New Delhi metallo-β-lactamase-1: Chemical synthesis, biological evaluation and molecular simulation. Chin. Chem. Lett. 2018, 29, 899–902. [Google Scholar] [CrossRef]
- Wang, Y.; Cheong, W.L.; Liang, Z.; So, L.Y.; Chan, K.F.; So, P.K.; Chen, Y.W.; Wong, W.L.; Wong, K.Y. Hydrophobic substituents on isatin derivatives enhance their inhibition against bacterial peptidoglycan glycosyltransferase activity. Bioorg. Chem. 2020, 97, 103710. [Google Scholar] [CrossRef]
- Ma, T.; Chen, R.; Xue, H.; Miao, Z.; Chen, L.; Zhang, H.; Shi, X. Di-isatin heteronuclear compounds and their antibacterial activity. J. Heterocycl. Chem. 2020, 57, 503–509. [Google Scholar] [CrossRef]
- Ugale, V.; Patel, H.; Patel, B.; Bari, S. Benzofurano-isatins: Search for antimicrobial agents. Arab. J. Chem. 2017, 10, S389–S396. [Google Scholar] [CrossRef] [Green Version]
- Kandile, N.G.; Zaky, H.T.; Mohamed, M.I.; Ismaeel, H.M.; Ahmed, N.A. Synthesis, characterization and in vitro antimicrobial evaluation of new compounds incorporating oxindole nucleus. J. Enzym. Inhib. Med. Chem. 2012, 27, 599–608. [Google Scholar] [CrossRef]
- Farag, A.A. Synthesis and Antimicrobial Activity of 5-(morpholinosulfonyl)isatin Derivatives Incorporating a Thiazole Moiety. Drug Res. 2014, 65, 373–379. [Google Scholar] [CrossRef]
- Zhang, X.M.; Guo, H.; Li, Z.S.; Song, F.H.; Wang, W.M.; Dai, H.Q.; Zhang, L.X.; Wang, J.G. Synthesis and evaluation of isatin-β-thiosemicarbazones as novel agents against antibiotic-resistant Gram-positive bacterial species. Eur. J. Med. Chem. 2015, 101, 419–430. [Google Scholar] [CrossRef]
- Yagnam, S.; Trivedi, R.; Krishna, S.; Giribabu, L.; Praveena, G.; Prakasham, R.S. Bioactive isatin (oxime)-triazole-thiazolidinedione ferrocene molecular conjugates: Design, synthesis and antimicrobial activities. J. Organomet. Chem. 2021, 937, 121716. [Google Scholar] [CrossRef]
- Haj, K.; Mohammad, E.T.; Hashemi, M.; Hassan, M.; Kobarfard, F.; Mohebbi, S. Synthesis and antibacterial activity of Schiff bases of 5-substituted isatins. Chin. Chem. Lett. 2016, 27, 221–225. [Google Scholar]
- Guo, H. Design, Synthesis, and Antibacterial Evaluation of Propylene-tethered 8-Methoxyl Ciprofloxacin-isatin Hybrids. J. Heterocycl. Chem. 2018, 55, 2434–2440. [Google Scholar] [CrossRef]
- Gao, F.; Ye, L.; Kong, F.; Huang, G.; Xiao, J. Design, synthesis and antibacterial activity evaluation of moxifloxacin-amide-1,2,3-triazole-isatin hybrids. Bioorg. Chem. 2019, 91, 103162. [Google Scholar] [CrossRef]
- Salem, M.A.; Ragab, A.; Askar, A.A.; El-Khalafawy, A.; Makhlouf, A.H. One-pot synthesis and molecular docking of some new spiropyranindol-2-one derivatives as immunomodulatory agents and in vitro antimicrobial potential with DNA gyrase inhibitor. Eur. J. Med. Chem. 2020, 188, 111977. [Google Scholar] [CrossRef]
- Bhagat, K.; Bhagat, J.; Gupta, M.K.; Singh, J.V.; Gulati, H.K.; Singh, A.; Kaur, K.; Kaur, G.; Sharma, S.; Rana, A.; et al. Design, synthesis, antimicrobial evaluation, and molecular modeling studies of novel indolinedione-coumarin molecular hybrids. ACS Omega 2019, 4, 8720–8730. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Singh, J.V.; Rana, A.; Bhagat, K.; Gulati, H.K.; Kumar, R.; Salwan, R.; Bhagat, K.; Kaur, G.; Singh, N.; et al. Monocarbonyl curcumin based molecular hybrids as potent antibacterial agents. ACS Omega 2019, 4, S1–S50. [Google Scholar] [CrossRef]
- Khatoon, S.; Aroosh, A.; Islam, A.; Kalsoom, S.; Ahmad, F.; Hameed, S.; Abbasi, S.W.; Yasinzai, M.; Naseer Novel, M.M. Coumarin-isatin hybrids as potent antileishmanial agents: Synthesis, in silico and in vitro evaluations. Bioorg. Chem. 2021, 110, 104816. [Google Scholar] [CrossRef]
- Freitas, L.A.B.; da Silva Santos, A.C.; de Cássia Silva, G.; do Nascimento Albuquerque, F.N.; Silva, E.D.; de Simone, C.A.; Pereira, V.R.A.; Alves, L.C.; Brayner, F.A.; Leite, A.C.L.; et al. Structural improvement of new thiazolyl-isatin derivatives produces potent and selective trypanocidal and leishmanicidal compounds. Chem.-Biol. Interact. 2021, 345, 109561. [Google Scholar] [CrossRef]



















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chowdhary, S.; Shalini; Arora, A.; Kumar, V. A Mini Review on Isatin, an Anticancer Scaffold with Potential Activities against Neglected Tropical Diseases (NTDs). Pharmaceuticals 2022, 15, 536. https://doi.org/10.3390/ph15050536
Chowdhary S, Shalini, Arora A, Kumar V. A Mini Review on Isatin, an Anticancer Scaffold with Potential Activities against Neglected Tropical Diseases (NTDs). Pharmaceuticals. 2022; 15(5):536. https://doi.org/10.3390/ph15050536
Chicago/Turabian StyleChowdhary, Shefali, Shalini, Amandeep Arora, and Vipan Kumar. 2022. "A Mini Review on Isatin, an Anticancer Scaffold with Potential Activities against Neglected Tropical Diseases (NTDs)" Pharmaceuticals 15, no. 5: 536. https://doi.org/10.3390/ph15050536
APA StyleChowdhary, S., Shalini, Arora, A., & Kumar, V. (2022). A Mini Review on Isatin, an Anticancer Scaffold with Potential Activities against Neglected Tropical Diseases (NTDs). Pharmaceuticals, 15(5), 536. https://doi.org/10.3390/ph15050536