
A Combination of Pharmacophore-Based Virtual Screening, Structure-Based Lead Optimization, and DFT Study for the Identification of S. epidermidis TcaR Inhibitors



Abstract
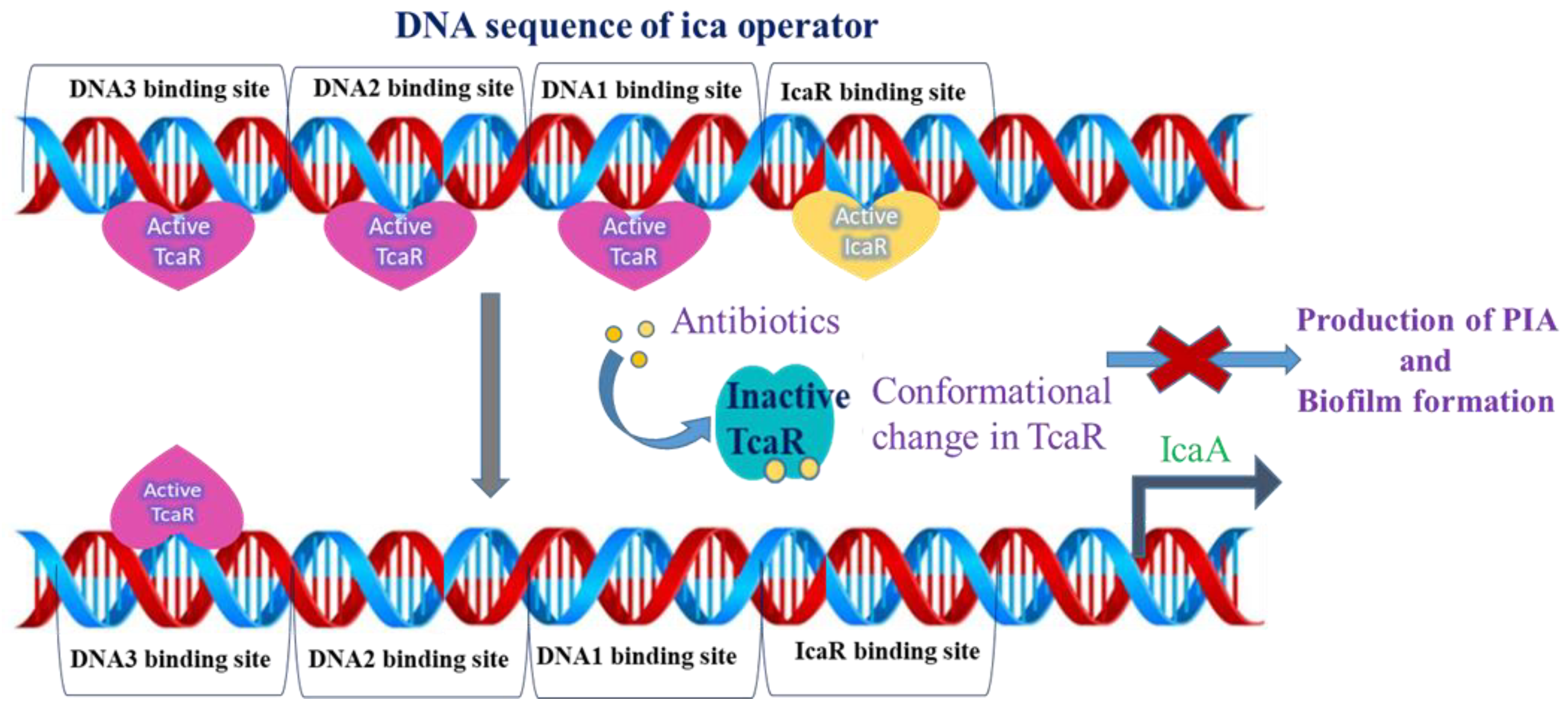
:1. Introduction
2. Results
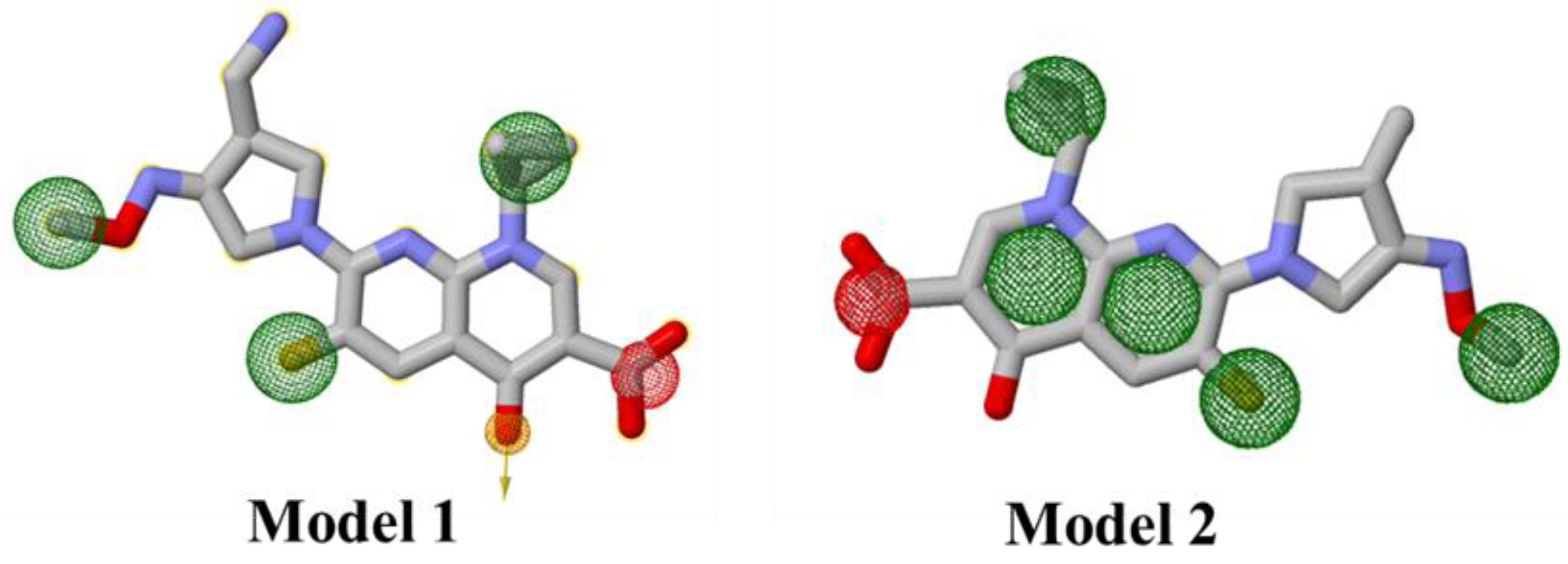
2.1. Ligand-Based Pharmacophore Modeling
2.1.1. Generation of Pharmacophore Model
2.1.2. Molecular Docking Simulations for the Identification of Hit Compounds
- Validation of Ligand Binding Mode
- Identification of Hit Compounds
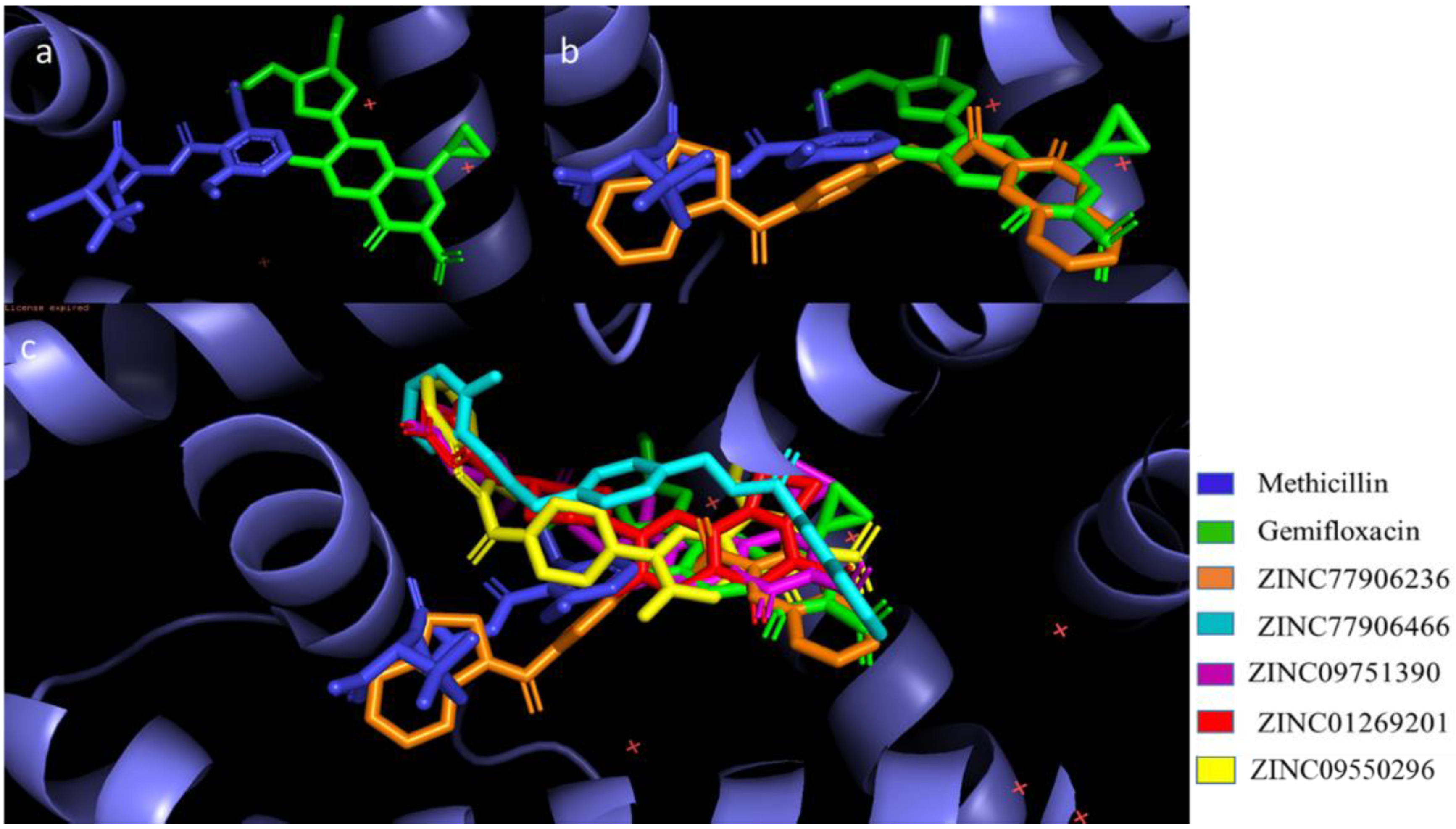
- Binding Mode Analysis of Final Hits
- Binding Mode Analysis of Hit Compound ZINC77906236
- Binding Mode Analysis of Hit Compound ZINC09550296
2.2. Structure-Based Lead Optimization Studies
2.2.1. SAR and ADMET Analysis of Selected Experimentally Known Inhibitors
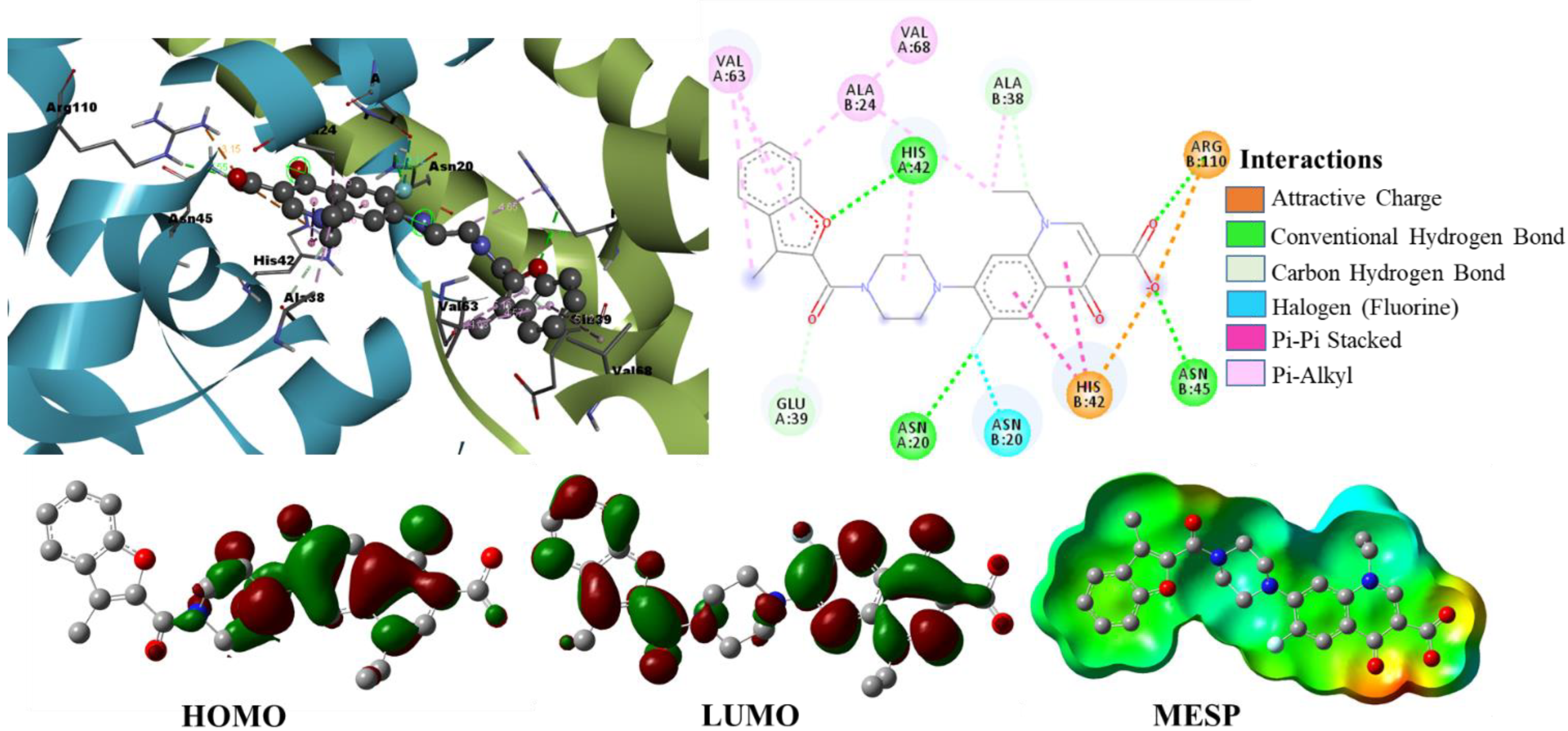
2.2.2. Molecular Docking Analysis of Selected Experimentally Known Inhibitors
2.2.3. Lead Optimization Studies
- SAR and ADMET Analysis of Designed Molecules
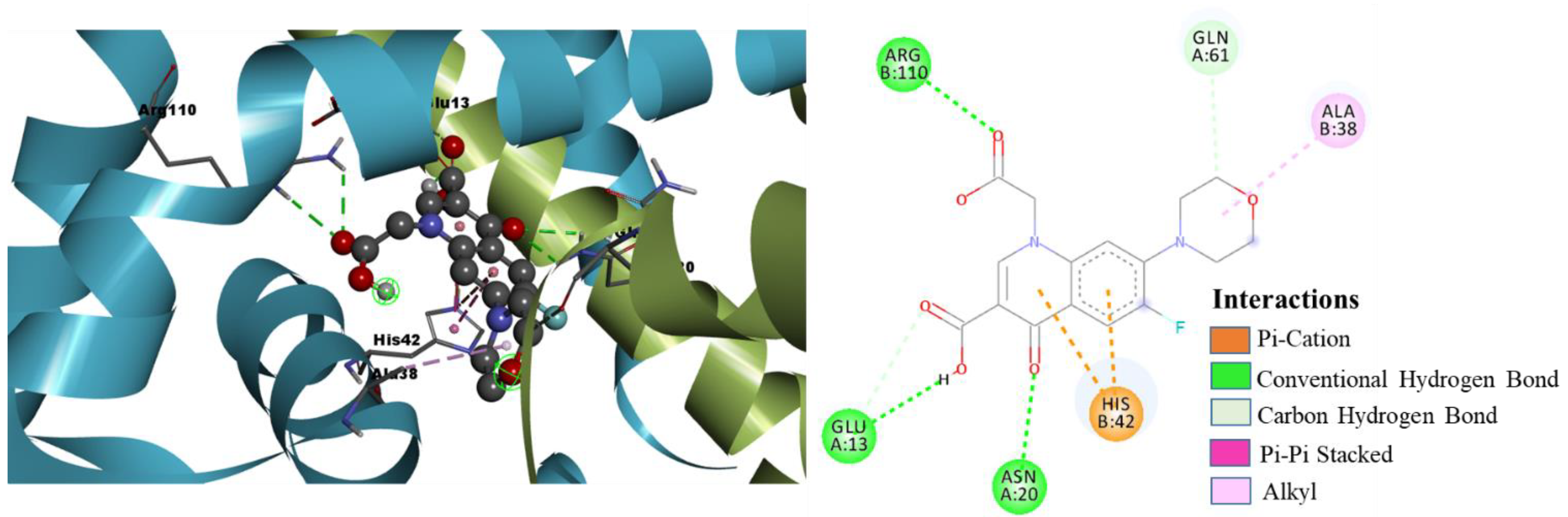
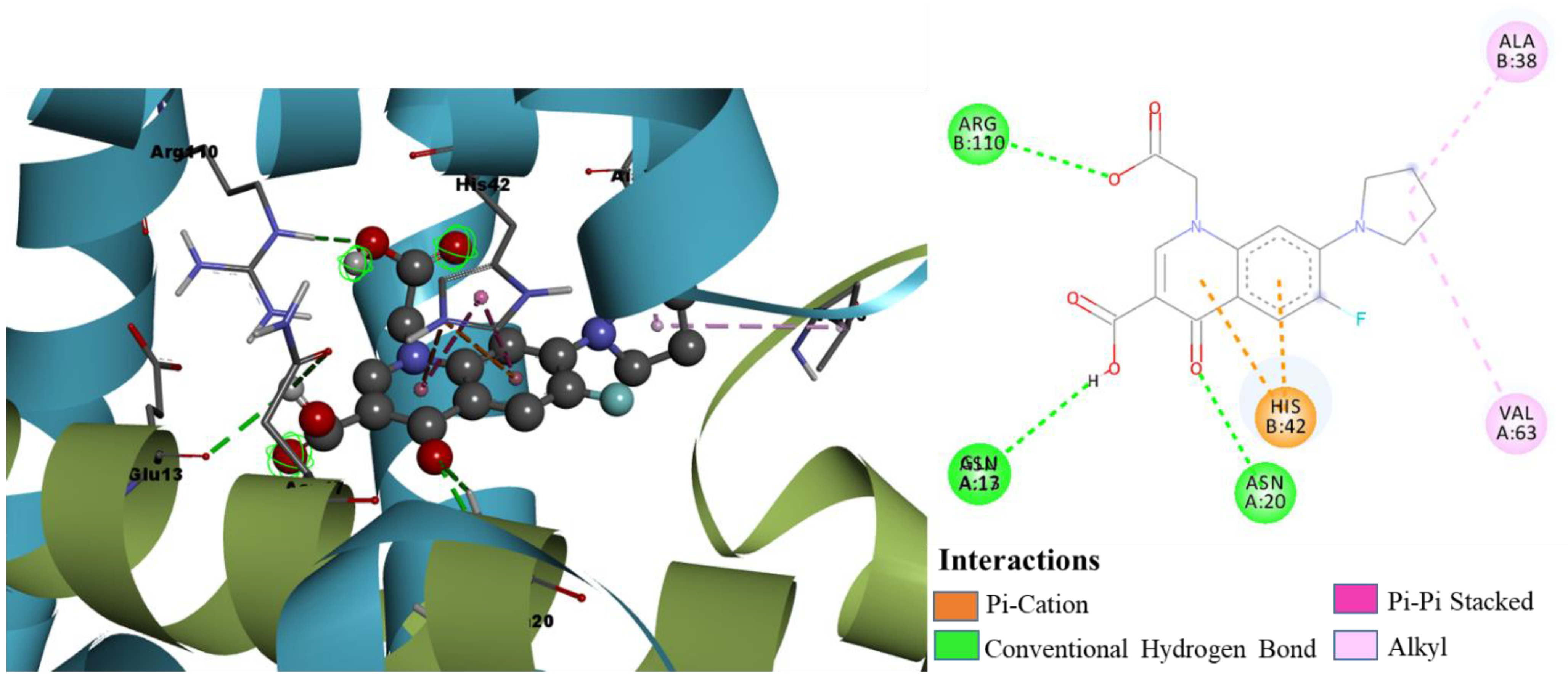
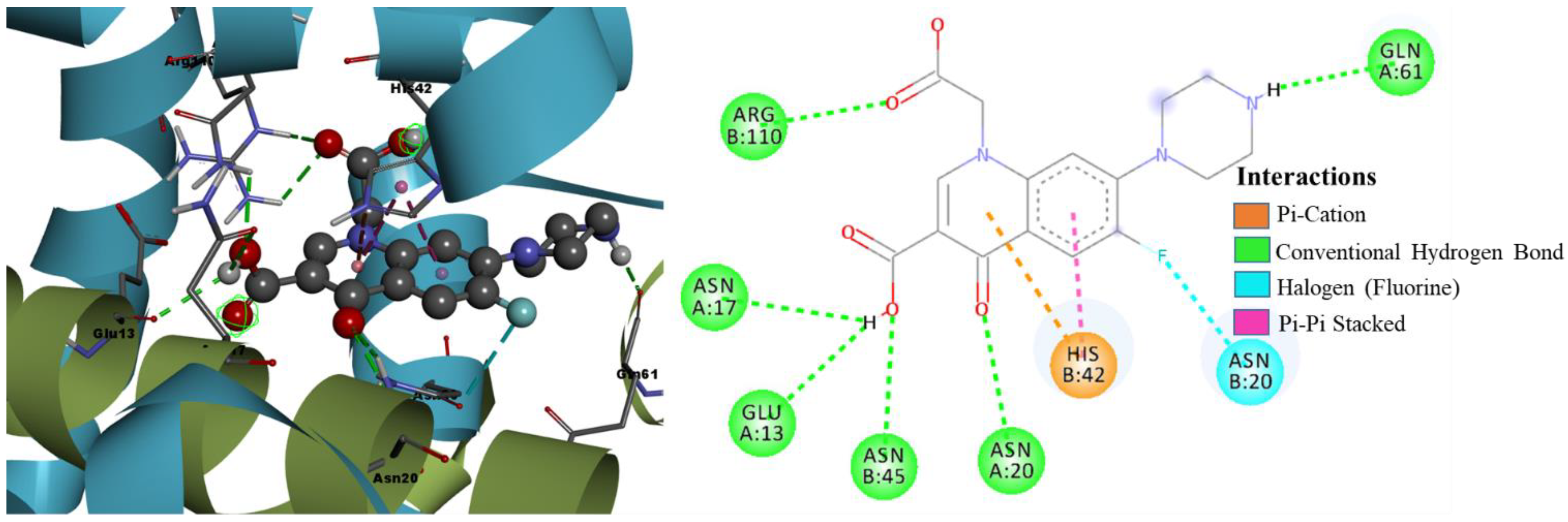
- Molecular Docking Analysis of Designed Molecules
2.3. Density Functional Theory Calculations
3. Discussion
4. Materials and Methods
4.1. Generation of Pharmacophore Models and Virtual Screening
4.2. Ligand Preparation and SAR and ADMET Analysis
4.3. Molecular Docking Simulations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Uçkay, I.; Pittet, D.; Vaudaux, P.; Sax, H.; Lew, D.; Waldvogel, F. Foreign body infections due to Staphylococcus epidermidis. Ann. Med. 2009, 41, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Otto, M. Staphylococcus epidermidis—The ‘accidental’ pathogen. Nat. Rev. Microbiol. 2009, 7, 555–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, K.L.; Fey, P.D.; Rupp, M.E. Coagulase—Negative staphylococcal infections. Infect. Dis. Clin. N. Am. 2009, 23, 73–98. [Google Scholar] [CrossRef] [PubMed]
- Costerton, J.W.; Stewart, P.S.; Greenberg, E.P. Bacterial biofilms: A common cause of persistent infections. Science 1999, 284, 1318–1322. [Google Scholar] [CrossRef] [Green Version]
- Dhar, Y.; Han, Y. Current developments in biofilm treatments: Wound and implant infections. Eng. Regen. 2020, 1, 64–75. [Google Scholar] [CrossRef]
- Bancroft, E.A. Antimicrobial resistance: It’s not just for hospitals. JAMA 2007, 298, 1803–1804. [Google Scholar] [CrossRef] [Green Version]
- Klevens, R.M.; Morrison, M.A.; Nadle, J.; Petit, S.; Gershman, K.; Ray, S.; Harrison, L.H.; Lynfield, R.; Dumyati, G.; Townes, J.M.; et al. Invasive Methicillin-Resistant Staphylococcus aureus Infections in the United States. JAMA 2007, 298, 1763–1771. [Google Scholar] [CrossRef] [Green Version]
- Darouiche, R.O. Treatment of Infections Associated with Surgical Implants. N. Engl. J. Med. 2004, 350, 1422–1429. [Google Scholar] [CrossRef]
- Davies, D. Understanding biofilm resistance to antibacterial agents. Nat. Rev. Drug Discov. 2003, 2, 114–122. [Google Scholar] [CrossRef]
- Vuong, C.; Voyich, J.M.; Fischer, E.R.; Braughton, K.R.; Whitney, A.R.; DeLeo, F.R.; Otto, M. Polysaccharide intercellular adhesin (PIA) protects Staphylococcus epidermidis against major components of the human innate immune system. Cell. Microbiol. 2004, 6, 269–275. [Google Scholar] [CrossRef]
- Dobinsky, S.; Kiel, K.; Rohde, H.; Bartscht, K.; Knobloch, J.K.M.; Horstkotte, M.A.; Mack, D. Glucose-related dissociation between icaADBC transcription and biofilm expression by Staphylococcus epidermidis: Evidence for an additional factor required for polysaccharide intercellular adhesin synthesis. J. Bacteriol. 2003, 185, 2879–2886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fluckiger, U.; Ulrich, M.; Steinhuber, A.; Döring, G.; Mack, D.; Landmann, R.; Goerke, C.; Wolz, C. Biofilm formation, icaADBC transcription, and polysaccharide intercellular adhesin synthesis by staphylococci in a device-related infection model. Infect. Immun. 2005, 73, 1811–1819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cafiso, V.; Bertuccio, T.; Santagati, M.; Campanile, F.; Amicosante, G.; Perilli, M.G.; Selan, L.; Artini, M.; Nicoletti, G.; Stefani, S. Presence of the ica operon in clinical isolates of Staphylococcus epidermidis and its role in biofilm production. Clin. Microbiol. Infect. 2004, 10, 1081–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandenberger, M.; Tschierske, M.; Giachino, P.; Wada, A.; Berger-Bächi, B. Inactivation of a novel three-cistronic operon tcaR-tcaA-tcaB increases teicoplanin resistance in Staphylococcus aureus. Biochim. Biophys. Acta 2000, 1523, 135–139. [Google Scholar] [CrossRef]
- Chang, Y.-M.; Jeng, W.-Y.; Ko, T.-P.; Yeh, Y.-J.; Chen, C.K.M.; Wang, A.H.J. Structural study of TcaR and its complexes with multiple antibiotics from Staphylococcus epidermidis. Proc. Natl. Acad. Sci. USA 2010, 107, 8617–8622. [Google Scholar] [CrossRef] [Green Version]
- Dickson, M.; Gagnon, J.P. Key factors in the rising cost of new drug discovery and development. Nat. Rev. Drug Discov. 2004, 3, 417–429. [Google Scholar] [CrossRef]
- Sproston, E.L.; Wimalarathna HM, L.; Sheppard, S.K. Trends in fluoroquinolone resistance in Campylobacter. Microb. Genom. 2018, 4, e000198. [Google Scholar] [CrossRef]
- Ferreira, M.; Gameiro, P. Fluoroquinolone-Transition Metal Complexes: A Strategy to Overcome Bacterial Resistance. Microorganisms 2021, 9, 1506. [Google Scholar] [CrossRef]
- Xu, Z.; Yang, Z.; Liu, Y.; Lu, Y.; Chen, K.; Zhu, W. Halogen Bond: Its Role beyond Drug–Target Binding Affinity for Drug Discovery and Development. J. Chem. Inf. Modeling 2014, 54, 69–78. [Google Scholar] [CrossRef]
- Koga, H.; Itoh, A.; Murayama, S.; Suzue, S.; Irikura, T. Structure-activity relationships of antibacterial 6,7- and 7,8-disubstituted 1-alkyl-1,4-dihydro-4-oxoquinoline-3-carboxylic acids. J. Med. Chem. 1980, 23, 1358–1363. [Google Scholar] [CrossRef]
- Wise, R.; Andrews, J.M.; Edwards, L.J. In vitro activity of Bay 09867, a new quinoline derivative, compared with those of other antimicrobial agents. Antimicrob. Agents Chemother. 1983, 23, 559–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham TD, M.; Ziora, Z.M.; Blaskovich, M.A.T. Quinolone antibiotics. MedChemComm 2019, 10, 1719–1739. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. IV. A new dynamical correlation functional and implications for exact-exchange mixing. J. Chem. Phys. 1996, 104, 1040–1046. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Hardy, D.; Amsterdam, D.; Mandell, L.A.; Rotstein, C. Comparative in vitro activities of ciprofloxacin, gemifloxacin, grepafloxacin, moxifloxacin, ofloxacin, sparfloxacin, trovafloxacin, and other antimicrobial agents against bloodstream isolates of gram-positive cocci. Antimicrob. Agents Chemother. 2000, 44, 802–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, R.N.; Pfaller, M.A.; Erwin, M.E. Evaluation of gemifloxacin (SB-265805, LB20304a): In vitro activity against over 6000 Gram-positive pathogens from diverse geographic areas. Int. J. Antimicrob. Agents 2000, 15, 227–230. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Daneshtalab, M.; Ahmed, A. Nonclassical Biological Activities of Quinolone Derivatives. J. Pharm. Pharm. Sci. 2011, 15, 52–72. [Google Scholar] [CrossRef] [Green Version]
- Dalhoff, A.; Schmitz, F.J. In vitro antibacterial activity and pharmacodynamics of new quinolones. Eur. J. Clin. Microbiol. Infect. Dis. 2003, 22, 203–221. [Google Scholar] [CrossRef]
- Ravi Kumar, A.; Sathaiah, G.; Chandra Shekhar, A.; Raju, K.; Shanthan Rao, P.; Narsaiah, B.; Kanaka Raju, Y.; Murthy, U.S.N.; Srimai, V.; Ramesh, M.; et al. Synthesis of 6-Fluoro-7-cyclic Amino-substituted Dicarboxylic Acid Quinolones and their Antibacterial Activity. J. Heterocycl. Chem. 2014, 51, E114–E122. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Mabkhot, Y.N.; Alatibi, F.; El-Sayed, N.N.E.; Al-Showiman, S.; Kheder, N.A.; Wadood, A.; Rauf, A.; Bawazeer, S.; Hadda, T.B. Antimicrobial Activity of Some Novel Armed Thiophene Derivatives and Petra/Osiris/Molinspiration (POM) Analyses. Molecules 2016, 21, 222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srimai, V.; Ramesh, M.; Satya Parameshwar, K.; Parthasarathy, T. Computer-aided design of selective Cytochrome P450 inhibitors and docking studies of alkyl resorcinol derivatives. Med. Chem. Res. 2013, 22, 5314–5323. [Google Scholar] [CrossRef]
- Scafuri, B.; Marabotti, A.; Carbone, V.; Minasi, P.; Dotolo, S.; Facchiano, A. A theoretical study on predicted protein targets of apple polyphenols and possible mechanisms of chemoprevention in colorectal cancer. Sci. Rep. 2016, 6, 32516. [Google Scholar] [CrossRef] [PubMed]
- Scafuri, B.; Bontempo, P.; Altucci, L.; De Masi, L.; Facchiano, A. Molecular Docking Simulations on Histone Deacetylases (HDAC)-1 and -2 to Investigate the Flavone Binding. Biomedicines 2020, 8, 568. [Google Scholar] [CrossRef] [PubMed]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-Pdb Viewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No | Compounds | Binding Energy (kcal/mol) | Estimated Inhibition Constant (Ki) |
|---|---|---|---|
| 1 | ZINC77906236 | −13.27 | 187.61 pM |
| 2 | ZINC03114214 | −13.07 | 260.55 pM |
| 3 | ZINC09550296 | −12.89 | 353.69 pM |
| 4 | ZINC77906466 | −12.74 | 460.70 pM |
| 5 | ZINC01958447 | −12.68 | 507.91 pM |
| 6 | ZINC09751390 | −12.38 | 843.81 pM |
| 7 | ZINC01269201 | −12.34 | 895.54 pM |
| 8 | ZINC21985520 | −12.13 | 1.29 nM |
| 9 | ZINC09751395 | −12.08 | 1.39 nM |
| 10 | ZINC02280291 | −12.07 | 1.41 nM |
| 11 | ZINC01440193 | −11.93 | 1.79 nM |
| 12 | ZINC01127091 | −11.91 | 1.85 nM |
| 13 | ZINC72332562 | −11.57 | 3.31 nM |
| 14 | ZINC00794058 | −11.56 | 3.37 nM |
| 15 | ZINC00686337 | −11.40 | 4.40 nM |
| 16 | ZINC09550295 | −11.36 | 4.71 nM |
| 17 | Gemifloxacin | −10.73 | 13.72 nM |
| 18 | Methicillin | −6.25 | 26.35 uM |
| Molecule | Binding Energy (kcal/mol) | Fitness Score | S(hb_ext) a | S(vdw_ext) b | S(vdw_int) c |
|---|---|---|---|---|---|
| 7a | −8.7 | 56.33 | 6.27 | 45.78 | −12.89 |
| 7b | −9.0 | 57.47 | 6.73 | 44.81 | −13.19 |
| 7c | −8.9 | 61.30 | 2.01 | 49.34 | −8.53 |
| 7d | −9.3 | 59.35 | 3.52 | 50.67 | −13.84 |
| 7e | −8.3 | 60.04 | 6.09 | 52.25 | −12.65 |
| 7f | −9.6 | 58.97 | 1.94 | 51.08 | −13.22 |
| 7g | −9.2 | 56.90 | 6.40 | 47.19 | −14.39 |
| 7h | −8.7 | 58.52 | 3.01 | 50.26 | −22.09 |
| 7i | −8.8 | 61.34 | 2.70 | 54.17 | −15.85 |
| 7j | −9.7 | 61.63 | 4.68 | 52.34 | −15.01 |
| 7k | −8.5 | 62.09 | 2.26 | 55.70 | −16.26 |
| 7l | −7.6 | 60.19 | 4.72 | 51.96 | −15.98 |
| 7n | −4.3 | 58.06 | 4.76 | 47.51 | −12.02 |
| 7o | −9.2 | 59.58 | 5.97 | 51.99 | −17.88 |
| 7p | −9.2 | 57.13 | 6.04 | 51.89 | −16.03 |
| Compound | H-Bond Interactions | Hydrophobic and Other Interactions |
|---|---|---|
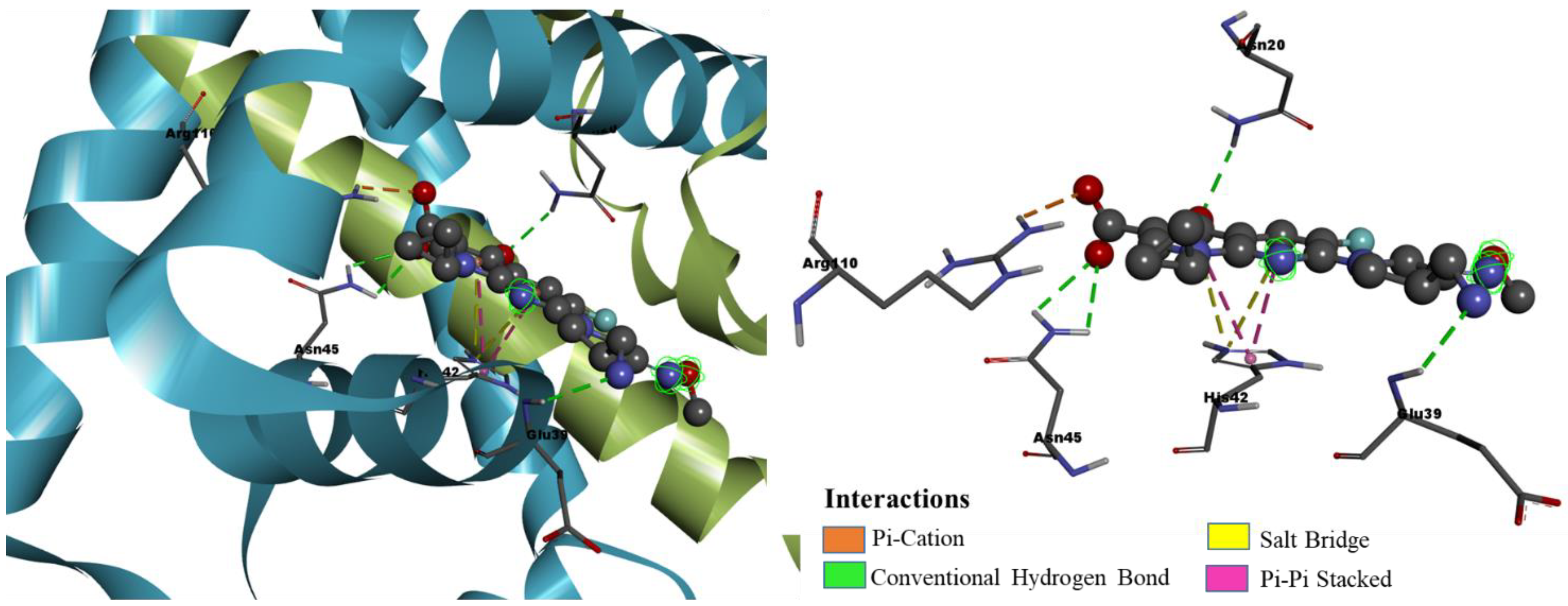
| ZINC77906236 | ARG110 (2.86, 2.76), ASN45 (2.79) | ILE16 (5.40), ALA24 (4.91, 4.68), ALA38 (4.09, 4.85), HIS42 (3.89, 4.27, 4.43, 3.97) |
| ZINC09550296 | ASN20 (2.57), HIS42 (2.79), ASN45 (2.56), ARG110 (2.55) | ALA24 (4.05), ALA38 (3.67, 3.67), GLU39 (3.50), HIS42 (4.65, 4.11, 4.16), VAL63 (4.68, 5.16, 4.57), VAL68 (5.12), ARG110 (3.15) |
| ZINC77906466 | ALA24 (2.96), ASN45 (2.70) | ASN20 (3.72), THR23 (3.87), HIS42 (2.93), VAL43 (5.37), ILE57 (5.37), VAL63 (5.05), VAL68 (5.0), ARG71 (5.25), ARG110 (3.59, 4.11) |
| ZINC09751390 | ASN20 (2.29), THR23 (2.38), HIS42 (2.62), ASN45 (2.68), ARG71 (2.69) | ALA38 (3.73, 4.65), HIS42 (3.83, 4.07, 4.30, 4.91), VAL63 (4.85), ARG110 (2.97) |
| ZINC01269201 | THR23 (2.91), HIS42 (2.24), ASN45 (2.72) | ALA38 (3.44), HIS42 (4.60, 4.40, 4.54, 4.74), VAL63 (3.72, 5.42), VAL68 (4.58), ARG110 (3.23) |
| Mol34 | ASN20 (3.00), HIS42 (3.09), ARG110 (2.69, 2.79) | ALA24 (4.31), HIS42 (3.90, 4.18, 3.09), ILE57 (5.48), VAL63 (4.85), VAL68 (4.89), ARG71 (5.20) |
| Gemifloxacin | ASN20 (2.54), GLU39 (2.76), ASN45 (2.69, 2.67) | HIS42 (3.65, 4.01, 3.98, 4.09), ARG110 (2.60) |
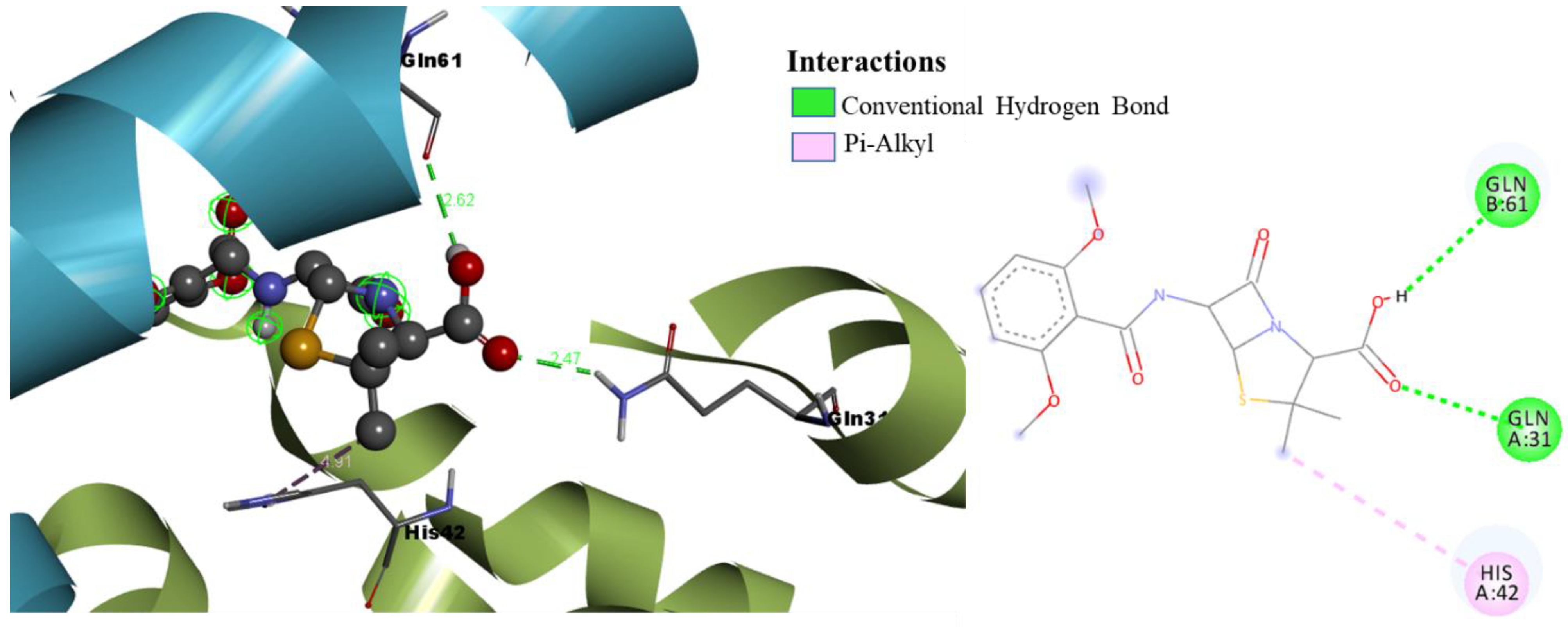
| Methicillin | GLN31 (2.47), GLN61 (2.62) | HIS42 (4.91) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vuppala, S.; Kim, J.; Joo, B.-S.; Choi, J.-M.; Jang, J. A Combination of Pharmacophore-Based Virtual Screening, Structure-Based Lead Optimization, and DFT Study for the Identification of S. epidermidis TcaR Inhibitors. Pharmaceuticals 2022, 15, 635. https://doi.org/10.3390/ph15050635
Vuppala S, Kim J, Joo B-S, Choi J-M, Jang J. A Combination of Pharmacophore-Based Virtual Screening, Structure-Based Lead Optimization, and DFT Study for the Identification of S. epidermidis TcaR Inhibitors. Pharmaceuticals. 2022; 15(5):635. https://doi.org/10.3390/ph15050635
Chicago/Turabian StyleVuppala, Srimai, Jaeyoung Kim, Bo-Sun Joo, Ji-Myung Choi, and Joonkyung Jang. 2022. "A Combination of Pharmacophore-Based Virtual Screening, Structure-Based Lead Optimization, and DFT Study for the Identification of S. epidermidis TcaR Inhibitors" Pharmaceuticals 15, no. 5: 635. https://doi.org/10.3390/ph15050635
APA StyleVuppala, S., Kim, J., Joo, B. -S., Choi, J. -M., & Jang, J. (2022). A Combination of Pharmacophore-Based Virtual Screening, Structure-Based Lead Optimization, and DFT Study for the Identification of S. epidermidis TcaR Inhibitors. Pharmaceuticals, 15(5), 635. https://doi.org/10.3390/ph15050635