
Synthesis, In Vitro Antiproliferative Activity, and In Silico Evaluation of Novel Oxiranyl-Quinoxaline Derivatives

 ,
,  , , and
, , and 
Abstract
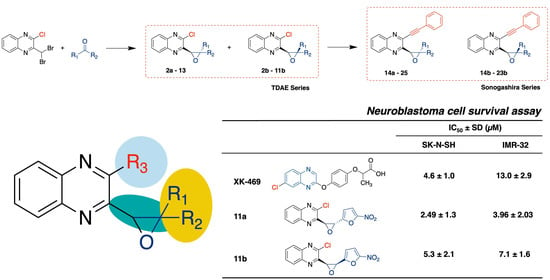
:
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. In Vitro Antiproliferative Activity Evaluation
2.3. In Silico Evaluation
2.3.1. Molecular Docking
2.3.2. ADMET Predictions
3. Materials and Methods
3.1. Chemistry
3.1.1. Generality
3.1.2. General Procedure for Compounds 2 to 13
3.1.3. General Procedure for Compounds 14 to 25
3.2. In Vitro Biological Evaluation
3.2.1. Culture
3.2.2. Drugs
3.2.3. MTT Assay
3.2.4. Data Analysis
3.2.5. Data Analysis
3.3. In Silico Evaluation
3.3.1. Molecular Docking
3.3.2. Pharmacokinetics Modeling with Simulation Plus Software Suite
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sokol, E.; Desai, A.V. The Evolution of Risk Classification for Neuroblastoma. Children 2019, 6, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.W.; Son, M.H.; Cho, H.W.; Ma, Y.E.; Yoo, K.H.; Sung, K.W.; Koo, H.H. Clinical significance of MYCN amplification in patients with high-risk neuroblastoma. Pediatr. Blood Cancer 2018, 65, e27257. [Google Scholar] [CrossRef] [PubMed]
- Whittle, S.B.; Smith, V.; Doherty, E.; Zhao, S.; McCarty, S.; Zage, P.E. Overview and recent advances in the treatment of neuroblastoma. Expert Rev. Anticancer Ther. 2017, 17, 369–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hobbie, W.L.; Li, Y.; Carlson, C.; Goldfarb, S.; Laskin, B.; Denburg, M.; Goldmuntz, E.; Mostoufi-Moab, S.; Wilkes, J.; Smith, K.; et al. Late effects in survivors of high-risk neuroblastoma following stem cell transplant with and without total body irradiation. Pediatr. Blood Cancer 2021, 69, e29537. [Google Scholar] [CrossRef]
- Dhillon, S. Dinutuximab: First Global Approval. Drugs 2015, 75, 923–927. [Google Scholar] [CrossRef]
- Markham, A. Naxitamab: First Approval. Drugs 2021, 81, 291–296. [Google Scholar] [CrossRef]
- PDQ Pediatric Treatment Editorial Board. Neuroblastoma Treatment (PDQ®): Health Professional Version; National Cancer Institute: Bethesda, MD, USA, 2002.
- LoRusso, P.M.; Parchment, R.; Demchik, L.; Knight, J.; Polin, L.; Dzubow, J.; Behrens, C.; Harrison, B.; Trainor, G.; Corbett, T.H. Preclinical antitumor activity of XK469 (NSC 656889). Investig. New Drugs 1999, 16, 287–296. [Google Scholar] [CrossRef]
- Ding, Z.; Parchment, R.E.; LoRusso, P.M.; Zhou, J.Y.; Li, J.; Lawrence, T.S.; Sun, Y.; Wu, G.S. The investigational new drug XK469 induces G(2)-M cell cycle arrest by p53-dependent and -independent pathways. Clin. Cancer Res. 2001, 7, 3336–3342. [Google Scholar]
- Alousi, A.M.; Boinpally, R.; Wiegand, R.; Parchment, R.; Gadgeel, S.; Heilbrun, L.K.; Wozniak, A.J.; DeLuca, P.; LoRusso, P.M. A phase 1 trial of XK469: Toxicity profile of a selective topoisomerase IIbeta inhibitor. Investig. New Drugs 2007, 25, 147–154. [Google Scholar] [CrossRef]
- Kakodkar, N.C.; Peddinti, R.; Kletzel, M.; Tian, Y.; Guerrero, L.J.; Undevia, S.D.; Geary, D.; Chlenski, A.; Yang, Q.; Salwen, H.R.; et al. The quinoxaline anti-tumor agent (R+)XK469 inhibits neuroblastoma tumor growth. Pediatr. Blood Cancer 2011, 56, 164–167. [Google Scholar] [CrossRef] [Green Version]
- Montana, M.; Montero, V.; Khoumeri, O.; Vanelle, P. Quinoxaline Derivatives as Antiviral Agents: A Systematic Review. Molecules 2020, 25, 2784. [Google Scholar] [CrossRef] [PubMed]
- Montana, M.; Montero, V.; Khoumeri, O.; Vanelle, P. Quinoxaline Moiety: A Potential Scaffold against Mycobacterium tuberculosis. Molecules 2021, 26, 4742. [Google Scholar] [CrossRef] [PubMed]
- Montana, M.; Mathias, F.; Terme, T.; Vanelle, P. Antitumoral activity of quinoxaline derivatives: A systematic review. Eur. J. Med. Chem. 2019, 163, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Ajani, O.O. Present status of quinoxaline motifs: Excellent pathfinders in therapeutic medicine. Eur. J. Med. Chem. 2014, 85, 688–715. [Google Scholar] [CrossRef] [PubMed]
- Pereira, J.A.; Pessoa, A.M.; Cordeiro, M.N.D.S.; Fernandes, R.; Prudêncio, C.; Noronha, J.P.; Vieira, M. Quinoxaline, its derivatives and applications: A State of the Art review. Eur. J. Med. Chem. 2015, 97, 664–672. [Google Scholar] [CrossRef] [Green Version]
- Suthar, S.K.; Chundawat, N.S.; Pal Singh, G.; Padrón, J.M.; Kunwar Jhala, Y. Quinoxaline: A Comprehension of current pharmacological advancement in medicinal chemistry. Eur. J. Med. Chem. Rep. 2022, 5, 100040. [Google Scholar] [CrossRef]
- Fisherman, J.S.; Osborn, B.L.; Chun, H.G.; Plowman, J.; Smith, A.C.; Christian, M.C.; Zaharko, D.S.; Shoemaker, R.H. Chloroquinoxaline sulfonamide: A sulfanilamide antitumor agent entering clinical trials. Investig. New Drugs 1993, 11, 1–9. [Google Scholar] [CrossRef]
- Gao, H.; Yamasaki, E.F.; Chan, K.K.; Shen, L.L.; Snapka, R.M. DNA sequence specificity for topoisomerase II poisoning by the quinoxaline anticancer drugs XK469 and CQS. Mol. Pharmacol. 2003, 63, 1382–1388. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Kim, N.; Kim, S.J.; Song, J.; Gong, Y.D.; Kim, S.Y. Anti-cancer effect of a quinoxaline derivative GK13 as a transglutaminase 2 inhibitor. J. Cancer Res. Clin. Oncol. 2013, 139, 1279–1294. [Google Scholar] [CrossRef]
- Budillon, A.; Carbone, C.; Di Gennaro, E. Tissue transglutaminase: A new target to reverse cancer drug resistance. Amino Acids 2013, 44, 63–72. [Google Scholar] [CrossRef] [Green Version]
- Montana, M.; Correard, F.; Khoumeri, O.; Esteve, M.A.; Terme, T.; Vanelle, P. Synthesis of new quinoxalines containing an oxirane ring by the TDAE strategy and in vitro evaluation in neuroblastoma cell lines. Molecules 2014, 19, 14987–14998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, A.R.; Varela, C.L.; Tavares-da Silva, E.J.; Roleira, F.M.F. Epoxide containing molecules: A good or a bad drug design approach. Eur. J. Med. Chem. 2020, 201, 112327. [Google Scholar] [CrossRef] [PubMed]
- Hajri, M.; Esteve, M.A.; Khoumeri, O.; Abderrahim, R.; Terme, T.; Montana, M.; Vanelle, P. Synthesis and evaluation of in vitro antiproliferative activity of new ethyl 3-(arylethynyl)quinoxaline-2-carboxylate and pyrido[4,3-b]quinoxalin-1(2H)-one derivatives. Eur. J. Med. Chem. 2016, 124, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Fersing, C.; Boudot, C.; Paoli-Lombardo, R.; Primas, N.; Pinault, E.; Hutter, S.; Castera-Ducros, C.; Kabri, Y.; Pedron, J.; Bourgeade-Delmas, S.; et al. Antikinetoplastid SAR study in 3-nitroimidazopyridine series: Identification of a novel non-genotoxic and potent anti-T. b. brucei hit-compound with improved pharmacokinetic properties. Eur. J. Med. Chem. 2020, 206, 112668. [Google Scholar] [CrossRef]
- Mathias, F.; Cohen, A.; Kabri, Y.; Negrão, N.W.; Crozet, M.D.; Docampo, R.; Azas, N.; Vanelle, P. Synthesis and in vitro evaluation of new 5-substituted 6-nitroimidazooxazoles as antikinetoplastid agents. Eur. J. Med. Chem. 2020, 191, 112146. [Google Scholar] [CrossRef]
- Bosson-Vanga, H.; Primas, N.; Franetich, J.F.; Lavazec, C.; Gomez, L.; Ashraf, K.; Tefit, M.; Soulard, V.; Dereuddre-Bosquet, N.; Le Grand, R.; et al. A New Thienopyrimidinone Chemotype Shows Multistage Activity against Plasmodium falciparum, Including Artemisinin-Resistant Parasites. Microbiol. Spectr. 2021, 9, e0027421. [Google Scholar] [CrossRef]
- Thiele, C.J. Neuroblastoma Cell Lines. In Human Cell Culture; Springer: Dordrecht, The Netherlands, 1998; Volume 1, pp. 21–53. [Google Scholar] [CrossRef]
- Kurowski, C.; Berthold, F. Presence of classical multidrug resistance and P-glycoprotein expression in human neuroblastoma cells. Ann. Oncol. 1998, 9, 1009–1014. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Wu, C.C.; Li, T.K.; Farh, L.; Lin, L.Y.; Lin, T.S.; Yu, Y.J.; Yen, T.J.; Chiang, C.W.; Chan, N.L. Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science 2011, 333, 459–462. [Google Scholar] [CrossRef] [Green Version]
- Jang, T.H.; Lee, D.S.; Choi, K.; Jeong, E.M.; Kim, I.G.; Kim, Y.W.; Chun, J.N.; Jeon, J.H.; Park, H.H. Crystal structure of transglutaminase 2 with GTP complex and amino acid sequence evidence of evolution of GTP binding site. PLoS ONE 2014, 9, e107005. [Google Scholar] [CrossRef] [Green Version]
- Hetényi, C.; van der Spoel, D. Efficient docking of peptides to proteins without prior knowledge of the binding site. Protein Sci. 2002, 11, 1729–1737. [Google Scholar] [CrossRef] [Green Version]
- Hetényi, C.; van der Spoel, D. Blind docking of drug-sized compounds to proteins with up to a thousand residues. FEBS Lett. 2006, 580, 1447–1450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- McClendon, A.K.; Osheroff, N. DNA topoisomerase II, genotoxicity, and cancer. Mutat. Res.-Fundam. Mol. Mech. Mutagen. 2007, 623, 83–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.D.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Wickham, H. Reshaping Data with the reshape Package. J. Stat. Softw. 2007, 21, 1–20. [Google Scholar] [CrossRef]
- Palakkan, A.A.; Davies, J. Bioassays: Summarising Multi Well Plate Cellular Assay, R package version 1.0.1 CRAN Repository; 2020. Available online: https://cran.rstudio.com/web/packages/bioassays/index.html (accessed on 31 October 2021).
- Rousseeuw, P.J.; Hubert, M. Robust statistics for outlier detection. Wiley Interdiscip. Rev.-Data Min. Knowl. Discov. 2011, 1, 73–79. [Google Scholar] [CrossRef]
- Ritz, C.; Baty, F.; Streibig, J.C.; Gerhard, D. Dose-Response Analysis Using R. PLoS ONE 2015, 10, e0146021. [Google Scholar] [CrossRef] [Green Version]
- Sebaugh, J.L. Guidelines for accurate EC50/IC50 estimation. Pharm. Stat. 2011, 10, 128–134. [Google Scholar] [CrossRef]
- Kassambara, A. Rstatix: Pipe-Friendly Framework for Basic Statistical Tests, R Package Version 0.7.0 CRAN Repository; 2021. Available online: https://rpkgs.datanovia.com/rstatix/ (accessed on 24 December 2021).
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018, 27, 14–25. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TDAE Series | Sonogashira Series | ||||||
|---|---|---|---|---|---|---|---|
| R1 | R2 | Compound | IC50 ± SD μM | Compound | IC50 ± SD μM | ||
| SK-N-SH | IMR-32 | SK-N-SH | IMR-32 | ||||
| H |  | 2a | 26.9 ± 18.83 | 17.34 ± 8.58 | 14a | 10.04 ± 8.26 | 9.18 ± 3.44 |
| 2b | 84.08 ± 35.27 | 66.93 ± 16.19 | 14b | 17.75 ± 7.26 | 15.08 ± 5.25 | ||
| H |  | 3a | 38.01 ± 7.13 | 29.47 ± 6.98 | 15a | 20.77 ± 10.06 | 15.27 ± 7.45 |
| H |  | 4a | >100 | >100 | 16a | >100 | >100 |
| 4b | 55.54 ± 27.05 | 42.12 ± 11.86 | 16b | >100 | >100 | ||
| H |  | 5a | 20.25 ± 4.45 | 22.69 ± 7.49 | 17a | >100 | >100 |
| 5b | 44.09 ± 12.49 | 40.12 ± 5.69 | 17b | >100 | >100 | ||
| H |  | 6a | 19.86 ± 5.96 | 12.98 ± 3.39 | 18a | >100 | >100 |
| H |  | 7a | 15.94 ± 5.83 | 13.08 ± 0.69 | 19a | >100 | 29.93 ± 14.82 |
| 7b | 60.29 ± 11.04 | 44.58 ± 3.31 | 19b | >100 | >100 | ||
| H |  | 8a | 36.21 ± 15.49 | 24.8 ± 11.81 | 20a | 12.08 ± 2.15 | 14.81 ± 2.93 |
| 8b | 33 ± 22.3 | 71.64 ± 25.6 | 20b | 9.5 ± 4.12 | 10.55 ± 4.79 | ||
| H |  | 9a | 39.29 ± 14.87 | 25.59 ± 5.37 | 21a | >100 | >100 |
| 9b | 66.24 ± 7.13 | 59.14 ± 15.68 | 21b | >100 | >100 | ||
| H |  | 10a | 74.29 ± 16.84 | >100 | 22a | >100 | 27.62 ± 13.18 |
| 10b | 47.93 ± 6.73 | >100 | 22b | >100 | >100 | ||
| H |  | 11a | 2.49 ± 1.33 | 3.96 ± 2.03 | 23a | 10.95 ± 0.62 | 14.31 ± 2.35 |
| 11b | 5.3 ± 2.12 | 7.12 ± 1.59 | 23b | 25.35 ± 6.87 | 25.65 ± 1.46 | ||
| H |  | 12a | 47.05 ± 22.91 | 38.26 ± 18.28 | 24a | 58.87 ± 7.79 | 63.2 ± 3.82 |
| COOEt |  | 13 | 55.77 ± 26.37 | 47.81 ± 7.52 | 25 | 10.31 ± 2.4 | 7.26 ± 2.19 |
| XK-469 | 4.6 ± 1.0 | 13.0 ± 2.9 | |||||
| Binding Energies (kcal/mol) | ||
|---|---|---|
| Compounds | h Topoisomerase II β | h Tissue Transglutaminase 2 |
| XK-469 | −7.489 | - |
| GK-13 | - | −7.750 |
| 11a | −6.993 | −7.147 |
| 11b | −6.564 | −6.368 |
| 14a | −8.294 | −7.758 |
| 25 | −7.527 | −6.507 |
| TDAE Series | Sonogashira Series | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | MW | LogP | H-BA | H-BD | Compound | MW | LogP | H-BA | H-BD |
| 2 | 282.731 | 3.280 | 3 | 0 | 14 | 348.407 | 4.317 | 3 | 0 |
| 3 | 296.758 | 3.778 | 3 | 0 | 15 | 362.434 | 4.831 | 3 | 0 |
| 4 | 350.729 | 4.113 | 3 | 0 | 16 | 416.405 | 5.172 | 3 | 0 |
| 5 | 317.176 | 3.982 | 3 | 0 | 17 | 382.852 | 5.026 | 3 | 0 |
| 6 | 317.176 | 3.854 | 3 | 0 | 18 | 382.852 | 4.845 | 3 | 0 |
| 7 | 300.722 | 3.630 | 3 | 0 | 19 | 366.397 | 4.679 | 3 | 0 |
| 8 | 300.722 | 3.549 | 3 | 0 | 20 | 366.397 | 4.606 | 3 | 0 |
| 9 | 307.741 | 3.192 | 4 | 0 | 21 | 373.417 | 4.184 | 4 | 0 |
| 10 | 327.729 | 2.912 | 5 | 0 | 22 | 393.404 | 4.170 | 5 | 0 |
| 11 | 317.69 | 2.281 | 6 | 0 | 23 | 383.366 | 3.550 | 6 | 0 |
| 12 | 278.697 | 2.064 | 5 | 0 | 24 | 344.372 | 3.145 | 5 | 0 |
| 13 | 350.761 | 2.207 | 7 | 0 | 25 | 416.436 | 3.505 | 7 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montero, V.; Montana, M.; Khoumeri, O.; Correard, F.; Estève, M.-A.; Vanelle, P. Synthesis, In Vitro Antiproliferative Activity, and In Silico Evaluation of Novel Oxiranyl-Quinoxaline Derivatives. Pharmaceuticals 2022, 15, 781. https://doi.org/10.3390/ph15070781
Montero V, Montana M, Khoumeri O, Correard F, Estève M-A, Vanelle P. Synthesis, In Vitro Antiproliferative Activity, and In Silico Evaluation of Novel Oxiranyl-Quinoxaline Derivatives. Pharmaceuticals. 2022; 15(7):781. https://doi.org/10.3390/ph15070781
Chicago/Turabian StyleMontero, Vincent, Marc Montana, Omar Khoumeri, Florian Correard, Marie-Anne Estève, and Patrice Vanelle. 2022. "Synthesis, In Vitro Antiproliferative Activity, and In Silico Evaluation of Novel Oxiranyl-Quinoxaline Derivatives" Pharmaceuticals 15, no. 7: 781. https://doi.org/10.3390/ph15070781
APA StyleMontero, V., Montana, M., Khoumeri, O., Correard, F., Estève, M. -A., & Vanelle, P. (2022). Synthesis, In Vitro Antiproliferative Activity, and In Silico Evaluation of Novel Oxiranyl-Quinoxaline Derivatives. Pharmaceuticals, 15(7), 781. https://doi.org/10.3390/ph15070781