
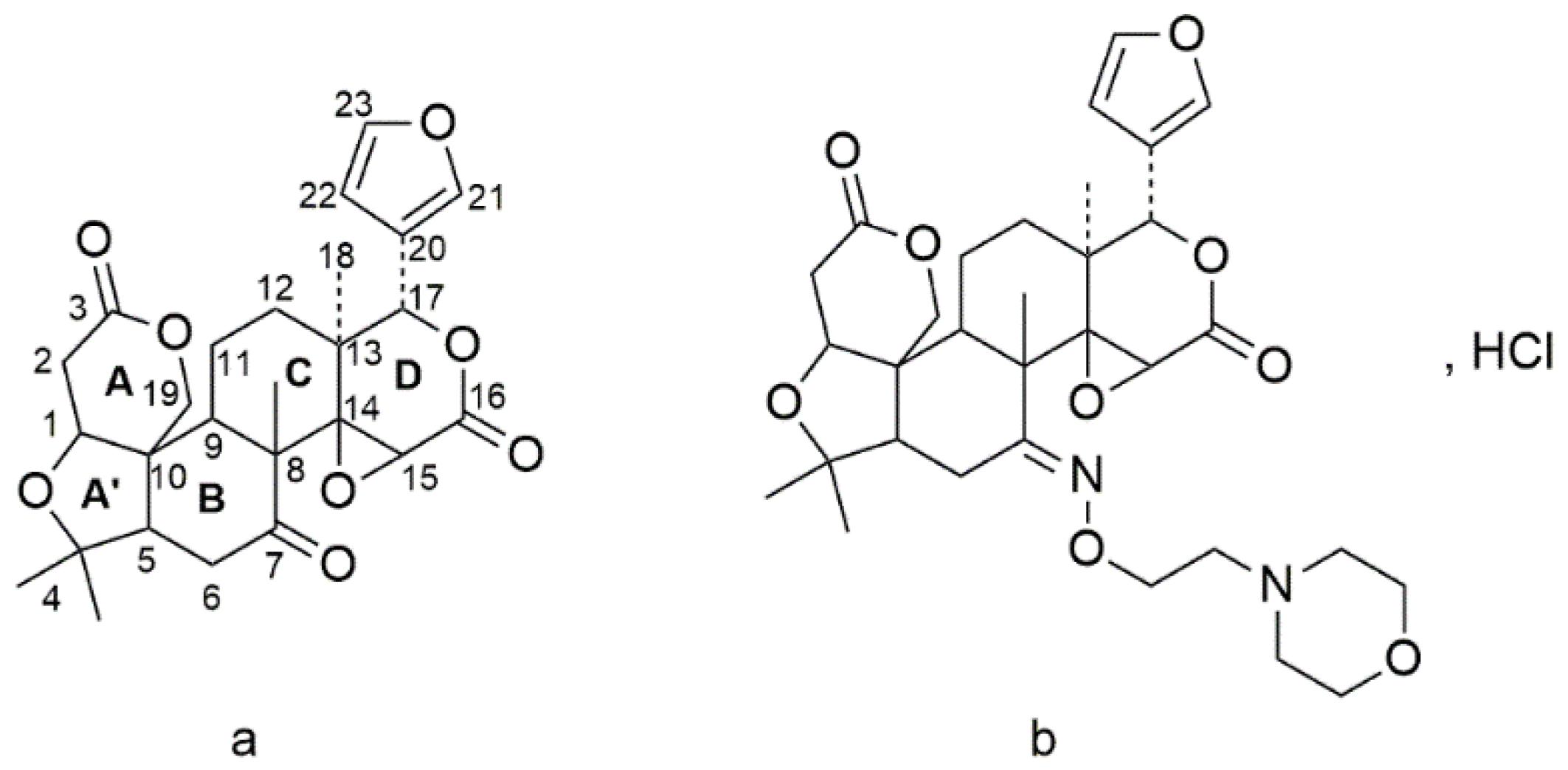
Preclinical Drug Pharmacokinetic, Tissue Distribution and Excretion Profiles of the Novel Limonin Derivate HY-071085 as an Anti-Inflammatory and Analgesic Candidate in Rats and Beagle Dogs


Abstract
:1. Introduction
2. Results
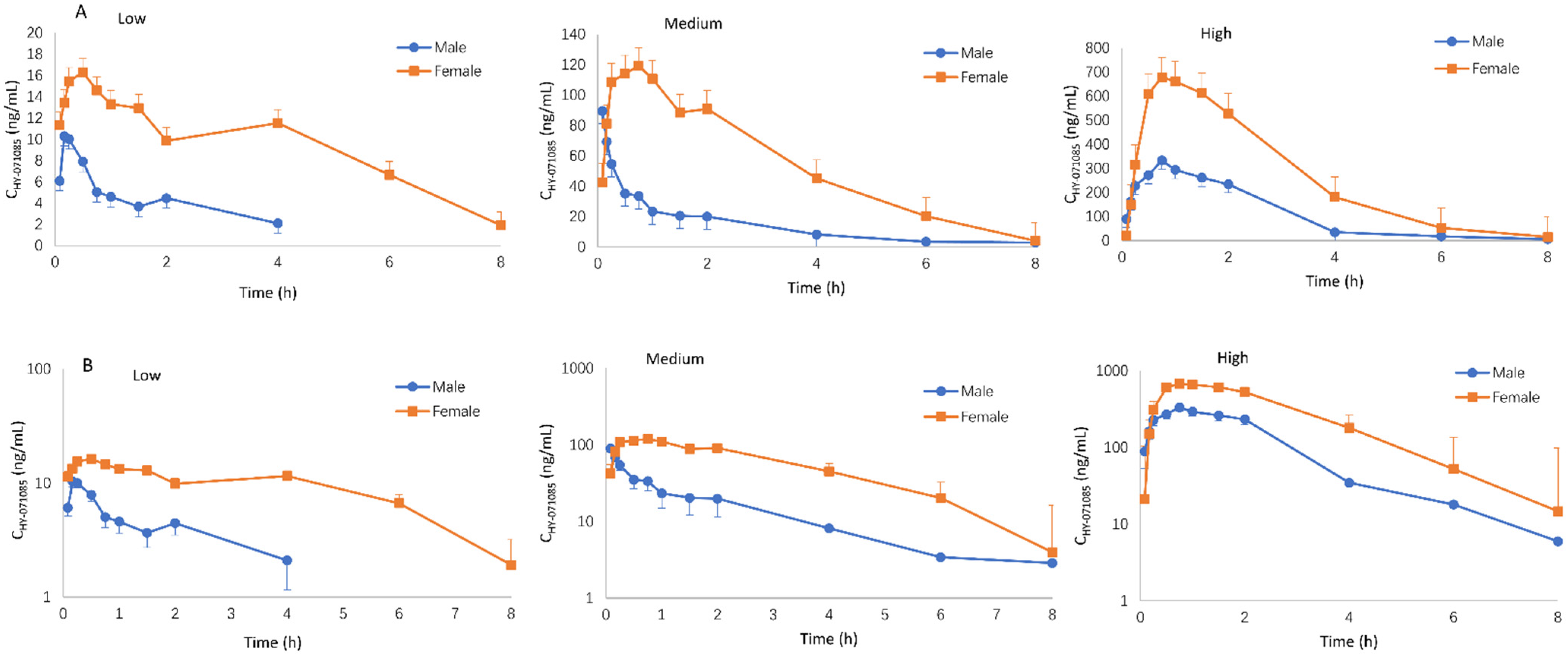
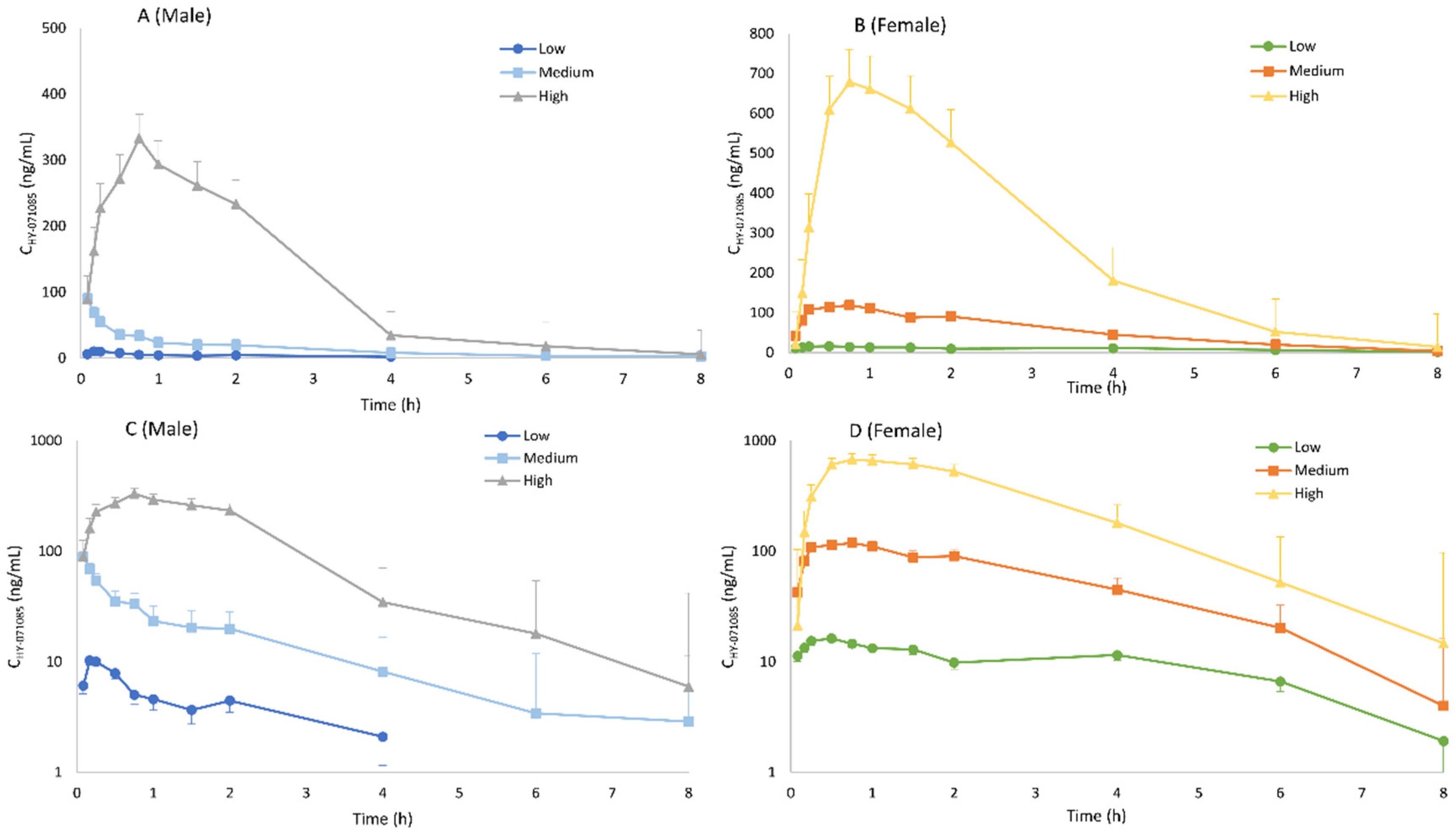
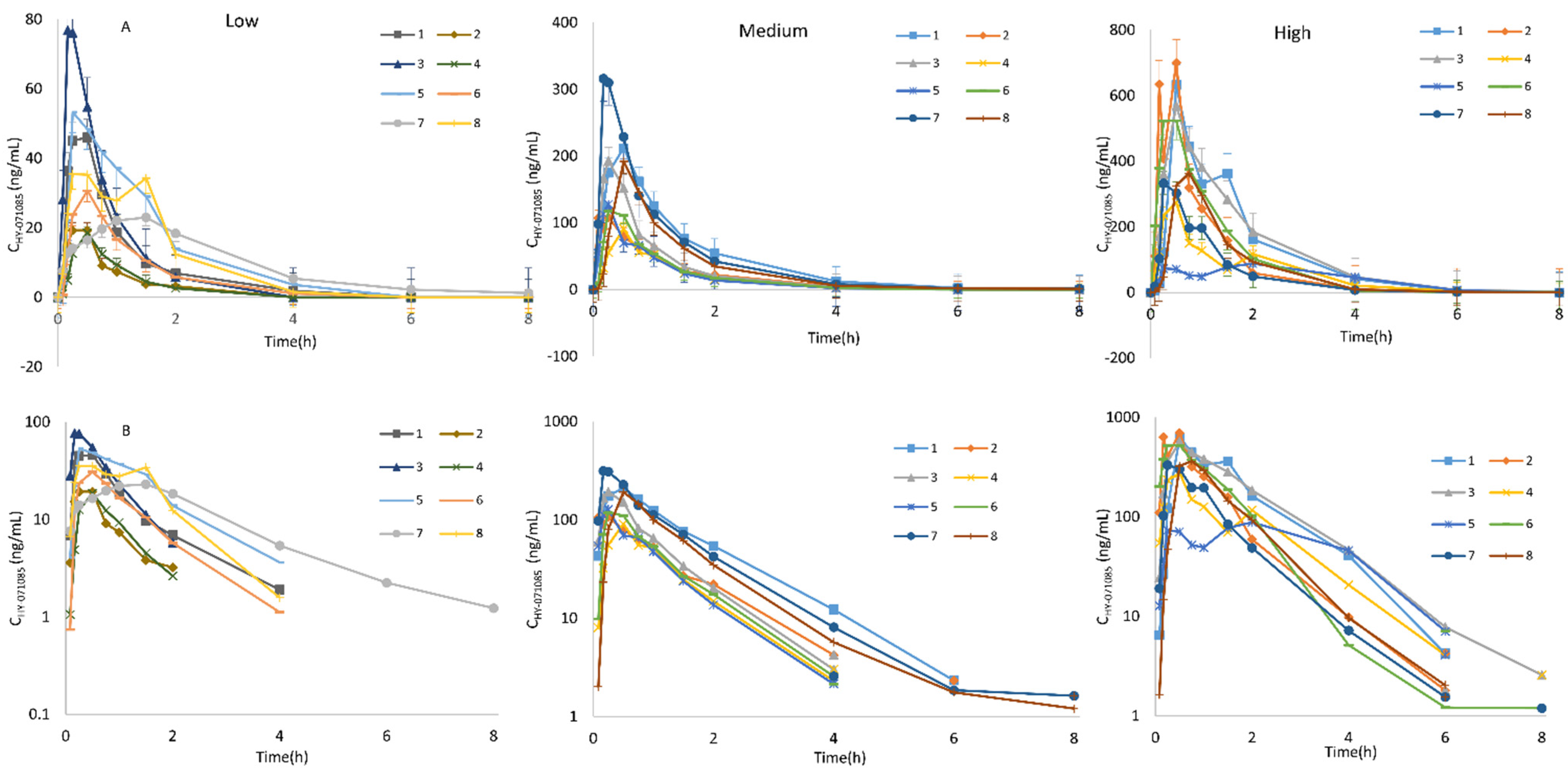
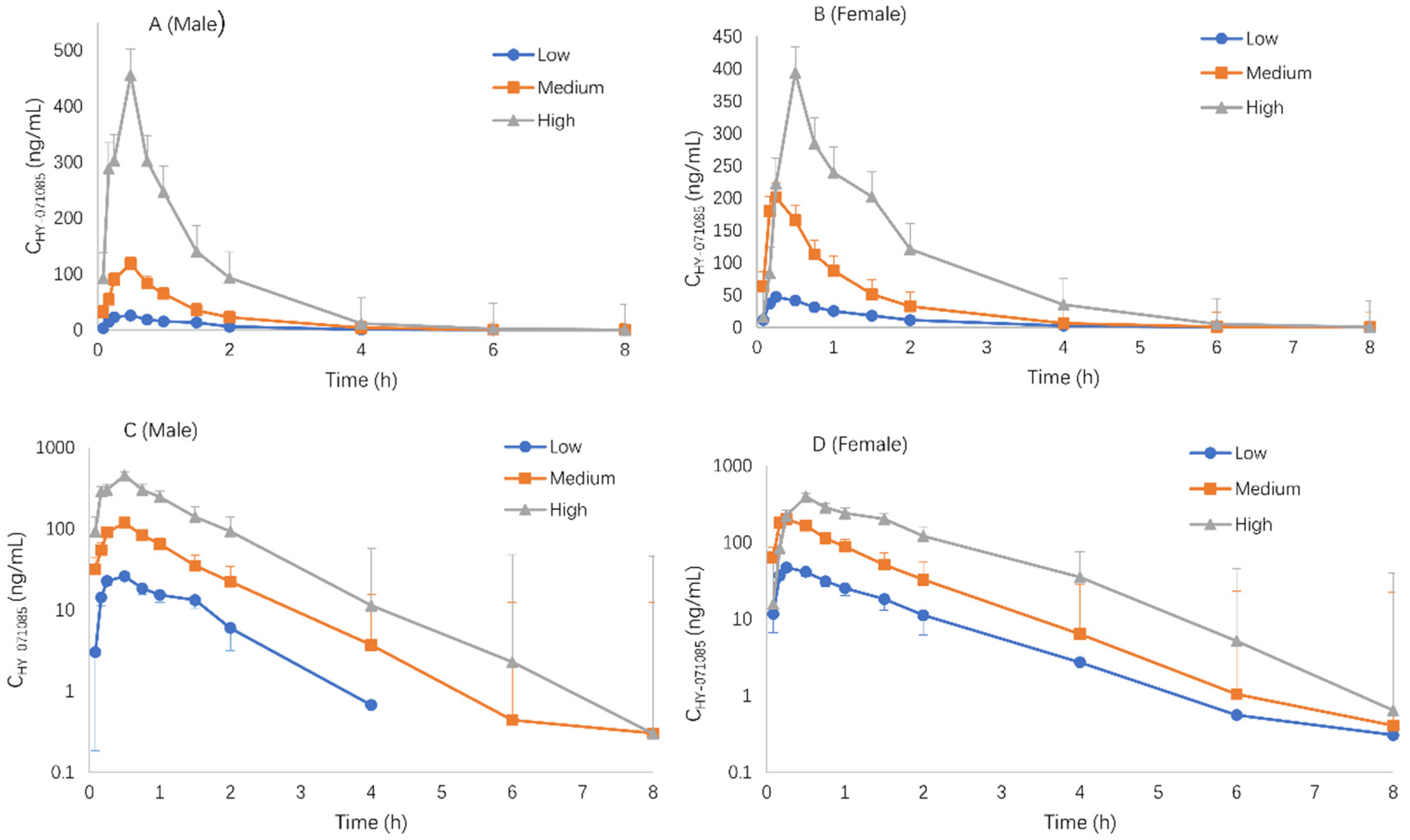
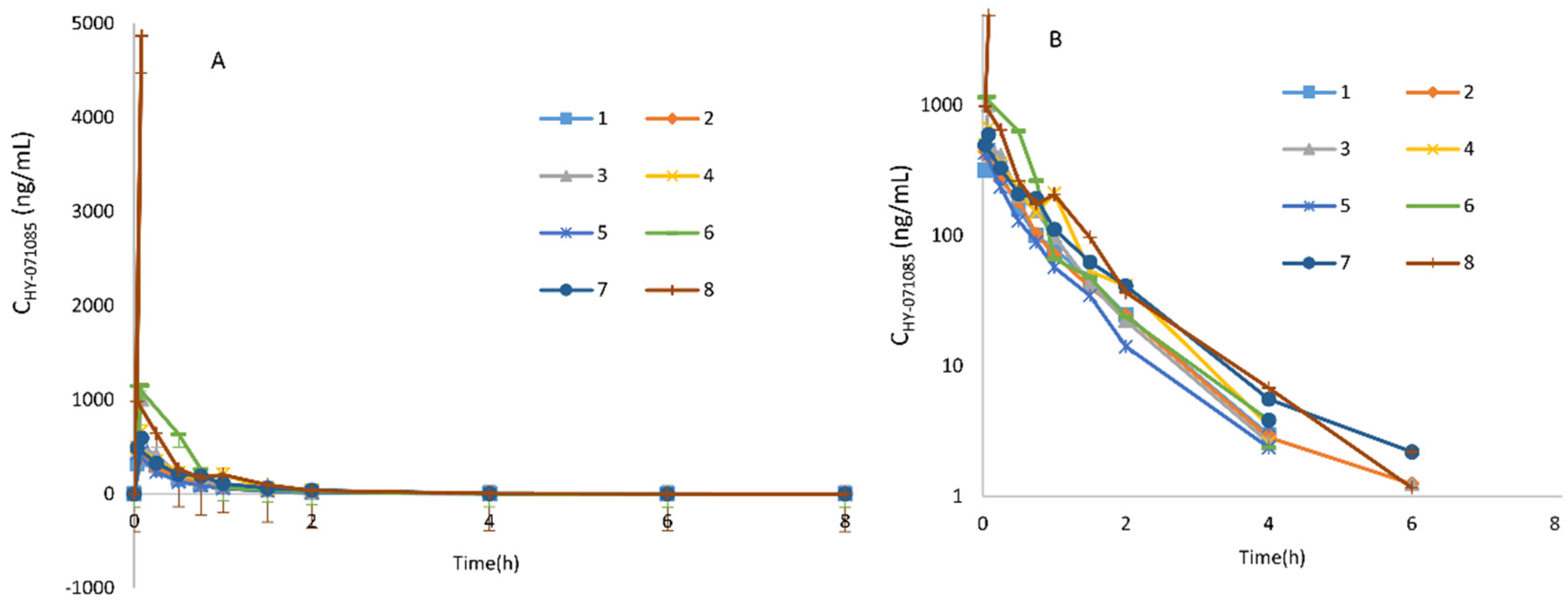
2.1. Pharmacokinetics after a Single Dose of HY-071085 in Rats
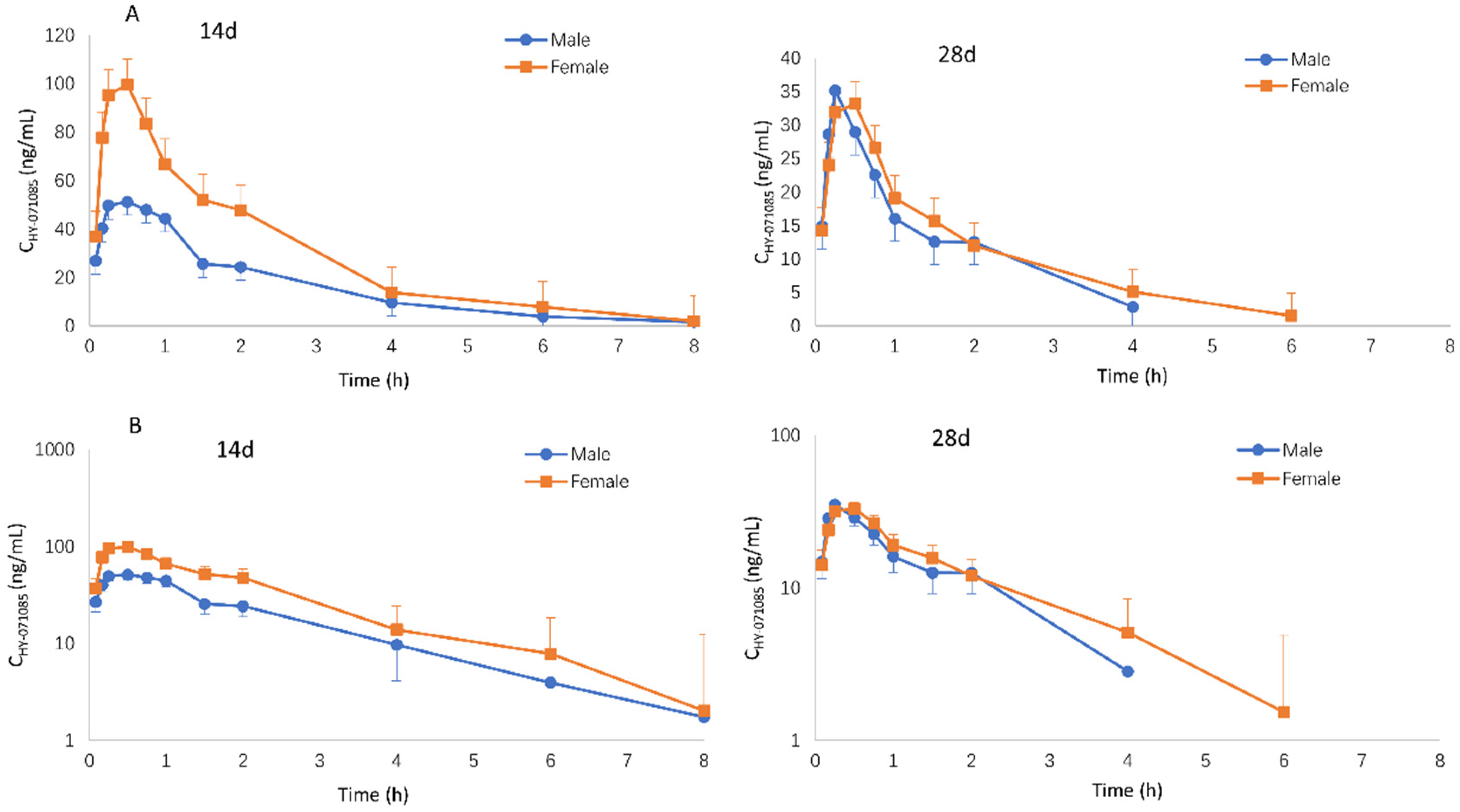
2.2. Pharmacokinetics after Repeated Doses of HY-071085 in Rats
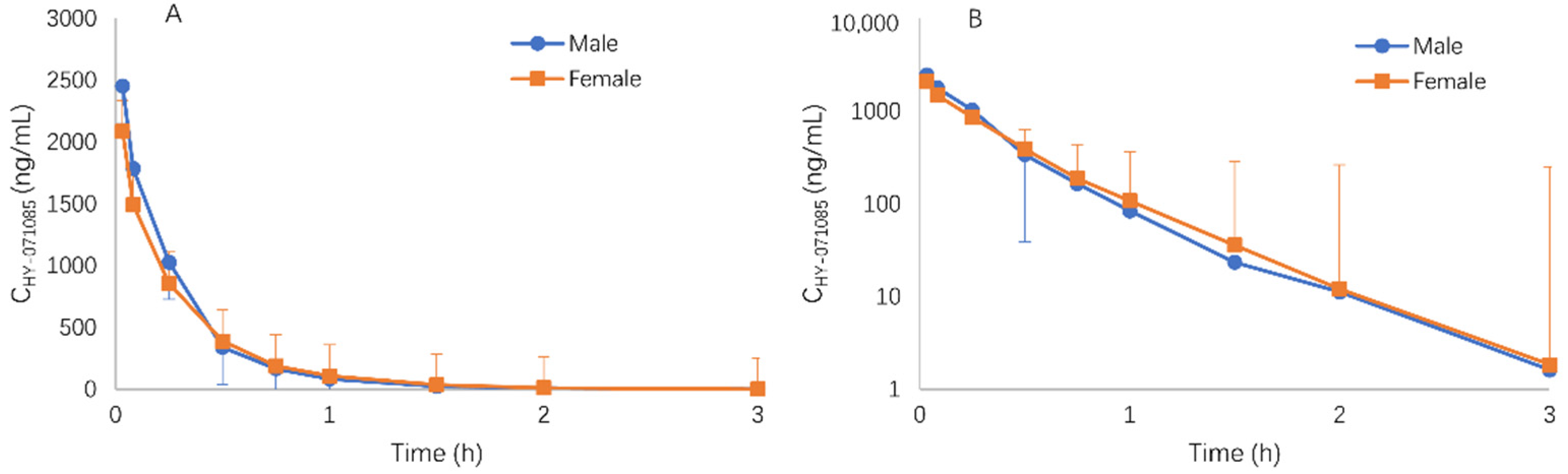
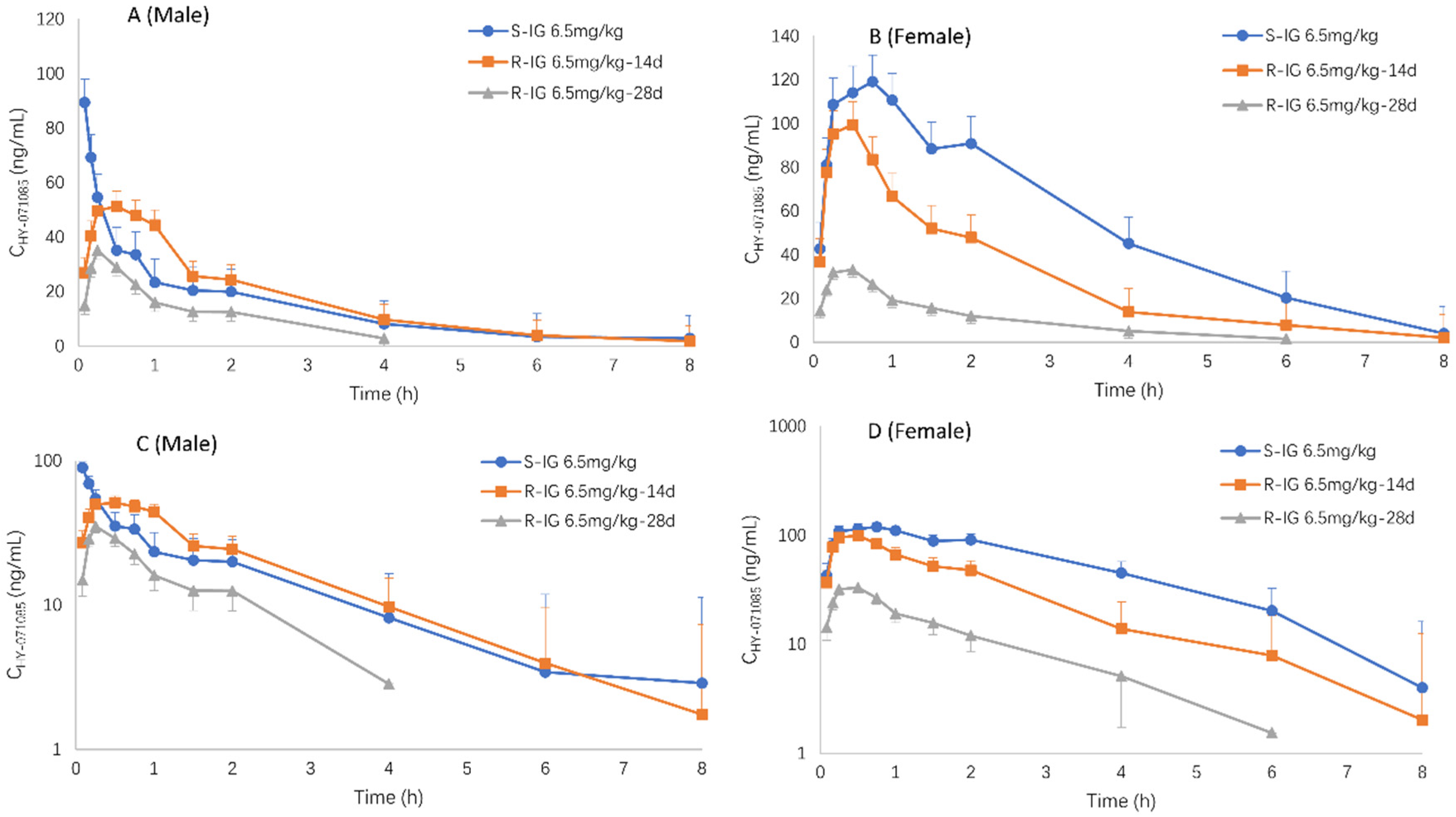
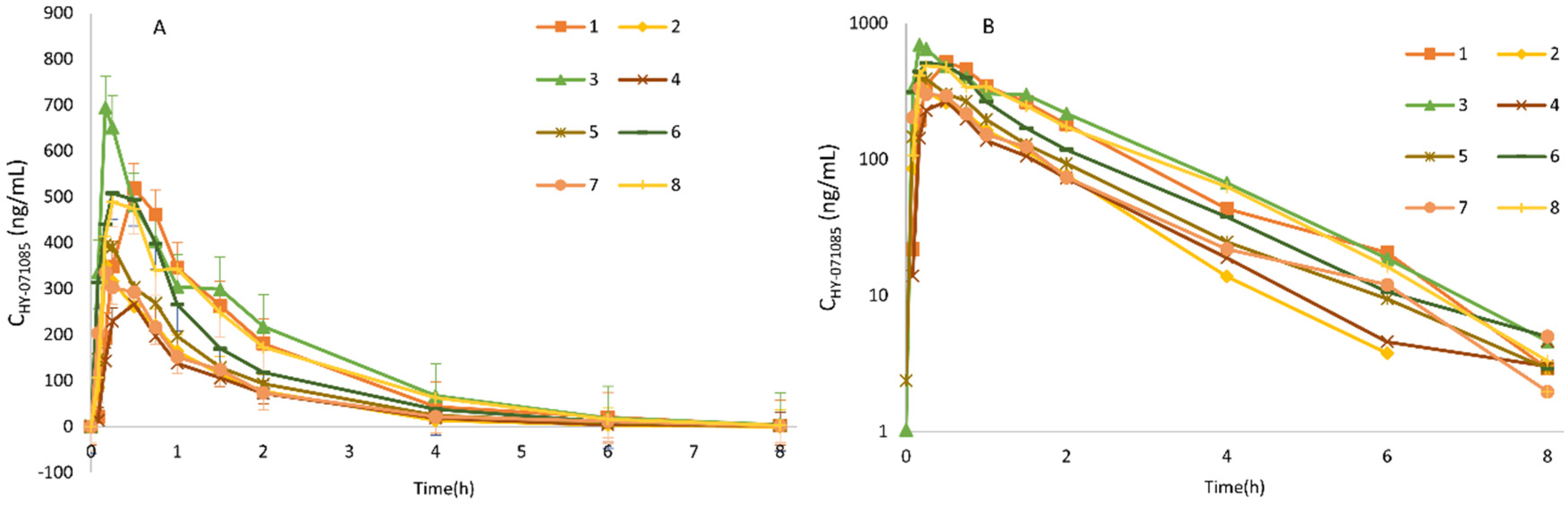
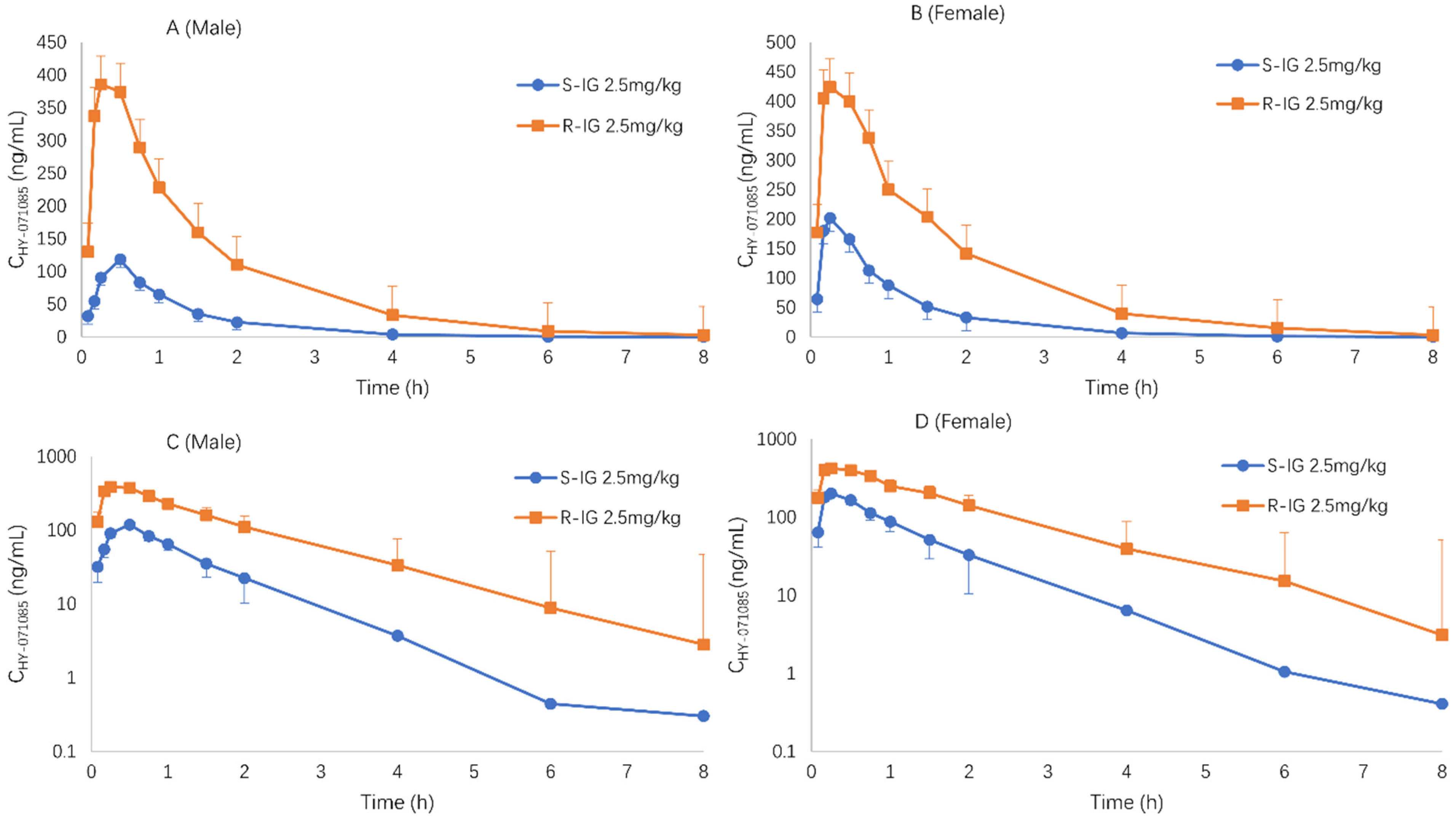
2.3. Pharmacokinetics after Single Doses of HY-071085 in Dogs
2.4. Pharmacokinetics after Repeated Doses of HY-071085 in Dogs
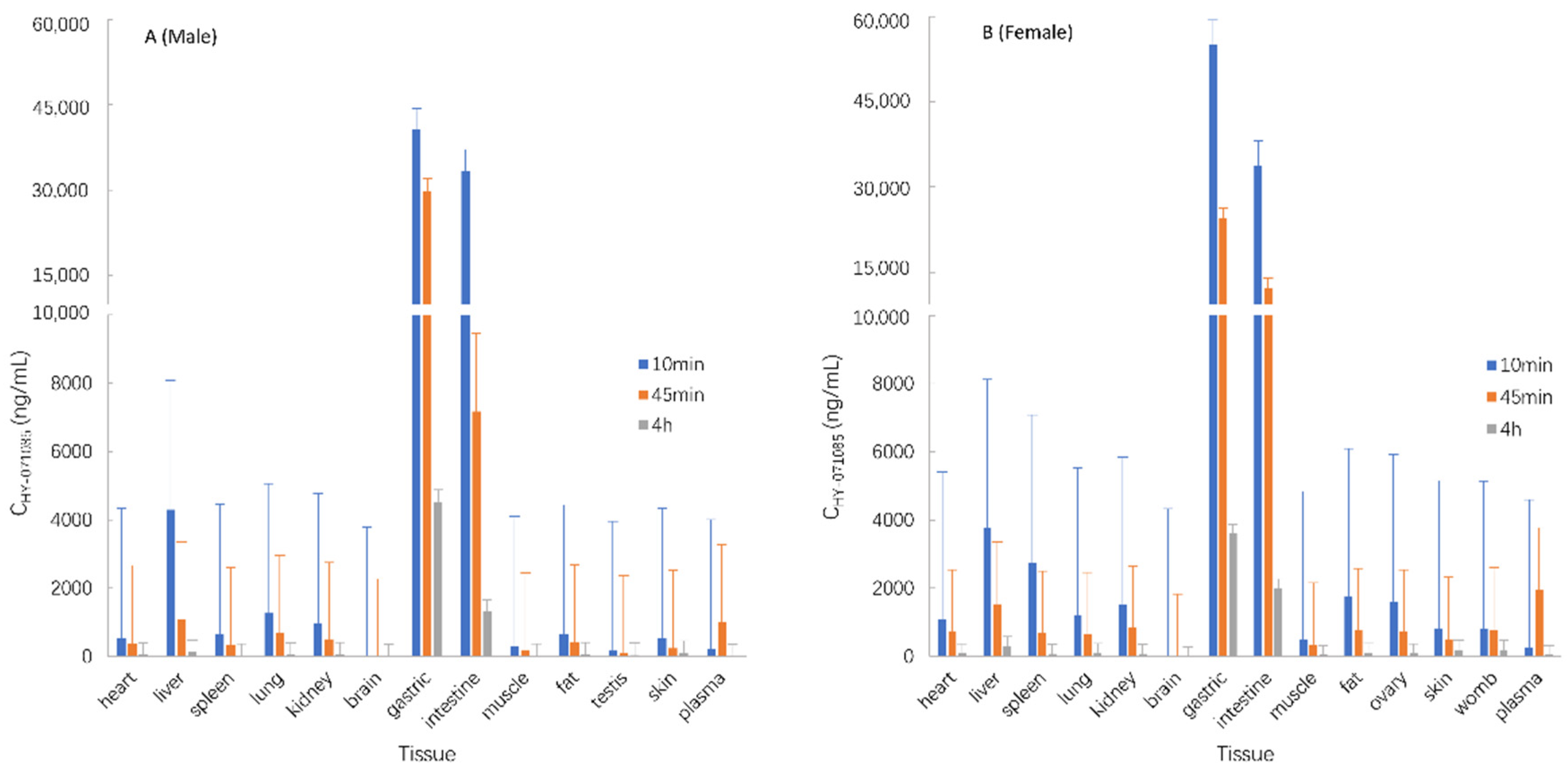
2.5. Tissue Distribution in Rats
2.6. Excretion in Rats
2.7. Plasma Protein Binding
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Instrumental Conditions for UPLC-MS/MS
4.3. Animals and Study Design
4.4. Preparation of the Calibration Standards and QC Samples
4.5. Sample Preparation
4.6. Validation of the Analytical Method
4.7. Pharmacokinetic Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fan, W.; Fan, L.; Wang, Z.; Yang, L. Limonoids from the Genus Melia (Meliaceae): Phytochemistry, Synthesis, Bioactivities, Pharmacokinetics, and Toxicology. Front. Pharmacol. 2022, 12, 795565. [Google Scholar] [CrossRef]
- Roy, A.; Saraf, S. Limonoids: Overview of Significant Bioactive Triterpenes Distributed in Plants Kingdom. Biol. Pharm. Bull. 2006, 29, 191–201. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, H.; Wu, J.X.; Tanaka, T.; Iinuma, M.; Kubo, M. Antinociceptive Activities of 70% Methanol Extract of Evodiae Fructus (Fruit of Evodia rutaecarpa var. bodinieri) and Its Alkaloidal Components. Biol. Pharm. Bull. 1997, 20, 243–248. [Google Scholar] [CrossRef]
- Matsuda, H.; Yoshikawa, M.; Iinuma, M.; Kubo, M. Antinociceptive and anti-inflammatory activities of limonin isolated from the fruits of Evodia rutaecarpa var. bodinieri. Planta Med. 1998, 64, 339–342. [Google Scholar] [CrossRef]
- Fan, S.; Zhang, C.; Luo, T.; Wang, J.; Tang, Y.; Chen, Z.; Yu, L. Limonin: A Review of Its Pharmacology, Toxicity, and Pharmacokinetics. Molecules 2019, 24, 3679. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.; Wang, J.; Chen, S.; Jiang, A.; Jiang, M.; Su, Y.; Yan, W.; Xu, Y.; Gong, G. A novel limonin derivate modulates inflammatory response by suppressing the TLR4/NF-κB signalling pathway. Biomed. Pharmacother. 2018, 100, 501–508. [Google Scholar] [CrossRef]
- Kim, W.; Fan, Y.-Y.; Smith, R.; Patil, B.; Jayaprakasha, G.K.; McMurray, D.N.; Chapkin, R.S. Dietary Curcumin and Limonin Suppress CD4+ T-Cell Proliferation and Interleukin-2 Production in Mice. J. Nutr. 2009, 139, 1042–1048. [Google Scholar] [CrossRef] [Green Version]
- Hassan, N.A.; El Bassossy, H.M.; Fahmy, A.; Mahmoud, M.F. Limonin alleviates macro- and micro-vascular complications of metabolic syndrome in rats: A comparative study with azelnidipine. Phytomedicine 2018, 43, 92–102. [Google Scholar] [CrossRef]
- Zhang, A.; Wang, H.; Sun, H.; Zhang, Y.; An, N.; Yan, G.; Meng, X.; Wang, X. Metabolomics strategy reveals therapeutical assessment of limonin on nonbacterial prostatitis. Food Funct. 2015, 6, 3540–3549. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, X.; Zhu, Q.; Gong, G.; Luo, D.; Jiang, A.; Yang, L.; Xu, Y. Synthesis and pharmacological evaluation of novel limonin derivatives as anti-inflammatory and analgesic agents with high water solubility. Bioorgan. Med. Chem. Lett. 2014, 24, 1851–1855. [Google Scholar] [CrossRef]
- Chidambara Murthy, K.N.; Jayaprakasha, G.K.; Kumar, V.; Rathore, K.S.; Patil, B.S. Citrus limonin and its glucoside inhibit colon adenocarcinoma cell proliferation through apoptosis. J. Agric. Food Chem. 2011, 59, 2314–2323. [Google Scholar] [CrossRef]
- Rahman, A.; Alam Siddiqui, S.; Jakhar, R.; Kang, S.C. Growth Inhibition of Various Human Cancer Cell Lines by Imperatorin and Limonin from Poncirus trifoliata Rafin. Seeds. Anti-Cancer Agents Med. Chem. 2015, 15, 236–241. [Google Scholar] [CrossRef]
- Yu, J.; Wang, L.; Walzem, R.L.; Miller, E.G.; Pike, L.M.; Patil, B.S. Antioxidant Activity of Citrus Limonoids, Flavonoids, and Coumarins. J. Agric. Food Chem. 2005, 53, 2009–2014. [Google Scholar] [CrossRef]
- Vikram, A.; Jesudhasan, P.R.; Jayaprakasha, G.; Pillai, S.D.; Jayaraman, A.; Patil, B.S. Citrus flavonoid represses Salmonella pathogenicity island 1 and motility in S. Typhimurium LT2. Int. J. Food Microbiol. 2011, 145, 28–36. [Google Scholar] [CrossRef]
- Battinelli, L.; Mengoni, F.; Lichtner, M.; Mazzanti, G.; Saija, A.; Mastroianni, C.M.; Vullo, V. Effect of Limonin and Nomilin on HIV-1 Replication on Infected Human Mononuclear Cells. Planta Med. 2003, 69, 910–913. [Google Scholar] [CrossRef]
- Mahmoud, M.F.; Gamal, S.; El-Fayoumi, H.M. Limonin attenuates hepatocellular injury following liver ischemia and reperfusion in rats via toll-like receptor dependent pathway. Eur. J. Pharmacol. 2014, 740, 676–682. [Google Scholar] [CrossRef]
- Yang, N.; Wang, J.; Liu, C.; Song, Y.; Zhang, S.; Zi, J.; Zhan, J.; Masilamani, M.; Cox, A.; Nowak-Wegrzyn, A.; et al. Berberine and limonin suppress IgE production by human B cells and peripheral blood mononuclear cells from food-allergic patients. Ann. Allergy Asthma Immunol. 2014, 113, 556–564.e4. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, J.; Zhou, L.; Yu, B.; Li, C.; Liu, Z.; Ju, W. Quantification of limonin in human urine using solid-phase extraction by LC–MS/MS. J. Chromatogr. B 2012, 907, 163–167. [Google Scholar] [CrossRef]
- Wang, S.C.; Yang, Y.; Liu, J.; Jiang, A.D.; Chu, Z.X.; Chen, S.Y.; Gong, G.-Q.; He, G.-W.; Xu, Y.-G.; Zhu, Q.-H. Discovery of novel limonin derivatives as potent anti-inflammatory and analgesic agents. Chin. J. Nat. Med. 2018, 16, 231–240. [Google Scholar] [CrossRef]
- Wang, S.; Han, X.; Yang, Y.; Zhou, C.; Luo, D.; He, W.; Zhu, Q.; Xu, Y. Discovery of deoxylimonin δ-lactam derivative with favorable anti-inflammation and antinociception efficacy from chemical modified limonin/deoxylimonin analogs. Bioorgan. Chem. 2020, 100, 103886. [Google Scholar] [CrossRef]
- Wang, S.; Han, X.; Yang, Y.; Chen, R.; Guo, Z.; Zhu, Q.; Xu, Y. A practical synthesis of amino limonin/deoxylimonin derivatives as effective mitigators against inflammation and nociception. RSC Med. Chem. 2020, 11, 843–847. [Google Scholar] [CrossRef]
- Zhao, J.; Han, X.; Zhao, X.; Wang, C.; Li, Q.; Chen, X.; Bi, K. A sensitive liquid chromatographic–mass spectrometric method for simultaneous determination of dehydroevodiamine and limonin from Evodia rutaecarpa in rat plasma. Anal. Bioanal. Chem. 2011, 401, 289–296. [Google Scholar] [CrossRef]
- Liang, Y.; Xie, L.; Liu, X.D.; Lu, T.; Wang, G.J.; Hu, Y.Z. Determination of limonin in rat plasma by liquid chromatography–electrospray mass spectrometry. J. Pharm. Biomed. Anal. 2005, 39, 1031–1035. [Google Scholar] [CrossRef]
- Liu, S.J.; Zhou, L.; Zhang, J.; Yu, B.Y.; Li, C.Y.; Liu, Z.X.; Ju, W.Z. Determination of limonin in dog plasma by liquid chromatography-tandem mass spectrometry and its application to a pharmacokinetic study. Biomed. Chromatogr. 2013, 27, 515–519. [Google Scholar] [CrossRef]
- Manners, G.D.; Jacob, R.A.; Breksa, A.P., III; Schoch, T.K.; Hasegawa, S. Bioavailability of citrus limonoids in humans. J. Agric. Food Chem. 2003, 51, 4156–4161. [Google Scholar] [CrossRef]
- Wang, P.; Sun, J.; Gao, E.; Zhao, Y.; Qu, W.; Yu, Z. Simultaneous determination of limonin, dictamnine, obacunone and fraxinellone in rat plasma by a validated UHPLC-MS/MS and its application to a pharmacokinetic study after oral administration of Cortex Dictamni extract. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2013, 928, 44–51. [Google Scholar] [CrossRef]
- Liang, Y.; Xie, L.; Liu, X.D.; Hu, Y.Z.; Lu, T.; Wang, G.J. Gender differences in limonin pharmacokinetics in rats. Eur. J. Drug Metab. Pharmacokinet. 2005, 30, 243–248. [Google Scholar] [CrossRef]
- Xia, Y.Y.; Xu, H.Y.; Cai, Y.Y.; Si, D.Y.; Liu, C.X. Simultaneous determination of evodiamine and evodine in Beagle dog plasma using liquid chromatography tandem mass spectrometry. J. Asian Nat. Prod. Res. 2013, 15, 235–243. [Google Scholar] [CrossRef]
- Xu, H.; Li, Q.; Yin, Y.; Lv, C.; Sun, W.; He, B.; Liu, R.; Chen, X.; Bi, K. Simultaneous determination of three alkaloids, four ginsenosides and limonin in the plasma of normal and headache rats after oral administration of Wu-Zhu-Yu decoction by a novel ultra fast liquid chromatography-tandem mass spectrometry method: Application to a comparative pharmacokinetics and ethological study. J. Mass. Spectrom. 2013, 48, 519–532. [Google Scholar]
- Martignoni, M.; Groothuis, G.M.; de Kanter, R. Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin. Drug Metab. Toxicol. 2006, 2, 875–894. [Google Scholar] [CrossRef]
- Zhou, S.; Kestell, P.; Tingle, M.D.; Paxton, J.W. Gender differences in the metabolism and pharmacokinetics of the experimental anticancer agent 5,6-dimethylxanthenone-4-acetic acid (DMXAA). Cancer Chemother. Pharmacol. 2002, 49, 126–132. [Google Scholar] [CrossRef]
- Li, M.; Wang, H.; Huan, X.; Cao, N.; Guan, H.; Zhang, H.; Cheng, X.; Wang, C. Simultaneous LC-MS/MS bioanalysis of alkaloids, terpenoids, and flavonoids in rat plasma through salting-out-assisted liquid-liquid extraction after oral administration of extract from Tetradium ruticarpum and Glycyrrhiza uralensis: A sample preparation strategy to broaden analyte coverage of herbal medicines. Anal. Bioanal. Chem. 2021, 413, 5871–5884. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Rat | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S-IG (3.25 mg/kg) | S-IG (6.5 mg/kg) | S-IG (13 mg/kg) | S-IV (3.25 mg/kg) | R-IG (6.5 mg/kg) | ||||||||
| Male | Female | Male | Female | Male | Female | Male | Female | Male 14 Days | Female 14 Days | Male 28 Days | Female 28 Days | |
| Cmax (ng/mL) | 11.33 ± 4.54 | 20.42 ± 9.08 | 107.36 ± 139.23 | 145.32 ± 64.54 | 380.80 ± 152.47 | 761.40 ± 321.71 * | - | - | 63.20 ± 11.17 | 124.94 ± 80.28 | 38.86 ± 12.71 | 37.24 ± 19.93 # |
| Tmax (h) | 0.22 ± 0.04 | 0.70 ± 0.73 | 0.45 ± 0.59 | 0.90 ± 0.68 | 0.85 ± 0.45 | 0.95 ± 0.51 | - | - | 0.65 ± 0.38 | 0.68 ± 0.75 | 0.60 ± 0.78 | 0.33 ± 0.16 |
| AUC0-tn (h·ng/mL) | 20.82 ± 3.01 | 73.74 ± 25.31 | 107.54 ± 54.17 | 420.99 ± 205.42 * | 854.77 ± 290.22 | 2079.61± 407.39 ** | 738.95 ± 146.40 | 682.45 ± 224.58 | 126.81 ± 22.45 | 228.83 ± 159.42 | 55.00 ± 8.82 | 65.83 ± 28.92 # |
| AUC0-∞ (h·ng/mL) | - | - | 125.13 ± 72.28 | 428.74 ± 204.08 * | - | - | 740.28 ± 146.42 | 683.92 ± 39.25 | 132.59 ± 22.26 | 232.87 ± 158.92 | 59.85 ± 10.53 | 70.15 ± 28.76 # |
| Cl/F (L/h/kg) | - | - | 70.66 ± 43.65 | 18.13 ± 8.07 * | - | - | 4.50 ± 0.71 | 4.76 ± 0.27 | 50.13 ± 8.32 | 37.82 ± 20.40 | 111.46 ± 20.58 | 103.69 ± 35.13 # |
| MRT (h) | 3.48 ± 1.98 | 4.06 ± 0.82 | 2.89 ± 1.87 | 2.57 ± 0.64 | 1.94 ± 0.39 | 2.29 ± 0.92 | 0.29 ± 0.04 | 0.34 ± 0.05 | 2.26 ± 0.33 | 2.18 ± 0.27 | 1.74 ± 0.86 | 2.54 ± 1.15 |
| Vss/F (L/kg) | - | - | 172.12 ± 70.35 | 36.31 ± 23.24 ** | - | - | 1.31 ± 0.18 | 1.63 ± 0.17 * | 108.46 ± 13.81 | 79.97 ± 51.00 | 168.72 ± 35.19 | 291.76 ± 251.13 |
| t1/2 (h) | 2.35 ± 1.27 | 2.16 ± 1.13 | 1.98 ± 1.04 | 1.30 ± 0.36 | 1.21 ± 0.31 | 0.90 ± 0.42 | 0.40 ± 0.10 | 0.34 ± 0.05 | 1.52 ± 0.23 | 1.38 ± 0.24 | 1.11 ± 0.41 | 1.75 ± 1.14 |
| Parameters | Dog | ||||
|---|---|---|---|---|---|
| S-IG (mg/kg) | S-IV (mg/kg) | R-IG (mg/kg) | |||
| 1.25 | 1.25 | 5 | 1.25 | 2.5 | |
| Cmax (ng/mL) | 37.9 ± 20.05 | 1202.70 ± 1506.40 | 435.68 ± 205.91 | 1202.70 ± 1506.40 | 445.00 ± 135.32 # |
| Tmax (h) | 0.49 ± 0.43 | 0.09 ± 0.03 | 0.69 ± 0.55 | 0.09 ± 0.03 | 0.27 ± 0.15 |
| AUC0-tn (h·ng/mL) | 51.13 ± 24.92 | 591.16 ± 538.62 | 606.04 ± 236.49 | 591.16 ± 538.62 | 723.23 ± 269.96 # |
| AUC0-∞ (h·ng/mL) | 54.06 ± 25.37 | 593.60 ± 538.37 | 609.98 ± 235.31 | 593.60 ± 538.37 | 728.57 ± 270.64 # |
| Cl/F (L/h/kg) | 31.00 ± 20.44 | 3.07 ± 1.45 | 9.38 ± 3.66 | 3.07 ± 1.45 | 1.94 ± 0.72 # |
| MRT (h) | 1.34 ± 0.53 | 0.62 ± 0.20 | 1.46 ± 0.49 | 0.62 ± 0.20 | 1.58 ± 0.14 # |
| Vss/F (L/kg) | 35.26 ± 19.11 | 2.07 ± 1.17 | 11.52 ± 6.03 | 2.07 ± 1.17 | 3.04 ± 1.19 # |
| t1/2 (h) | 0.88 ± 0.44 | 0.70 ± 0.09 | 0.83 ± 0.13 | 0.70 ± 0.09 | 1.08 ± 0.13 # |
| Tissue | HY-071085 Prototype |
|---|---|
| Feces | 0.812 ± 0.611% |
| Urine | 0.254 ± 0.164% |
| Bile | 0.206 ± 0.206% |
| Total | 1.27% |
| Concentration (ng/mL) | 50 | 100 | 200 | 400 | |
|---|---|---|---|---|---|
| Dog | Mean% | 37.81 | 32.86 | 42.18 | 45.46 |
| SD% | 2.60 | 7.84 | 4.60 | 4.38 | |
| Human | Mean% | 100.00 | 73.28 | 67.48 | 61.56 |
| SD% | 0.00 | 6.39 | 1.48 | 3.17 | |
| Rat | Mean% | 73.93 | 75.25 | 77.17 | 78.86 |
| SD% | 1.35 | 1.83 | 2.77 | 0.41 |
| Serial number | Sex | Age | Drug | Route | n | Dose (mg/kg) | Sampling Time |
|---|---|---|---|---|---|---|---|
| 1~5 | M | 6 weeks | HY-071085 | S-IG | 5 | 3.25 | Blood: 5, 10, 15, 30, 45, 60, 90, 120, 360, 480 min |
| 6~10 | F | 6 weeks | HY-071085 | S-IG | 5 | 3.25 | |
| 11~15 | M | 6 weeks | HY-071085 | S-IG | 5 | 6.5 | |
| 16~20 | F | 6 weeks | HY-071085 | S-IG | 5 | 6.5 | |
| 21~25 | M | 6 weeks | HY-071085 | S-IG | 5 | 13 | |
| 26~30 | F | 6 weeks | HY-071085 | S-IG | 5 | 13 | |
| 31~35 | M | 6 weeks | HY-071085 | S-IV | 5 | 3.25 | Blood: 2, 5, 15, 30, 45, 60, 90, 120, 180, 240, 360 min |
| 36~40 | F | 6 weeks | HY-071085 | S-IV | 5 | 3.25 | |
| 41~45 | M | 6 weeks | HY-071085 | R-IG | 5 | 6.5 | Blood 14 days and 28 days: 0.083, 0.167, 0.25, 0.75, 1, 1.5, 2, 4, 6, 8 h |
| 46~50 | F | 6 weeks | HY-071085 | R-IG | 5 | 6.5 | |
| 51~55, 61~65, 71~75 | M | 6 weeks | HY-071085 | S-IG | 15 | 13 | Tissue: 10 min, 45 min, 4 h |
| 56~60, 66~70, 76~80 | F | 6 weeks | HY-071085 | S-IG | 15 | 13 | |
| 81~85 | M | 6 weeks | HY-071085 | S-IG | 5 | 6.5 | Urine, Feces: 0~6 h, 6~12 h, 12~24 h |
| 86~90 | F | 6 weeks | HY-071085 | S-IG | 5 | 6.5 | |
| 91~95 | M | 6 weeks | HY-071085 | S-IG | 5 | 6.5 | Bile: 0~2 h, 2~4 h, 4~6 h, 6~8 h |
| 96~100 | F | 6 weeks | HY-071085 | S-IG | 5 | 6.5 |
| Group | Sex | Age | Drug | Single-Dose Crossover Cycle | R-IG Dose (mg/kg) | Blood Sampling Time | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| I Dose (mg/kg) | II Dose (mg/kg) | III Dose (mg/kg) | IV Dose (mg/kg) | |||||||
| A | 1 | F | 7–9 months | HY-071085 | S-IG (1.25) | S-IG (5) | S-IG (2.5) | S-IV (1.25) | 2.5 | S-IG: 5, 10, 15, 30, 45, 60, 90, 120, 240, 360, 480 min S-IV: 2, 5, 15, 30, 45, 60, 90, 120, 240, 360, 480 min R-IG 7 d: 5, 10, 15, 30, 45, 60, 90, 120, 240, 360, 480 min |
| 2 | M | 2.5 | ||||||||
| B | 3 | F | 7–9 months | HY-071085 | S-IG (2.5) | S-IV (1.25) | S-IG (5) | S-IG (1.25) | 2.5 | |
| 4 | M | 2.5 | ||||||||
| C | 5 | F | 7–9 months | HY-071085 | S-IG (5) | S-IG (1.25) | S-IV (1.25) | S-IG (2.5) | 2.5 | |
| 6 | M | 2.5 | ||||||||
| D | 7 | F | 7–9 months | HY-071085 | S-IV (1.25) | S-IG (2.5) | S-IG (1.25) | S-IG (5) | 2.5 | |
| 8 | M | 2.5 | ||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, L.; Liu, W.; Zhao, X.; Yu, F.; Xu, Y.; Su, M. Preclinical Drug Pharmacokinetic, Tissue Distribution and Excretion Profiles of the Novel Limonin Derivate HY-071085 as an Anti-Inflammatory and Analgesic Candidate in Rats and Beagle Dogs. Pharmaceuticals 2022, 15, 801. https://doi.org/10.3390/ph15070801
Dong L, Liu W, Zhao X, Yu F, Xu Y, Su M. Preclinical Drug Pharmacokinetic, Tissue Distribution and Excretion Profiles of the Novel Limonin Derivate HY-071085 as an Anti-Inflammatory and Analgesic Candidate in Rats and Beagle Dogs. Pharmaceuticals. 2022; 15(7):801. https://doi.org/10.3390/ph15070801
Chicago/Turabian StyleDong, Liping, Wenjuan Liu, Xiaoyuan Zhao, Feng Yu, Yungen Xu, and Mengxiang Su. 2022. "Preclinical Drug Pharmacokinetic, Tissue Distribution and Excretion Profiles of the Novel Limonin Derivate HY-071085 as an Anti-Inflammatory and Analgesic Candidate in Rats and Beagle Dogs" Pharmaceuticals 15, no. 7: 801. https://doi.org/10.3390/ph15070801
APA StyleDong, L., Liu, W., Zhao, X., Yu, F., Xu, Y., & Su, M. (2022). Preclinical Drug Pharmacokinetic, Tissue Distribution and Excretion Profiles of the Novel Limonin Derivate HY-071085 as an Anti-Inflammatory and Analgesic Candidate in Rats and Beagle Dogs. Pharmaceuticals, 15(7), 801. https://doi.org/10.3390/ph15070801