
Curcumin-Based β-Diketo Ligands for Ga3+: Thermodynamic Investigation of Potential Metal-Based Drugs


 , , , ,
, , , ,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Synthesis
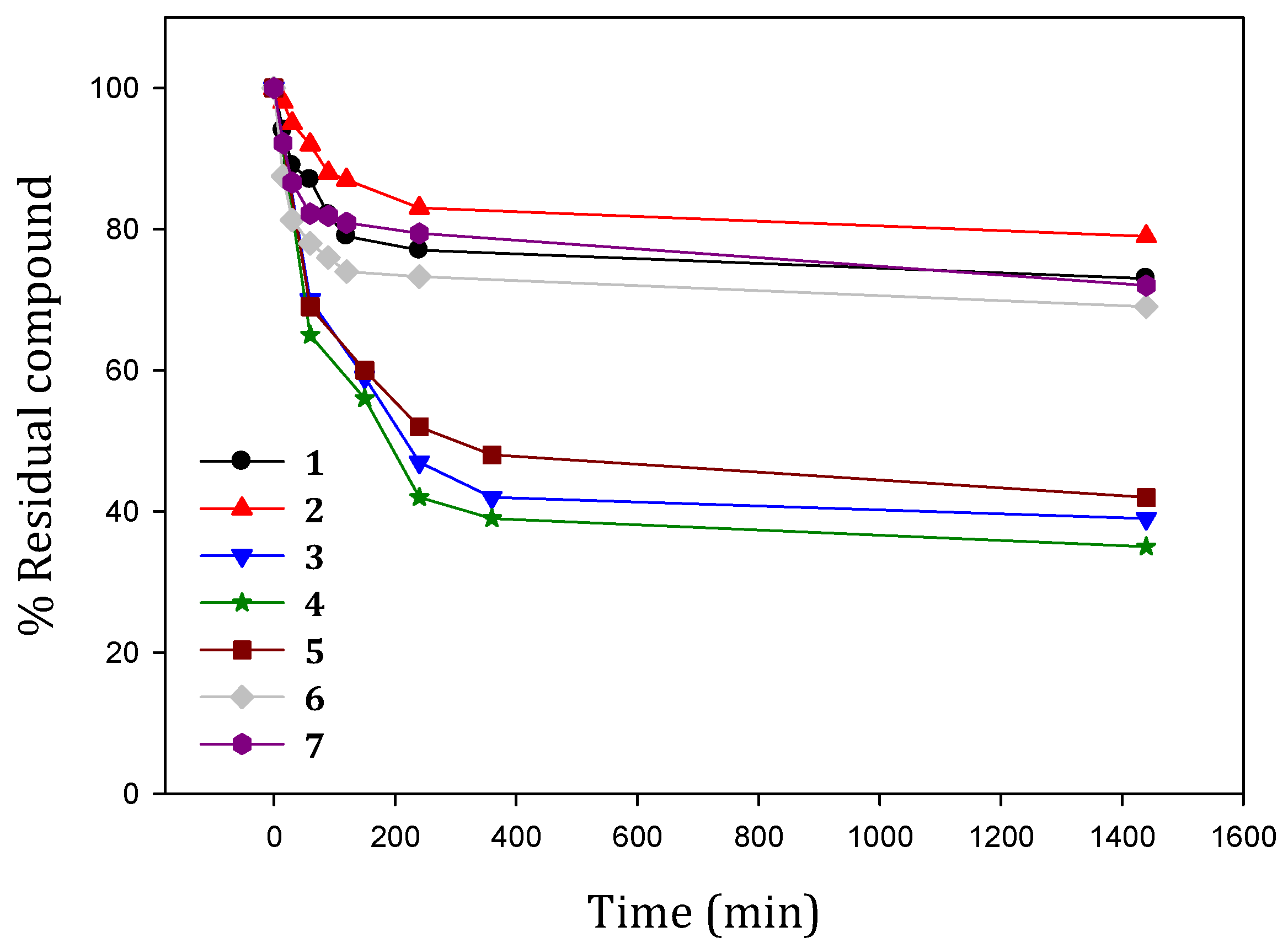
2.2. Stability in Physiological Conditions
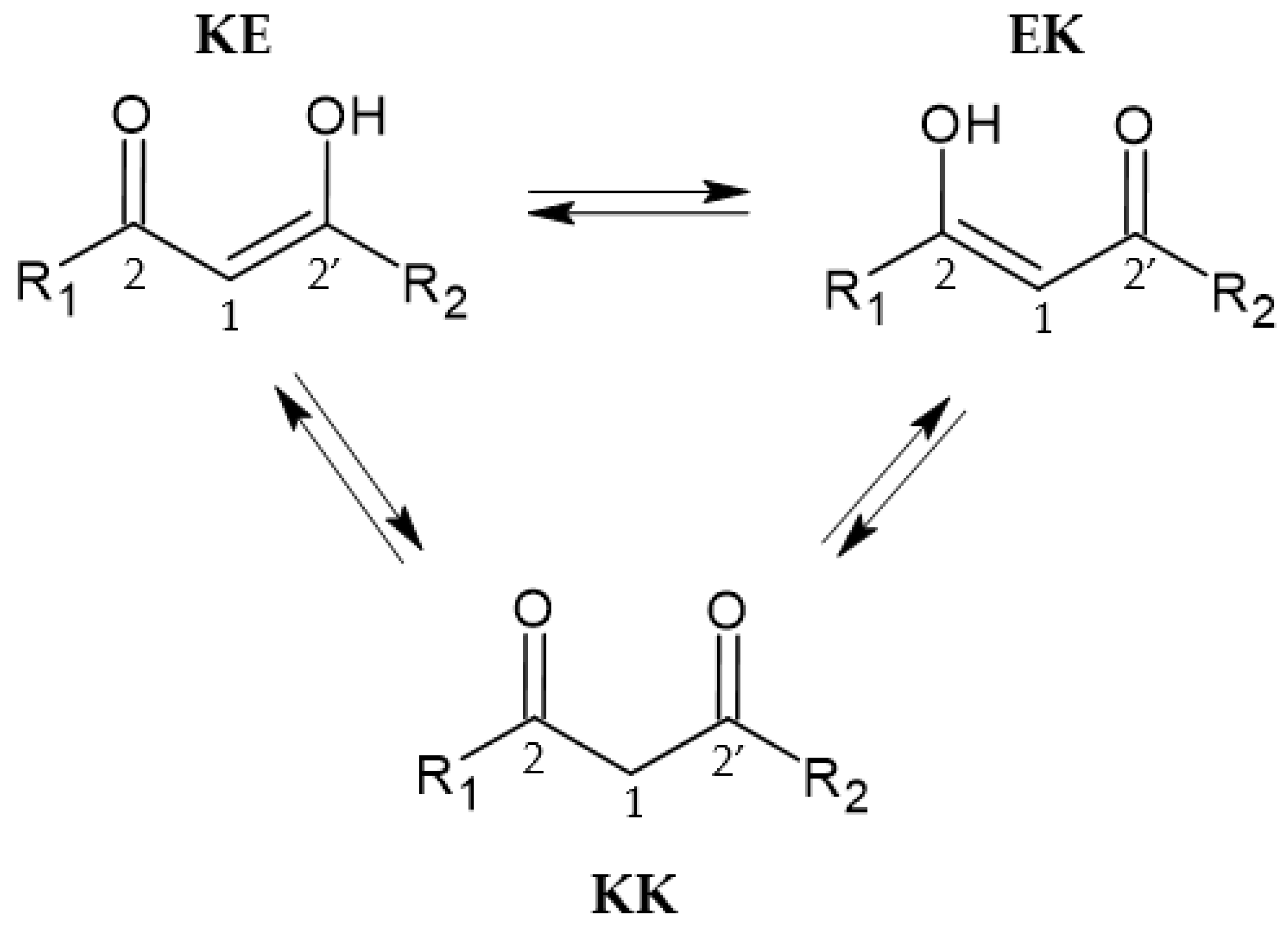
2.3. Tautomeric Equilibria in a Solution and Acid–Base Behavior of Ligands
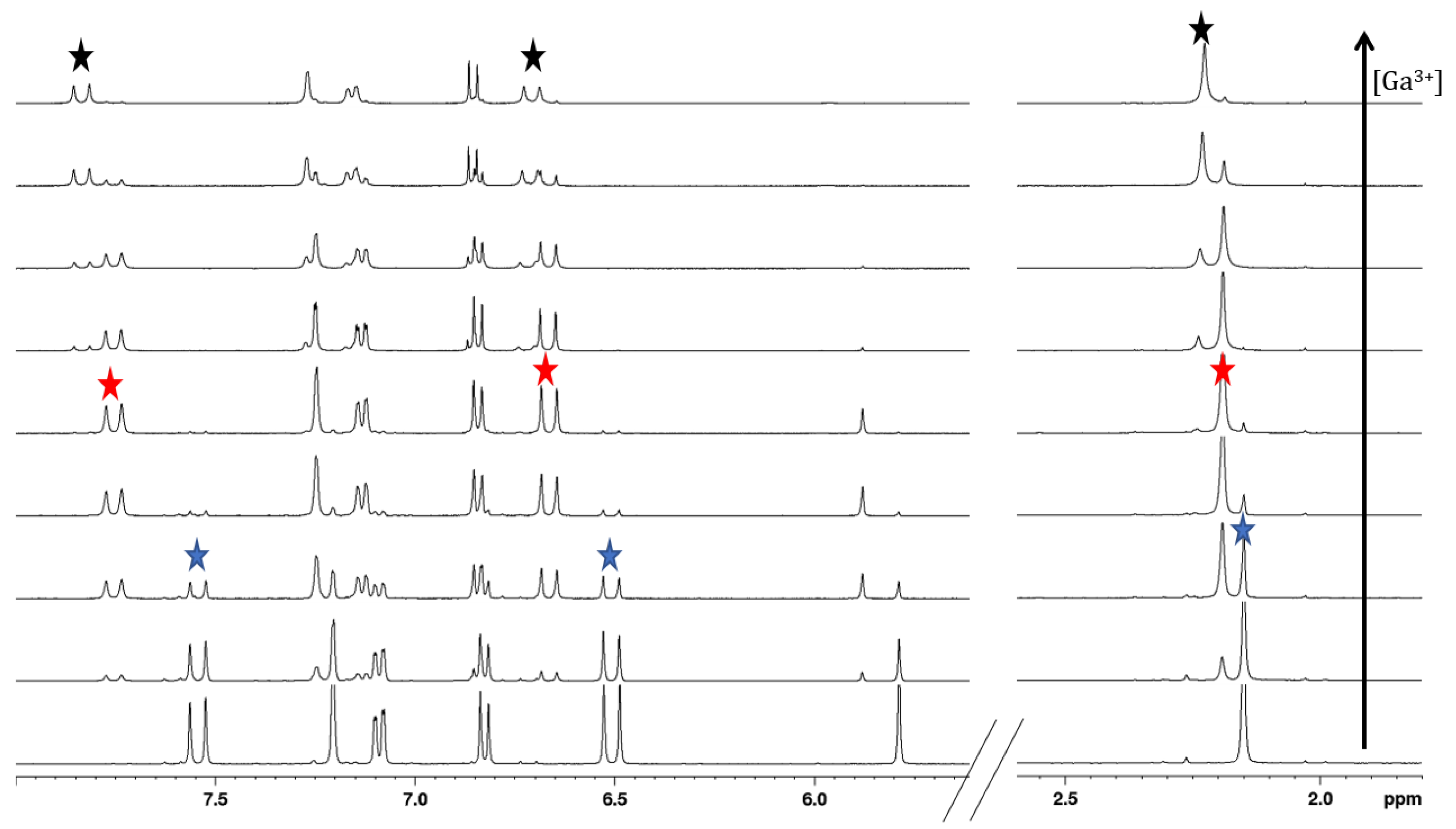
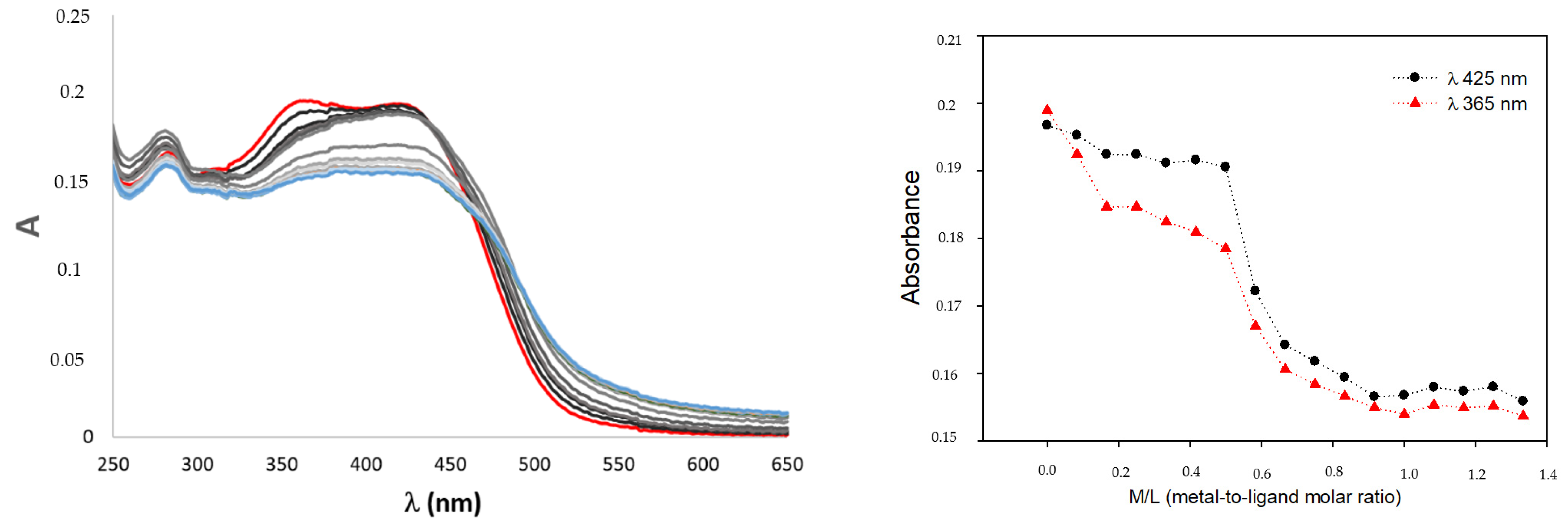
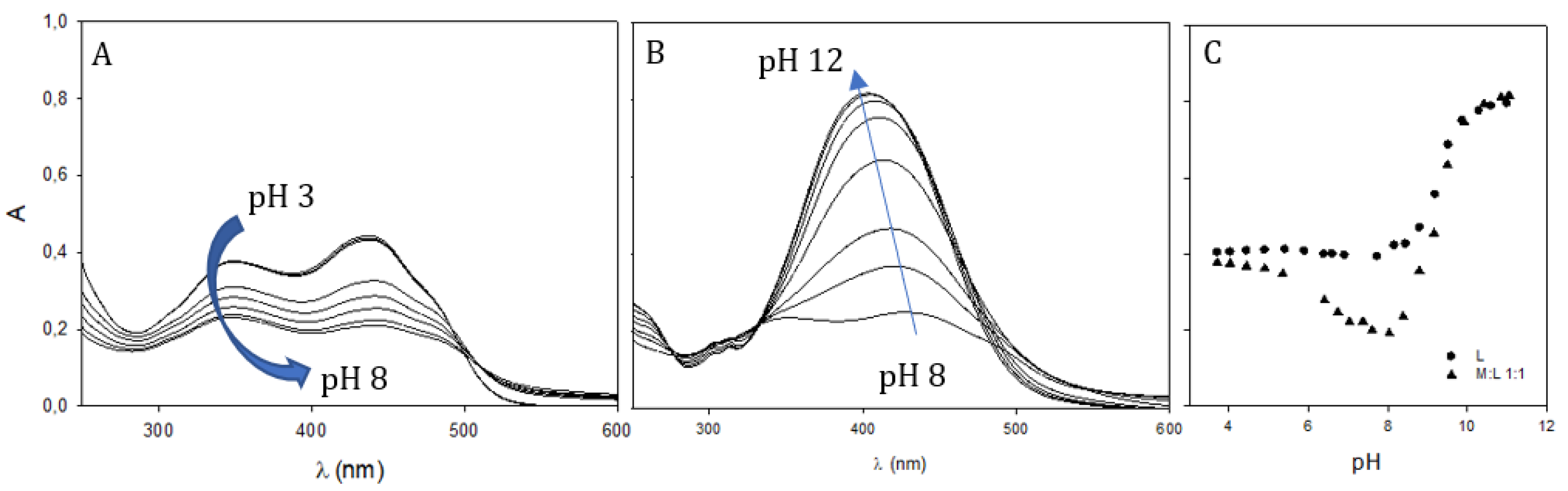
2.4. Gallium Complexing Ability
2.5. Transmetalation Reactions of Selected Curcumin Derivatives 4 and 5
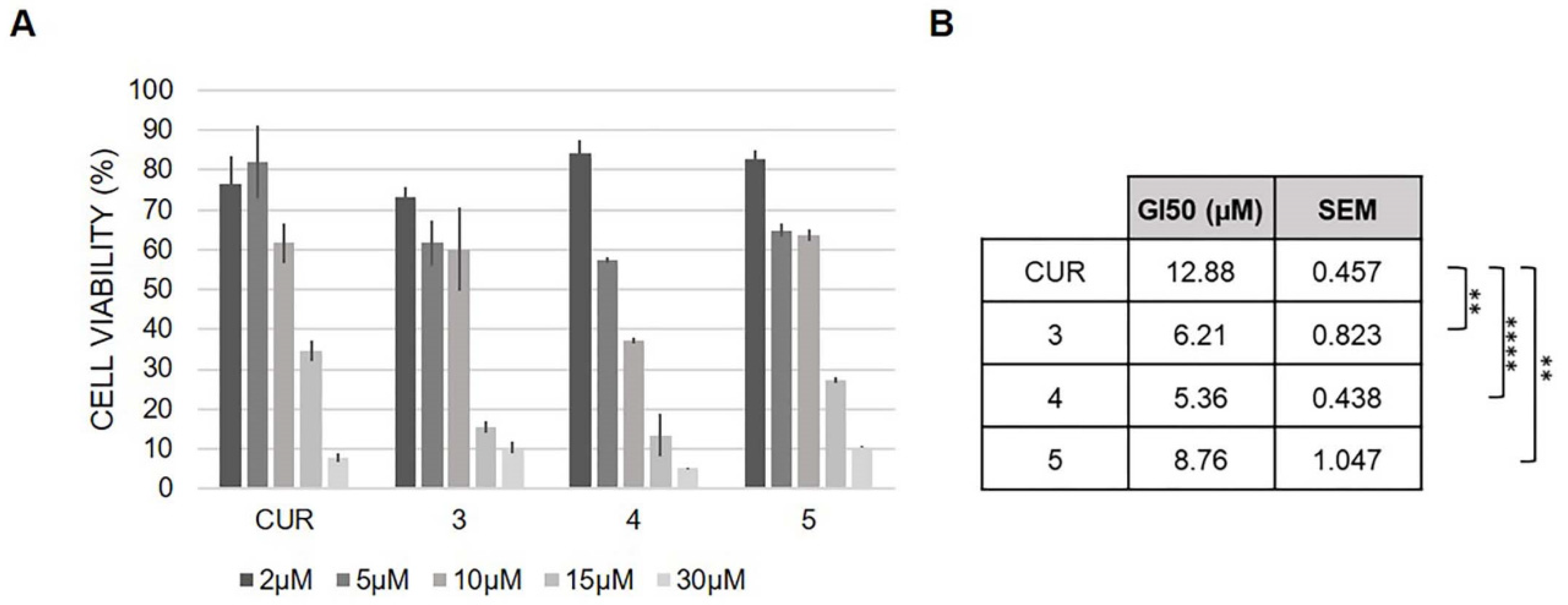
2.6. Antioxidant Activity and Cell Viability Assays
3. Materials and Methods
3.1. General Procedures
3.2. Synthesis
- 4-(3-bromopropyl)-3-methoxybenzaldehyde
- (3Z,5E)-4-hydroxy-6-(3-methoxy-4-methylphenyl)hexa-3,5-dien-2-one (1)
- (3Z,5E)-6-[4-(3-bromopropoxy)-3-methoxyphenyl]-4-hydroxyhexa-3,5-dien-2-one (2)
- (2-((4Z,6E)-5-hydroxy-7-(4-hydroxyphenyl)-4-((E)-3-(4-hydroxy-3-ethoxyphenyl) acryloyl) hepta-4,6-dien-1-yl)isoindoline-1,3-dione (3)
- (2-((4Z,6E)-5-hydroxy-7-(3-methoxy-4-hydroxyphenyl)-4-((E)-3-(4-hydroxy-3-ethoxyphenyl) acryloyl) hepta-4,6-dien-1-yl)isoindoline-1,3-dione (4)
- 2-((4Z,6E)-5-hydroxy-7-(4-hydroxyphenyl)-4-((E)-3-(4-hydroxy-3-ethoxyphenyl) acryloyl) hepta-4,6-dien-1-yl)isoindoline-1,3-dione (5)
- (6E)-7-(4-hydroxyphenyl)-4-[(2E)-3-(4-hydroxyphenyl)prop-2-enoyl]-5-oxohept-6-enoic acid (6)
- (6E)-7-(3-methoxy-4-hydroxyphenyl)-4-[(2E)-3-(3-methoxy-4-hydroxyphenyl)prop-2-enoyl]-5-oxohept-6-enoic acid (7)
3.3. Kinetic Stability of Ligands in Physiological Conditions
3.4. UV–Visible Spectroscopy
3.5. Nuclear Magnetic Resonance
3.6. Antioxidant Activity (DPPH Assay)
3.7. Cell Viability Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Razavi, B.M.; Ghasemzadeh Rahbardar, M.; Hosseinzadeh, H. A review of therapeutic potentials of turmeric (Curcuma longa) and its active constituent, curcumin, on inflammatory disorders, pain, and their related patents. Phyther. Res. 2021, 35, 6489–6513. [Google Scholar] [CrossRef] [PubMed]
- Ojo, O.A.; Adeyemo, T.R.; Rotimi, D.; Batiha, G.E.-S.; Mostafa-Hedeab, G.; Iyobhebhe, M.E.; Elebiyo, T.C.; Atunwa, B.; Ojo, A.B.; Lima, C.M.G.; et al. Anticancer Properties of Curcumin Against Colorectal Cancer: A Review. Front. Oncol. 2022, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kharat, M.; Du, Z.; Zhang, G.; McClements, D.J. Physical and Chemical Stability of Curcumin in Aqueous Solutions and Emulsions: Impact of pH, Temperature, and Molecular Environment. J. Agric. Food Chem. 2017, 65, 1525–1532. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, G.; Kaviyil, J.E.; Paul, W.; Sasi, R.; Joseph, R. Gallium-Curcumin Nanoparticle Conjugates as an Antibacterial Agent against Pseudomonas aeruginosa: Synthesis and Characterization. ACS Omega 2022, 7, 6795–6809. [Google Scholar] [CrossRef] [PubMed]
- Panzarini, E.; Mariano, S.; Tacconi, S.; Carata, E.; Tata, A.M.; Dini, L. Novel therapeutic delivery of nanocurcumin in central nervous system related disorders. Nanomaterials 2021, 11, 2. [Google Scholar] [CrossRef]
- Noureddin, S.A.; El-Shishtawy, R.M.; Al-Footy, K.O. Curcumin analogues and their hybrid molecules as multifunctional drugs. Eur. J. Med. Chem. 2019, 182, 111631. [Google Scholar] [CrossRef]
- Yanagisawa, D.; Kato, T.; Taguchi, H.; Shirai, N.; Hirao, K.; Sogabe, T.; Tomiyama, T.; Gamo, K.; Hirahara, Y.; Kitada, M.; et al. Keto form of curcumin derivatives strongly binds to Aβ oligomers but not fibrils. Biomaterials 2021, 270, 120686. [Google Scholar] [CrossRef]
- Manolova, Y.; Deneva, V.; Antonov, L.; Drakalska, E.; Momekova, D.; Lambov, N. The effect of the water on the curcumin tautomerism: A quantitative approach. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 132, 815–820. [Google Scholar] [CrossRef]
- Puglisi, A.; Giovannini, T.; Antonov, L.; Cappelli, C. Interplay between conformational and solvent effects in UV-visible absorption spectra: Curcumin tautomers as a case study. Phys. Chem. Chem. Phys. 2019, 21, 15504–15514. [Google Scholar] [CrossRef]
- Parimita, S.P.; Ramshankar, Y.V.; Suresh, S.; Row, T.N.G. Redetermination of curcumin: (1E,4Z,6E)-5-hydroxy-1,7-bis(4-hydroxy-3-methoxy-phenyl)hepta-1,4,6-trien-3-one. Acta Crystallogr. Sect. E Struct. Rep. Online 2007, 63, 860–862. [Google Scholar] [CrossRef]
- Prasad, D.; Praveen, A.; Mahapatra, S.; Mogurampelly, S.; Chaudhari, S.R. Existence of β-diketone form of curcuminoids revealed by NMR spectroscopy. Food Chem. 2021, 360, 130000. [Google Scholar] [CrossRef] [PubMed]
- Enumo, A.; Irineu, C.; Pereira, D.; Parize, A.L. Temperature Evaluation of Curcumin Keto−Enolic Kinetics and Its Interaction with Two Pluronic Copolymers. J. Phys. Chem. B 2019, 123, 5641–5650. [Google Scholar] [CrossRef] [PubMed]
- Wanninger, S.; Lorenz, V.; Subhan, A.; Edelmann, F.T. Metal complexes of curcumin—Synthetic strategies, structures and medicinal applications. Chem. Soc. Rev. 2015, 44, 4986–5002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrari, E.; Benassi, R.; Saladini, M.; Orteca, G.; Gazova, Z.; Siposova, K. In vitro study on potential pharmacological activity of curcumin analogues and their copper complexes. Chem. Biol. Drug Des. 2017, 89, 411–419. [Google Scholar] [CrossRef]
- Balasubramanian, K. Quantum chemical insights into Alzheimer’s disease: Curcumin’s chelation with Cu(II), Zn(II), and Pd(II) as a mechanism for its prevention. Int. J. Quantum Chem. 2016, 116, 1107–1119. [Google Scholar] [CrossRef]
- Halevas, E.; Pekou, A.; Papi, R.; Mavroidi, B.; Hatzidimitriou, A.G.; Zahariou, G.; Litsardakis, G.; Sagnou, M.; Pelecanou, M.; Pantazaki, A.A. Synthesis, physicochemical characterization and biological properties of two novel Cu(II) complexes based on natural products curcumin and quercetin. J. Inorg. Biochem. 2020, 208, 111083. [Google Scholar] [CrossRef]
- Prasad, S.; Dubourdieu, D.; Srivastava, A.; Kumar, P.; Lall, R.; Bachmeier, E. Molecular Sciences Metal-Curcumin Complexes in Therapeutics: An Approach to Enhance Pharmacological Effects of Curcumin. Int. J. Mole. Sci. 2021, 22, 7094. [Google Scholar] [CrossRef]
- dos, S.S.; Firmino, G.; André, S.C.; Hastenreiter, Z.; Campos, V.K.; Abdel-Salam, M.A.L.; de Souza-Fagundes, E.M.; Lessa, J.A. In vitro assessment of the cytotoxicity of gallium(III) complexes with Isoniazid-Derived Hydrazones: Effects on clonogenic survival of HCT-116 cells. Inorg. Chim. Acta 2019, 497, 119079. [Google Scholar] [CrossRef]
- Qi, J.; Liu, T.; Zhao, W.; Zheng, X.; Wang, Y. Synthesis, crystal structure and antiproliferative mechanisms of gallium(III) complexes with benzoylpyridine thiosemicarbazones. RSC Adv. 2020, 10, 18553–18559. [Google Scholar] [CrossRef]
- Chitambar, C.R.; Purpi, D.P.; Woodliff, J.; Yang, M.; Wereley, J.P. Development of Gallium Compounds for Treatment of Lymphoma: Gallium Maltolate, a Novel Hydroxypyrone Gallium Compound, Induces Apoptosis and Circumvents Lymphoma Cell Resistance to Gallium Nitrate. J. Pharmacol. Exp. Ther. 2007, 322, 1228–1236. [Google Scholar] [CrossRef] [Green Version]
- Chitambar, C.R. Gallium-containing anticancer compounds. Future Med. Chem. 2012, 4, 1257–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellahioui, Y.; Prashar, S.; Gómez-Ruiz, S. Anticancer applications and recent investigations of metallodrugs based on gallium, tin and titanium. Inorganics 2017, 5, 4. [Google Scholar] [CrossRef] [Green Version]
- Lessa, J.A.; Parrilha, G.L.; Beraldo, H. Gallium complexes as new promising metallodrug candidates. Inorg. Chim. Acta 2012, 393, 53–63. [Google Scholar] [CrossRef]
- Collery, P.; Keppler, B.; Madoulet, C.; Desoize, B. Gallium in cancer treatment. Crit. Rev. Oncol. Hematol. 2002, 42, 283–296. [Google Scholar] [CrossRef]
- Jahangoshaei, P.; Hassani, L.; Mohammadi, F.; Hamidi, A.; Mohammadi, K. Investigating the effect of gallium curcumin and gallium diacetylcurcumin complexes on the structure, function and oxidative stability of the peroxidase enzyme and their anticancer and antibacterial activities. J. Biol. Inorg. Chem. 2015, 20, 1135–1146. [Google Scholar] [CrossRef]
- Belluti, S.; Orteca, G.; Semeghini, V.; Rigillo, G.; Parenti, F.; Ferrari, E.; Imbriano, C. Potent anti-cancer properties of phthalimide-based curcumin derivatives on prostate tumor cells. Int. J. Mol. Sci. 2019, 20, 28. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, E.; Pignedoli, F.; Imbriano, C.; Marverti, G.; Basile, V.; Venturi, E.; Saladini, M. Newly synthesized curcumin derivatives: Crosstalk between chemico-physical properties and biological activity. J. Med. Chem. 2011, 54, 8066–8077. [Google Scholar] [CrossRef]
- Pabon, H.J.J. A synthesis of curcumin and related compounds. Recl. Trav. Chim. Pays Bas 2010, 83, 379–386. [Google Scholar] [CrossRef]
- Ferrari, E.; Benassi, R.; Sacchi, S.; Pignedoli, F.; Asti, M.; Saladini, M. Curcumin derivatives as metal-chelating agents with potential multifunctional activity for pharmaceutical applications. J. Inorg. Biochem. 2014, 139, 38–48. [Google Scholar] [CrossRef]
- Payton, F.; Sandusky, P.; Alworth, W.L. NMR study of the solution structure of curcumin. J. Nat. Prod. 2007, 70, 143–146. [Google Scholar] [CrossRef]
- Benassi, R.; Ferrari, E.; Grandi, R.; Lazzari, S.; Saladini, M. Synthesis and characterization of new β-diketo derivatives with iron chelating ability. J. Inorg. Biochem. 2007, 101, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Rapalli, V.K.; Kaul, V.; Gorantla, S.; Waghule, T.; Dubey, S.K.; Pandey, M.M.; Singhvi, G. UV Spectrophotometric method for characterization of curcumin loaded nanostructured lipid nanocarriers in simulated conditions: Method development, in-vitro and ex-vivo applications in topical delivery. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2020, 224, 117392. [Google Scholar] [CrossRef] [PubMed]
- Dias Pereira, C.I.; Fabiano de Freitas, C.; Braga, T.L.; Braga, G.; Gonçalves, R.S.; Tessaro, A.L.; Graton Mikcha, J.M.; Hioka, N.; Caetano, W. Concise behavior of Curcumin in water-ethanol: Critical Water Aggregation Percentage and multivariate analysis of protolytic equilibria. Dyes Pigment. 2022, 197, 143–7208. [Google Scholar] [CrossRef]
- Gans, P.; Sabatini, A.; Vacca, A. To improve accuracy of the calculated pKa values. Ann. Chim. 1999, 89, 45–49. [Google Scholar]
- Rigamonti, L.; Orteca, G.; Asti, M.; Basile, V.; Imbriano, C.; Saladini, M.; Ferrari, E. New curcumin-derived ligands and their affinity towards Ga3+, Fe3+ and Cu2+: Spectroscopic studies on complex formation and stability in solution. New J. Chem. 2018, 42, 7680–7690. [Google Scholar] [CrossRef]
- Gans, P.; Sabatini, A.; Vacca, A. Investigation of equilibria in solution. Determination of equilibrium constants with the HYPERQUAD suite of programs. Talanta 1996, 43, 1739–1753. [Google Scholar] [CrossRef]
- Harris, W.R.; Carrano, C.J.; Raymond, K.N. Coordination Chemistry of Microbial Iron Transport Compounds. 16. Isolation, Characterization, and Formation Constants of Ferric Aerobactin. J. Am. Chem. Soc. 1979, 101, 2722–2727. [Google Scholar] [CrossRef]
- Borgias, B.; Hugi, A.D.; Raymond, K.N. Isomerization and Solution Structures of Desferrioxamine B Complexes of Al3+ and Ga3+. Inorg. Chem. 1989, 28, 3538–3545. [Google Scholar] [CrossRef]
- Notni, J.; Hermann, P.; Havlíčková, J.; Kotek, J.; Kubíček, V.; Plutnar, J.; Loktionova, N.; Riss, P.J.; Rösch, F.; Lukeš, I. A triazacyclononane-based bifunctional phosphinate ligand for the preparation of multimeric 68Ga tracers for positron emission tomography. Chemistry 2010, 16, 7174–7185. [Google Scholar] [CrossRef]
- Toporivska, Y.; Mular, A.; Piasta, K.; Ostrowska, M.; Illuminati, D.; Baldi, A.; Albanese, V.; Pacifico, S.; Fritsky, I.O.; Remelli, M.; et al. Thermodynamic Stability and Speciation of Ga(III) and Zr(IV) Complexes with High-Denticity Hydroxamate Chelators. Inorg. Chem. 2021, 60, 13332–13347. [Google Scholar] [CrossRef]
- Asti, M.; Ferrari, E.; Croci, S.; Atti, G.; Rubagotti, S.; Iori, M.; Capponi, P.C.; Zerbini, A.; Saladini, M.; Versari, A. Synthesis and Characterization of 68 Ga-Labeled Curcumin and Curcuminoid Complexes as Potential Radiotracers for Imaging of Cancer and Alzheimer’s Disease. Inorg. Chem. 2014, 53, 4922–4933. [Google Scholar] [CrossRef] [PubMed]
- Rubagotti, S.; Croci, S.; Ferrari, E.; Orteca, G.; Iori, M.; Capponi, P.C.; Versari, A.; Asti, M. Uptake of Ga-curcumin derivatives in different cancer cell lines: Toward the development of new potential 68Ga-labelled curcuminoids-based radiotracers for tumour imaging. J. Inorg. Biochem. 2017, 173, 113–119. [Google Scholar] [CrossRef] [PubMed]
- El Deeb, S.; Ma, B.N.; Baecker, D.; Gust, R. Studies on the stability of the anticancer-active [N,N’-bis(salicylidene)-1,2-phenylenediamine]chloridoiron(III) complex under pharmacological-like conditions. Inorg. Chim. Acta 2019, 487, 76–80. [Google Scholar] [CrossRef]
- Zacchi, P.; Schioppa, T.; Stein, J.; Farrag, K.; Aksan, A.; Aksan, S.; Schroeder, O. Flipside of the Coin: Iron Deficiency and Colorectal Cancer. Front. Immunol. 2021, 12, 635899. [Google Scholar] [CrossRef]
- Pandrangi, S.L.; Chittineedi, P.; Chikati, R.; Lingareddy, J.R.; Nagoor, M.; Ponnada, S.K. Role of dietary iron revisited: In metabolism, ferroptosis and pathophysiology of cancer. Am. J. Cancer Res. 2022, 12, 974. [Google Scholar]
- Anderson, G.J.; Frazer, D.M. Current understanding of iron homeostasis. Am. J. Clin. Nutr. 2017, 106, 1559S–1566S. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, E.; Asti, M.; Benassi, R.; Pignedoli, F.; Saladini, M. Metal binding ability of curcumin derivatives: A theoretical vs. experimental approach. Dalt. Trans. 2013, 42, 5304–5313. [Google Scholar] [CrossRef]
- Belluti, S.; Basile, V.; Benatti, P.; Ferrari, E.; Marverti, G.; Imbriano, C. Concurrent inhibition of enzymatic activity and NF-Y-mediated transcription of Topoisomerase-IIa by bis-DemethoxyCurcumin in cancer cells. Cell Death Dis. 2013, 4, e756-11. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.R.; Britigan, B.E.; Narayanasamy, P. Dual Inhibition of Klebsiella pneumoniae and Pseudomonas aeruginosa Iron Metabolism Using Gallium Porphyrin and Gallium Nitrate. ACS Infect. Dis. 2019, 5, 1559–1569. [Google Scholar] [CrossRef]
- Payen, V.L.; Mina, E.; Van Hée, V.F.; Porporato, P.E.; Sonveaux, P. Monocarboxylate transporters in cancer. Mol. Metab. 2020, 33, 48–66. [Google Scholar] [CrossRef]
- Lee, J.Y.; Lee, I.K.; Chang, W.J.; Ahn, S.M.; Lim, S.H.; Kim, H.S.; Yoo, K.H.; Jung, K.S.; Song, H.N.; Cho, J.H.; et al. MCT4 as a potential therapeutic target for metastatic gastric cancer with peritoneal carcinomatosis. Oncotarget 2016, 7, 43492–43503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paolino, D.; Tudose, A.; Celia, C.; Di Marzio, L.; Cilurzo, F.; Mircioiu, C. Mathematical Models as Tools to Predict the Release Kinetic of Fluorescein from Lyotropic Colloidal Liquid Crystals. Material 2019, 12, 693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakobyan, S.; Boily, J.F.; Ramstedt, M. Proton and gallium(III) binding properties of a biologically active salicylidene acylhydrazide. J. Inorg. Biochem. 2014, 138, 9–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gama, S.; Frontauria, M.; Ueberschaar, N.; Brancato, G.; Milea, D.; Sammartano, S.; Plass, W. Cite this. New J. Chem. 2018, 42, 8062. [Google Scholar] [CrossRef]
- Belluti, S.; Semeghini, V.; Rigillo, G.; Ronzio, M.; Benati, D.; Torricelli, F.; Reggiani Bonetti, L.; Carnevale, G.; Grisendi, G.; Ciarrocchi, A.; et al. Alternative splicing of NF-YA promotes prostate cancer aggressiveness and represents a new molecular marker for clinical stratification of patients. J. Exp. Clin. Cancer Res. 2021, 40, 362. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | KE (%) | KK (%) |
|---|---|---|
| 1 | 96 | 4 |
| 2 | 88 * | 12 |
| 3 | 45 | 55 |
| 4 | 47 | 53 |
| 5 | 70 | 30 |
| 6 | 25 | 75 |
| 7 | 25 | 75 |
| logβMLH | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
| logβ011 | 10.212 (2) | 10.903 (8) | 10.88 (2) | 10.78 (4) | 10.44 (3) | 11.42 (2) | 12.22 (2) |
| logβ012 | 18.509 (3) | 20.29 (2) | 20.44 (6) | 20.45 (2) | 21.12 (6) | 21.61 (5) | |
| logβ013 | 28.84 (4) | 29.44 (6) | 29.58 (4) | 29.76 (7) | 29.98 (4) | ||
| logβ014 | 34.62 (5) | 34.92 (9) | |||||
| pKa1 | 8.297 (5) | 10.903 (8) * | 8.55 (4) | 9.00 (10) | 9.13 (6) | 4.86 (12) | 4.92 (13) |
| pKa2 | 10.212 (2) * | 9.41 (4) * | 9.66 (12) * | 10.01 (5) | 8.64 (13) | 8.37 (9) | |
| pKa3 | 10.88 (2) | 10.78 (4) | 10.44 (3) | 9.70 (8) * | 9.39 (7) * | ||
| pKa4 | 11.42 (2) | 12.22 (2) |
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | |
| HxL | H2L | HL | H3L | H3L | H3L | H4L | H4L |
| logβ110 | 11.78 (4) | 8.129 (9) | |||||
| logβ111 | 17.59 (3) | ||||||
| logβ112 | 28.28 (3) | 28.48 (5) | 28.54 (3) | 27.79 (5) | 28.29 (5) | ||
| logβ113 | 34.09 (5) | 36.51 (6) | |||||
| logβ120 | 20.30 (5) | 15.185 (4) | |||||
| logβ122 | 34.77(3) | ||||||
| logβ124 | 54.63 (5) | 55.33 (5) | 57.00 (1) | ||||
| logβ130 | 28.37 (7) | 20.45 (3) | |||||
| logβ133 | 49.7 (4) | ||||||
| logK11 | 7.38 (3) | 8.129 (9) | 7.99 | 8.04 | 8.09 | 6.67 | 6.68 |
| logK12 | 14.35 (3) | 15.185 | 14.05 | 14.45 | 16.1 | ||
| logK13 | 19.1 (4) | 20.45 | |||||
| pGa | 19.0 | 13.1 | 20.8 | 21.5 | 23.2 | 14.0 | 15.3 |
| Ligand | Prevailing Tautomer (MeOD-d4) | Stability * | pGa | Prevailing Species (pH 5.5) | Prevailing Species (pH 7.4) |
|---|---|---|---|---|---|
| 1 | KE | 77% (3) | 19.0 | [Ga(HL)3] | [Ga(L)3]3− |
| 2 | KE | 83% (1) | 13.1 | [Ga(L)3] | [Ga(L)3] |
| 3 | KK | 47% (6) | 20.8 | [Ga(H2L)2]+ | [Ga(H2L)2]+ |
| 4 | KK | 42% (7) | 21.5 | [Ga(H2L)2]+ | [Ga(H2L)2]+ |
| 5 | KE | 52% (5) | 23.4 | [Ga(H2L)2]+ | [Ga(H2L)2]+ |
| 6 | KK | 73% (4) | 14.0 | [Ga(H2L)2]+ | [Ga(H2L)2]+ ** |
| 7 | KK | 79% (2) | 15.3 | [Ga(H2L)2]+ | [Ga(H2L)2]+ ** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mari, M.; Carrozza, D.; Malavasi, G.; Venturi, E.; Avino, G.; Capponi, P.C.; Iori, M.; Rubagotti, S.; Belluti, S.; Asti, M.; et al. Curcumin-Based β-Diketo Ligands for Ga3+: Thermodynamic Investigation of Potential Metal-Based Drugs. Pharmaceuticals 2022, 15, 854. https://doi.org/10.3390/ph15070854
Mari M, Carrozza D, Malavasi G, Venturi E, Avino G, Capponi PC, Iori M, Rubagotti S, Belluti S, Asti M, et al. Curcumin-Based β-Diketo Ligands for Ga3+: Thermodynamic Investigation of Potential Metal-Based Drugs. Pharmaceuticals. 2022; 15(7):854. https://doi.org/10.3390/ph15070854
Chicago/Turabian StyleMari, Matteo, Debora Carrozza, Gianluca Malavasi, Ettore Venturi, Giulia Avino, Pier Cesare Capponi, Michele Iori, Sara Rubagotti, Silvia Belluti, Mattia Asti, and et al. 2022. "Curcumin-Based β-Diketo Ligands for Ga3+: Thermodynamic Investigation of Potential Metal-Based Drugs" Pharmaceuticals 15, no. 7: 854. https://doi.org/10.3390/ph15070854
APA StyleMari, M., Carrozza, D., Malavasi, G., Venturi, E., Avino, G., Capponi, P. C., Iori, M., Rubagotti, S., Belluti, S., Asti, M., & Ferrari, E. (2022). Curcumin-Based β-Diketo Ligands for Ga3+: Thermodynamic Investigation of Potential Metal-Based Drugs. Pharmaceuticals, 15(7), 854. https://doi.org/10.3390/ph15070854