
Effects of Nanosecond Pulsed Electric Field on Immune Checkpoint Receptors in Melanoma Cells

 , ,
, ,  ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Biophysical Characterization of nsPEF-Treated C32 and A375 Cells
2.2. The Effect of nsPEF Treatment on the Secretion of Cytokines
2.3. The Effect of nsPEF Treatment on the Expression of Immune Checkpoint Receptors and the Secretion of Cytokines
2.4. Morphological Changes of Cell Membranes following Treatment with nsPEFs
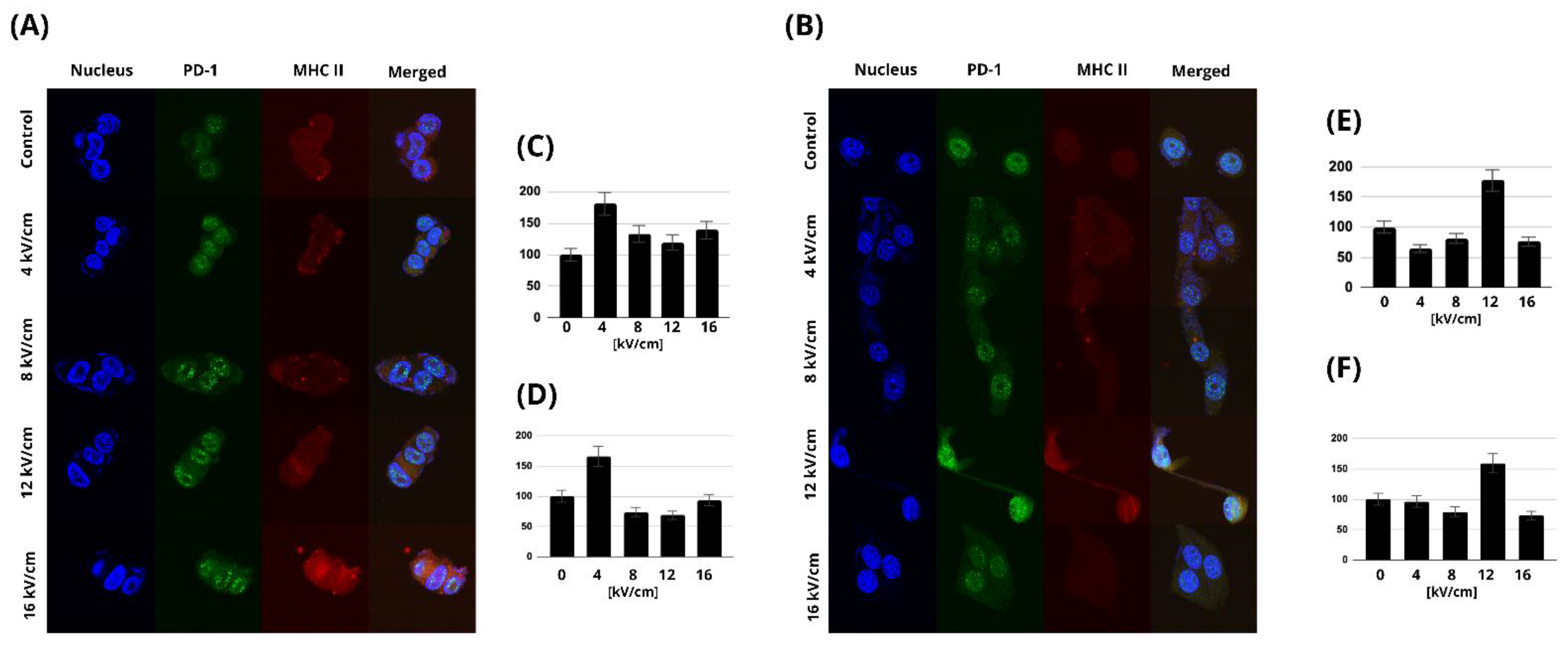
2.5. MHC Class II and PD-1 Expression
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. MTT Viability Assay
4.3. PrestoBlue™ Viability Assay
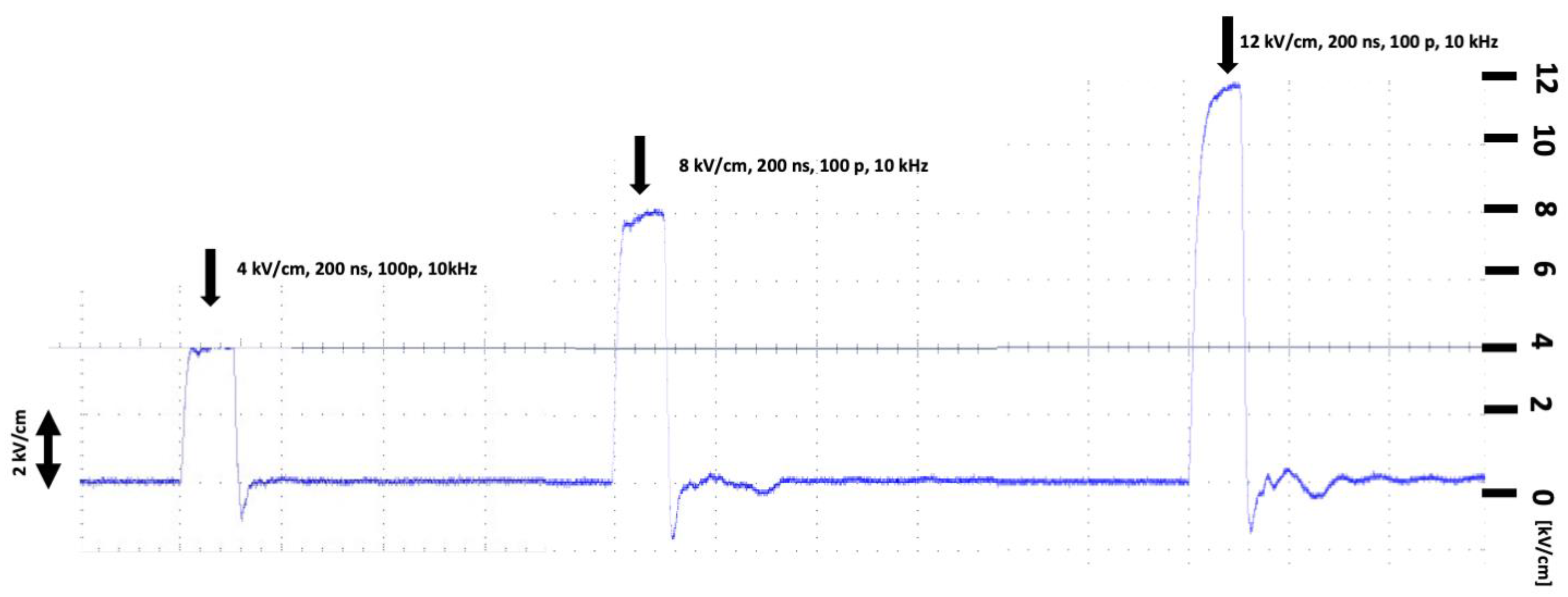
4.4. Electric Field Treatment
4.5. YO-PRO-1 Uptake Studies
4.6. Holotomographic Microscopy Studies
4.7. Confocal Microscopy
4.8. Western Blot
4.9. ELISA Assay
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Soumya, R.S.; Neethu, S.; Niju, T.S.; Renjini, A.; Aneesh, R.P. Advanced earlier melanoma detection algorithm using colour correlogram. In Proceedings of the 2016 International Conference on Communication Systems and Networks (ComNet), Thiruvananthapuram, India, 21–23 July 2016; pp. 190–194. [Google Scholar] [CrossRef]
- Vanka, L.P.; Chakravarty, S. Melanoma Detection from Skin Lesions using Convolution Neural Network. In Proceedings of the 2022 IEEE India Council International Subsections Conference (INDISCON), Bhubaneswar, India, 15–17 July 2022. [Google Scholar] [CrossRef]
- Uong, A.; Zon, L.I. Melanocytes in development and cancer. J. Cell. Physiol. 2010, 222, 38–41. [Google Scholar] [CrossRef] [PubMed]
- Marzagalli, M.; Ebelt, N.D.; Manuel, E.R. Unraveling the crosstalk between melanoma and immune cells in the tumor microenvironment. Semin. Cancer Biol. 2019, 59, 236–250. [Google Scholar] [CrossRef] [PubMed]
- Houghton, A.N.; Gold, J.S.; Blachere, N.E. Immunity against cancer: Lessons learned from melanoma. Curr. Opin. Immunol. 2001, 13, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Rausch, M.P.; Hastings, K.T. Immune Checkpoint Inhibitors in the Treatment of Melanoma: From Basic Science to Clinical Application. Cutan. Melanoma Etiol. Ther. 2017, 9, 121–142. [Google Scholar] [CrossRef]
- Singh, S.; Numan, A.; Agrawal, N.; Tambuwala, M.M.; Singh, V.; Kesharwani, P. Role of immune checkpoint inhibitors in the revolutionization of advanced melanoma care. Int. Immunopharmacol. 2020, 83, 106417. [Google Scholar] [CrossRef]
- Luther, C.; Swami, U.; Zhang, J.; Milhem, M.; Zakharia, Y. Advanced stage melanoma therapies: Detailing the present and exploring the future. Crit. Rev. Oncol. Hematol. 2019, 133, 99–111. [Google Scholar] [CrossRef]
- Dine, J.; Gordon, R.; Shames, Y.; Kasler, M.K.; Barton-Burke, M. Immune Checkpoint Inhibitors: An Innovation in Immunotherapy for the Treatment and Management of Patients with Cancer. Asia-Pac. J. Oncol. Nurs. 2017, 4, 127–135. [Google Scholar] [CrossRef]
- Curdy, N.; Lanvin, O.; Laurent, C.; Fournié, J.J.; Franchini, D.M. Regulatory Mechanisms of Inhibitory Immune Checkpoint Receptors Expression. Trends Cell Biol. 2019, 29, 777–790. [Google Scholar] [CrossRef]
- Sauer, N.; Szlasa, W.; Jonderko, L.; Oślizło, M.; Kunachowicz, D.; Kulbacka, J.; Karłowicz-Bodalska, K. LAG-3 as a Potent Target for Novel Anticancer Therapies of a Wide Range of Tumors. Int. J. Mol. Sci. 2022, 23, 9958. [Google Scholar] [CrossRef]
- Andrews, L.P.; Marciscano, A.E.; Drake, C.G.; Vignali, D.A.A. LAG3 (CD223) as a cancer immunotherapy target. Immunol. Rev. 2017, 276, 80–96. [Google Scholar] [CrossRef]
- Wiener, Z.; Kohalmi, B.; Pocza, P.; Jeager, J.; Tolgyesi, G.; Toth, S.; Gorbe, E.; Papp, Z.; Falus, A. TIM-3 is expressed in melanoma cells and is upregulated in TGF-beta stimulated mast cells. J. Investig. Dermatol. 2007, 127, 906–914. [Google Scholar] [CrossRef] [PubMed]
- Kleffel, S.; Posch, C.; Barthel, S.R.; Mueller, H.; Schlapbach, C.; Guenova, E.; Elco, C.P.; Lee, N.; Juneja, V.R.; Zhan, Q.; et al. Melanoma Cell-Intrinsic PD-1 Receptor Functions Promote Tumor Growth. Cell 2015, 162, 1242–1256. [Google Scholar] [CrossRef] [PubMed]
- Zak, K.M.; Grudnik, P.; Magiera, K.; Dömling, A.; Dubin, G.; Holak, T.A. Structural Biology of the Immune Checkpoint Receptor PD-1 and Its Ligands PD-L1/PD-L2. Structure 2017, 25, 1163–1174. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Lostao, L.; Anel, A.; Pardo, J. How Do Cytotoxic Lymphocytes Kill Cancer Cells? Clin. Cancer Res. 2015, 21, 5047–5056. [Google Scholar] [CrossRef]
- Li, X.; Wang, R.; Fan, P.; Yao, X.; Qin, L.; Peng, Y.; Ma, M.; Asley, N.; Chang, X.; Feng, Y.; et al. A comprehensive analysis of key immune checkpoint receptors on tumor-infiltrating t cells from multiple types of cancer. Front. Oncol. 2019, 9, 1066. [Google Scholar] [CrossRef]
- Yao, H.; Wang, H.; Li, C.; Fang, J.Y.; Xu, J. Cancer Cell-Intrinsic PD-1 and Implications in Combinatorial Immunotherapy. Front. Immunol. 2018, 9, 1774. [Google Scholar] [CrossRef]
- Lin, Z.; Chen, X.; Li, Z.; Luo, Y.; Fang, Z.; Xu, B.; Han, M. PD-1 Antibody Monotherapy for Malignant Melanoma: A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0160485. [Google Scholar] [CrossRef]
- Merelli, B.; Massi, D.; Cattaneo, L.; Mandalà, M. Targeting the PD1/PD-L1 axis in melanoma: Biological rationale, clinical challenges and opportunities. Crit. Rev. Oncol. Hematol. 2014, 89, 140–165. [Google Scholar] [CrossRef]
- Wang, M.; Du, Q.; Jin, J.; Wei, Y.; Lu, Y.; Li, Q. LAG3 and its emerging role in cancer immunotherapy. Clin. Transl. Med. 2021, 11, e365. [Google Scholar] [CrossRef]
- Souri, Z.; Wierenga, A.P.; Kroes, W.G.; van der Velden, P.A.; Verdijk, R.M.; Eikmans, M.; Luyten, G.P.; Jager, M.J. Lag3 and its ligands show increased expression in high-risk uveal melanoma. Cancers 2021, 13, 4445. [Google Scholar] [CrossRef]
- Gao, X.; Zhu, Y.; Li, G.; Huang, H.; Zhang, G.; Wang, F.; Sun, J.; Yang, Q.; Zhang, X.; Lu, B. TIM-3 expression characterizes regulatory T cells in tumor tissues and is associated with lung cancer progression. PLoS ONE 2012, 7, e30676. [Google Scholar] [CrossRef]
- Wu, W.; Shi, Y.; Li, S.; Zhang, Y.; Liu, Y.; Wu, Y.; Chen, Z. Blockade of Tim-3 signaling restores the virus-specific CD8+ T-cell response in patients with chronic hepatitis B. Eur. J. Immunol. 2012, 42, 1180–1191. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Zhu, C.; Kuchroo, V.K. Tim-3 and its role in regulating anti-tumor immunity. Immunol. Rev. 2017, 276, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Fourcade, J.; Sun, Z.; Pagliano, O.; Chauvin, J.M.; Sander, C.; Janjic, B.; Tarhini, A.A.; Tawbi, H.A.; Kirkwood, J.M.; Moschos, S.; et al. PD-1 and Tim-3 regulate the expansion of tumor antigen-specific CD8+ T cells induced by melanoma vaccines. Cancer Res. 2014, 74, 1045–1055. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef]
- Huang, A.C.; Postow, M.A.; Orlowski, R.J.; Mick, R.; Bengsch, B.; Manne, S.; Xu, W.; Harmon, S.; Giles, J.R.; Wenz, B.; et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 2017, 545, 60–65. [Google Scholar] [CrossRef]
- Grosso, J.F.; Goldberg, M.V.; Getnet, D.; Bruno, T.C.; Yen, H.R.; Pyle, K.J.; Hipkiss, E.; Vignali, D.A.; Pardoll, D.M.; Drake, C.G. Functionally distinct LAG-3 and PD-1 subsets on activated and chronically stimulated CD8 T cells. J. Immunol. 2009, 182, 6659–6669. [Google Scholar] [CrossRef]
- Wezgowiec, J.; Derylo, M.B.; Teissie, J.; Orio, J.; Rols, M.P.; Kulbacka, J.; Saczko, J.; Kotulska, M. Electric field-assisted delivery of photofrin to human breast carcinoma cells. J. Membr. Biol. 2013, 246, 725–735. [Google Scholar] [CrossRef]
- Gehl, J.; Sersa, G.; Matthiessen, L.W.; Muir, T.; Soden, D.; Occhini, A.; Quaglino, P.; Curatolo, P.; Campana, L.G.; Kunte, C.; et al. Updated standard operating procedures for electrochemotherapy of cutaneous tumours and skin metastases. Acta Oncol. 2018, 57, 874–882. [Google Scholar] [CrossRef]
- Miklavčič, D.; Serša, G.; Brecelj, E.; Gehl, J.; Soden, D.; Bianchi, G.; Ruggieri, P.; Rossi, C.R.; Campana, L.G.; Jarm, T. Electrochemotherapy: Technological advancements for efficient electroporation-based treatment of internal tumors. Med. Biol. Eng. Comput. 2012, 50, 1213–1225. [Google Scholar] [CrossRef] [PubMed]
- Esmaeili, N.; Friebe, M. Electrochemotherapy: A Review of Current Status, Alternative IGP Approaches, and Future Perspectives. J. Healthc. Eng. 2019, 2019, 2784516. [Google Scholar] [CrossRef] [PubMed]
- Nuccitelli, R. Application of Pulsed Electric Fields to Cancer Therapy. Bioelectricity 2019, 1, 30–34. [Google Scholar] [CrossRef]
- Pisani, S.; Bertino, G.; Prina-Mello, A.; Locati, L.D.; Mauramati, S.; Genta, I.; Dorati, R.; Conti, B.; Benazzo, M. Electroporation in Head-and-Neck Cancer: An Innovative Approach with Immunotherapy and Nanotechnology Combination. Cancers 2022, 14, 5363. [Google Scholar] [CrossRef] [PubMed]
- Rols, M.P.; Golzio, M.; Kolosnjaj-Tabi, J. Electric Field Based Therapies in Cancer Treatment. Cancers 2020, 12, 3420. [Google Scholar] [CrossRef]
- Haemmerich, D.; Laeseke, P.F. Thermal tumour ablation: Devices, clinical applications and future directions. Int. J. Hyperth. 2005, 21, 755–760. [Google Scholar] [CrossRef]
- Tanabe, K.K.; Curley, S.A.; Dodd, G.D.; Siperstein, A.E.; Goldberg, S.N. Radiofrequency ablation: The experts weigh in. Cancer 2004, 100, 641–650. [Google Scholar] [CrossRef]
- Nishi, T.; Dev, S.B.; Yoshizato, K.; Kuratsu, J.; Ushio, Y. Treatment of cancer using pulsed electric field in combination with chemotherapeutic agents or genes. Hum. Cell 1997, 10, 81–86. [Google Scholar]
- Kiełbik, A.; Szlasa, W.; Novickij, V.; Szewczyk, A.; Maciejewska, M.; Saczko, J.; Kulbacka, J. Effects of high-frequency nanosecond pulses on prostate cancer cells. Sci. Rep. 2021, 11, 15835. [Google Scholar] [CrossRef]
- Szlasa, W.; Michel, O.; Sauer, N.; Novickij, V.; Lewandowski, D.; Kasperkiewicz, P.; Tarek, M.; Saczko, J.; Kulbacka, J. Nanosecond pulsed electric field suppresses growth and reduces multi-drug resistance effect in pancreatic cancer. Sci. Rep. 2023, 13, 351. [Google Scholar] [CrossRef]
- Lázár-Molnár, E.; Yan, Q.; Cao, E.; Ramagopal, U.; Nathenson, S.G.; Almo, S.C. Crystal structure of the complex between programmed death-1 (PD-1) and its ligand PD-L2. Proc. Natl. Acad. Sci. USA 2008, 105, 10483–10488. [Google Scholar] [CrossRef] [PubMed]
- Ming, Q.; Celias, D.P.; Wu, C.; Cole, A.R.; Singh, S.; Mason, C.; Dong, S.; Tran, T.H.; Amarasinghe, G.K.; Ruffell, B.; et al. LAG3 ectodomain structure reveals functional interfaces for ligand and antibody recognition. Nat. Immunol. 2022, 23, 1031–1041. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, Y.; Chen, J.; Wu, Y.; Zhang, J.; Wang, J. Nanosecond pulsed electric field inhibits malignant melanoma growth by inducing the change of systemic immunity. Med. Oral Patol. Oral Cir. Bucal 2019, 24, e555–e561. [Google Scholar] [CrossRef] [PubMed]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef]
- Wu, M.; Huang, Q.; Xie, Y.; Wu, X.; Ma, H.; Zhang, Y.; Xia, Y. Improvement of the anticancer efficacy of PD-1/PD-L1 blockade via combination therapy and PD-L1 regulation. J. Hematol. Oncol. 2022, 15, 24. [Google Scholar] [CrossRef]
- Kulbacka, J. Nanosecond pulsed electric fields (nsPEFs) impact and enhanced Photofrin II® delivery in photodynamic reaction in cancer and normal cells. Photodiagnosis Photodyn. Ther. 2015, 12, 621–629. [Google Scholar] [CrossRef]
- Szlasa, W.; Kiełbik, A.; Szewczyk, A.; Rembiałkowska, N.; Novickij, V.; Tarek, M.; Saczko, J.; Kulbacka, J. Oxidative Effects during Irreversible Electroporation of Melanoma Cells—In Vitro Study. Molecules 2021, 26, 154. [Google Scholar] [CrossRef]
- Xu, T.; Wu, Y.K.; Luo, X.; Guo, L.J. Plasmonic nanoresonators for high-resolution colour filtering and spectral imaging. Nat. Commun. 2010, 1, 59. [Google Scholar] [CrossRef]
- Johnson, D.B.; Estrada, M.V.; Salgado, R.; Sanchez, V.; Doxie, D.B.; Opalenik, S.R.; Vilgelm, A.E.; Feld, E.; Johnson, A.S.; Greenplate, A.R.; et al. Melanoma-specific MHC-II expression represents a tumour-autonomous phenotype and predicts response to anti-PD-1/PD-L1 therapy. Nat. Commun. 2016, 7, 10582. [Google Scholar] [CrossRef]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in Previously Untreated Melanoma without BRAF Mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef]
- Lim, S.O.; Li, C.W.; Xia, W.; Cha, J.H.; Chan, L.C.; Wu, Y.; Chang, S.S.; Lin, W.C.; Hsu, J.M.; Hsu, Y.H.; et al. Deubiquitination and Stabilization of PD-L1 by CSN5. Cancer Cell 2016, 30, 925–939. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Chen, X.; Cui, G.; Yin, S.; Chen, L.; Jiang, J.; Hu, Z.; Xie, H.; Zheng, S.; Zhou, L. Nanosecond Pulsed Electric Field Inhibits Cancer Growth Followed by Alteration in Expressions of NF-κB and Wnt/β-Catenin Signaling Molecules. PLoS ONE 2013, 8, e74322. [Google Scholar] [CrossRef]
- Wei, Y.; Li, Z. LAG3-PD-1 Combo Overcome the Disadvantage of Drug Resistance. Front. Oncol. 2022, 12, 831407. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.Y.; Eppolito, C.; Lele, S.; Shrikant, P.; Matsuzaki, J.; Odunsi, K. LAG3 and PD1 co-inhibitory molecules collaborate to limit CD8+ T cell signaling and dampen antitumor immunity in a murine ovarian cancer model. Oncotarget 2015, 6, 27359. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Ju, X.; Zhang, H.; Zhou, Z.; Chen, M.; Wang, Q. Tumor-associated macrophages induce PD-L1 expression in gastric cancer cells through IL-6 and TNF-ɑ signaling. Exp. Cell Res. 2020, 396, 112315. [Google Scholar] [CrossRef] [PubMed]
- Numata, Y.; Akutsu, N.; Ishigami, K.; Koide, H.; Wagatsuma, K.; Motoya, M.; Sasaki, S.; Nakase, H. Synergistic effect of IFN-γ and IL-1β on PD-L1 expression in hepatocellular carcinoma. Biochem. Biophys. Rep. 2022, 30, 101270. [Google Scholar] [CrossRef]
- Haabeth, O.A.W.; Lorvik, K.B.; Yagita, H.; Bogen, B.; Corthay, A. Interleukin-1 is required for cancer eradication mediated by tumor-specific Th1 cells. Oncoimmunology 2016, 5, e1039763. [Google Scholar] [CrossRef]
- Jia, X.H.; Geng, L.Y.; Jiang, P.P.; Xu, H.; Nan, K.J.; Yao, Y.; Jiang, L.L.; Sun, H.; Qin, T.J.; Guo, H. The biomarkers related to immune related adverse events caused by immune checkpoint inhibitors. J. Exp. Clin. Cancer Res. 2020, 39, 284. [Google Scholar] [CrossRef]
- Valpione, S.; Pasquali, S.; Campana, L.G.; Piccin, L.; Mocellin, S.; Pigozzo, J.; Chiarion-Sileni, V. Sex and interleukin-6 are prognostic factors for autoimmune toxicity following treatment with anti-CTLA4 blockade. J. Transl. Med. 2018, 16, 94. [Google Scholar] [CrossRef]
- Pennica, D.; Nedwin, G.E.; Hayflick, J.S.; Seeburg, P.H.; Derynck, R.; Palladino, M.A.; Kohr, W.J.; Aggarwal, B.B.; Goeddel, D.V. Human tumour necrosis factor: Precursor structure, expression and homology to lymphotoxin. Nature 1984, 312, 724–729. [Google Scholar] [CrossRef] [PubMed]
- Brouckaert, P.G.G.; Leroux-Roels, G.G.; Guisez, Y.; Tavernier, J.; Fiers, W. In vivo anti-tumour activity of recombinant human and murine TNF, alone and in combination with murine IFN-γ, on a syngeneic murine melanoma. Int. J. Cancer 1986, 38, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Verhoef, C.; Wilt, J.H.W.; Grünhagen, D.J.; Geel, A.N.; Hagen, T.L.M.; Eggermont, A.M.M. Isolated limb perfusion with melphalan and TNF-α in the treatment of extremity sarcoma. Curr. Treat. Options Oncol. 2007, 8, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Ben-Baruch, A. Tumor Necrosis Factor α: Taking a Personalized Road in Cancer Therapy. Front. Immunol. 2022, 13, 903679. [Google Scholar] [CrossRef] [PubMed]
- Seynhaeve, A.L.; Hoving, S.; Schipper, D.; Vermeulen, C.E.; Aan de Wiel-Ambagtsheer, G.; van Tiel, S.T.; Eggermont, A.M.; Ten Hagen, T.L. Tumor Necrosis Factor α Mediates Homogeneous Distribution of Liposomes in Murine Melanoma that Contributes to a Better Tumor Response. Cancer Res. 2007, 67, 9455–9462. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sauer, N.; Szlasa, W.; Szewczyk, A.; Novickij, V.; Saczko, J.; Baczyńska, D.; Daczewska, M.; Kulbacka, J. Effects of Nanosecond Pulsed Electric Field on Immune Checkpoint Receptors in Melanoma Cells. Pharmaceuticals 2023, 16, 1362. https://doi.org/10.3390/ph16101362
Sauer N, Szlasa W, Szewczyk A, Novickij V, Saczko J, Baczyńska D, Daczewska M, Kulbacka J. Effects of Nanosecond Pulsed Electric Field on Immune Checkpoint Receptors in Melanoma Cells. Pharmaceuticals. 2023; 16(10):1362. https://doi.org/10.3390/ph16101362
Chicago/Turabian StyleSauer, Natalia, Wojciech Szlasa, Anna Szewczyk, Vitalij Novickij, Jolanta Saczko, Dagmara Baczyńska, Małgorzata Daczewska, and Julita Kulbacka. 2023. "Effects of Nanosecond Pulsed Electric Field on Immune Checkpoint Receptors in Melanoma Cells" Pharmaceuticals 16, no. 10: 1362. https://doi.org/10.3390/ph16101362
APA StyleSauer, N., Szlasa, W., Szewczyk, A., Novickij, V., Saczko, J., Baczyńska, D., Daczewska, M., & Kulbacka, J. (2023). Effects of Nanosecond Pulsed Electric Field on Immune Checkpoint Receptors in Melanoma Cells. Pharmaceuticals, 16(10), 1362. https://doi.org/10.3390/ph16101362