
Prediction of Phytochemicals for Their Potential to Inhibit New Delhi Metallo β-Lactamase (NDM-1)


 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
2.1. Cross-Validation
2.2. Pharmacokinetics and Screening of Receptor–Ligand Interactions
2.3. Predicted Ki Values of Potent Metabolites and Their Comparison with Reported Synthetic Inhibitors
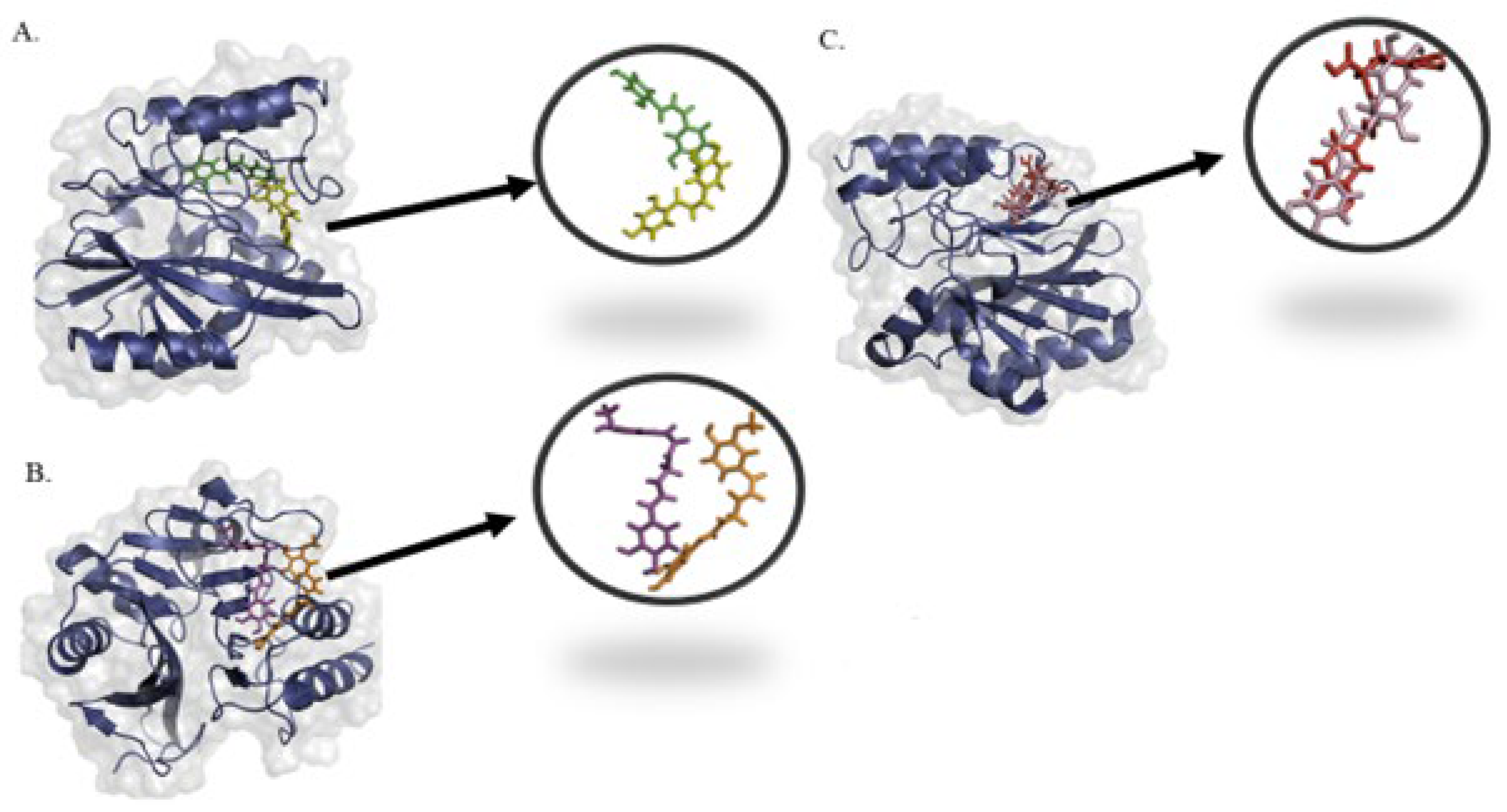
2.4. Molecular Interactions and Binding Mode of Potential Metabolites with NDM-1
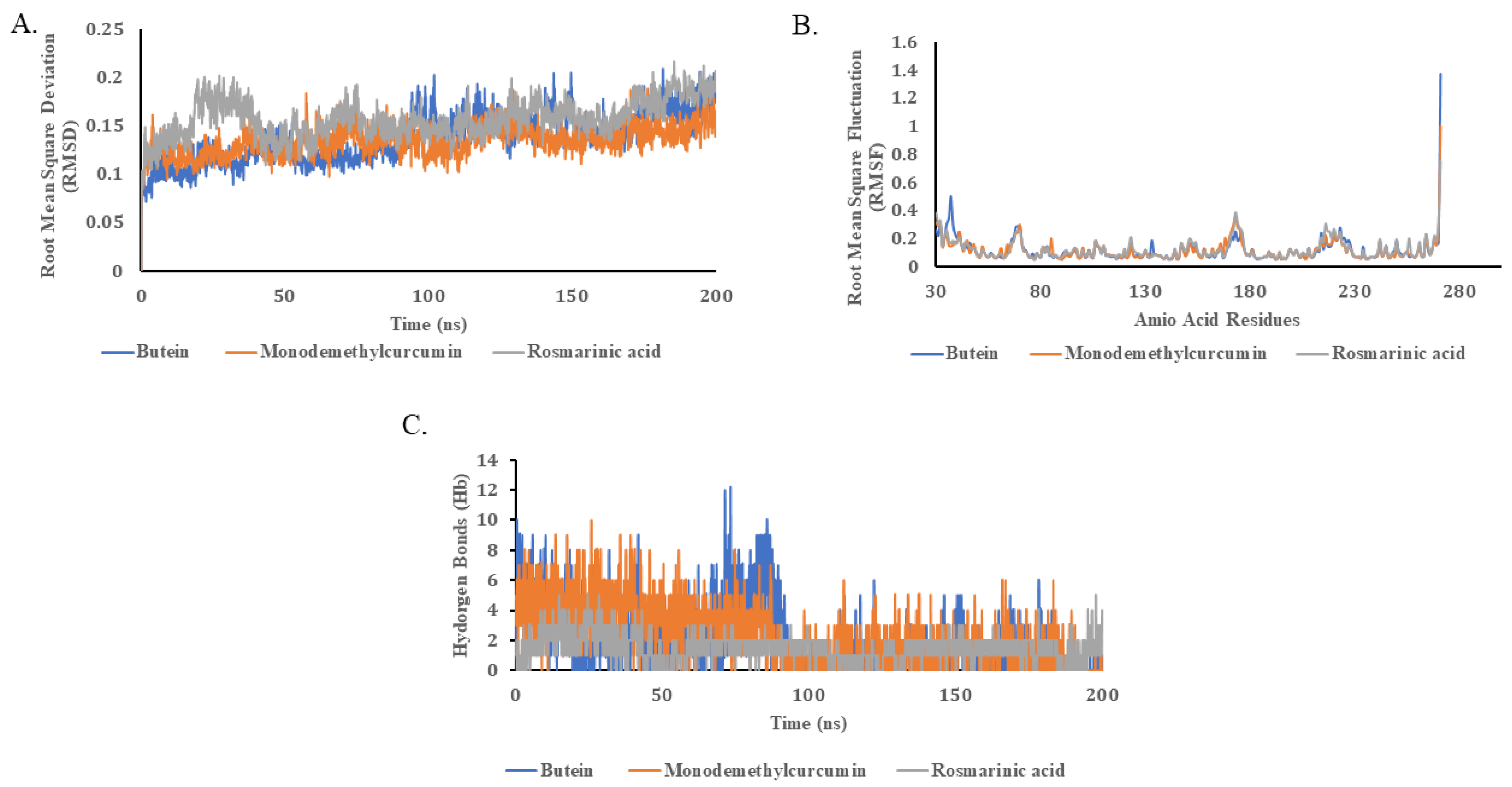
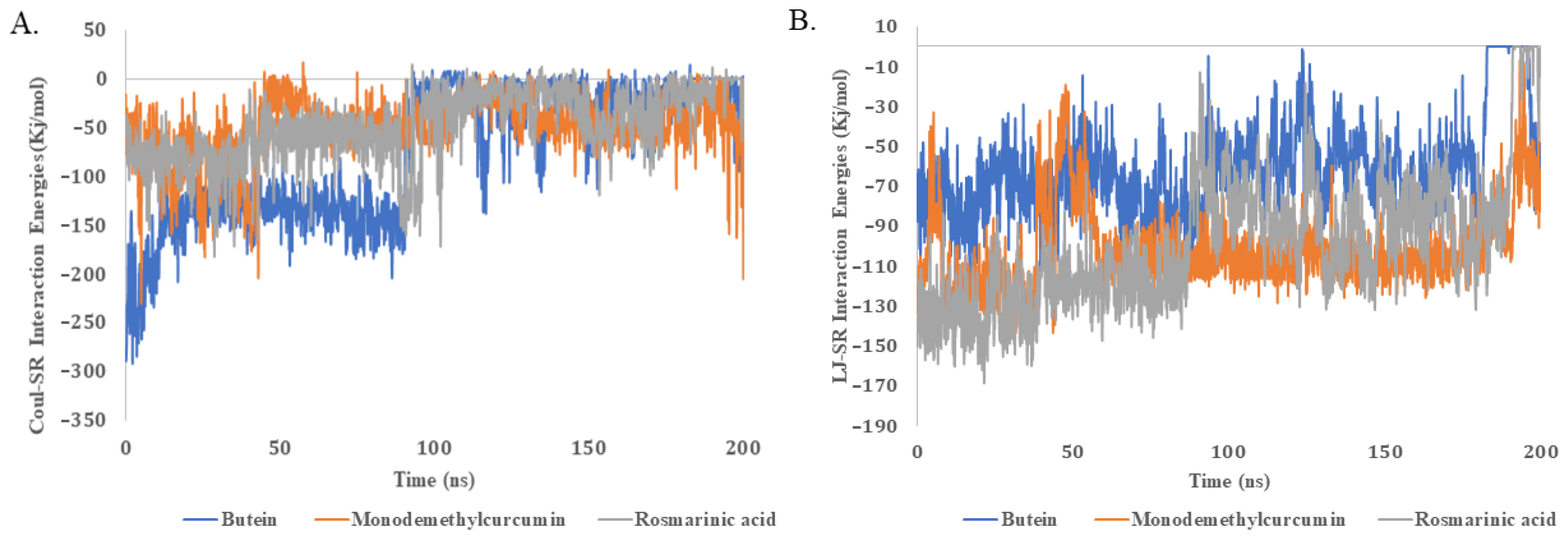
2.5. Molecular Dynamics Simulations
3. Discussion
4. Materials and Methods
4.1. Ligand Selection and Preparation
4.2. Drug-Likeness Prediction Using ADMETlab2.0
4.3. Protein Selection and Preparation
4.4. Validation of Target Protein–Ligand Complexes
4.5. Receptor–Ligand Docking and Evaluation of Docking Results
4.6. Prediction of Inhibition Constant of Selected Compounds Using AutoDock 4.2
4.7. Molecular Dynamics (MD) Simulations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Woodford, N.; Ellington, M.J. The emergence of antibiotic resistance by mutation. Clin. Microbiol. Infect. 2007, 13, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Palzkill, T. Metallo-β-lactamase structure and function. Ann. N. Y. Acad. Sci. 2013, 1277, 91–104. [Google Scholar] [CrossRef] [PubMed]
- González-Bello, C.; Rodríguez, D.; Pernas, M.; Rodriguez, A.; Colchón, E. β-Lactamase inhibitors to restore the efficacy of antibiotics against superbugs. J. Med. Chem. 2019, 63, 1859–1881. [Google Scholar] [CrossRef] [PubMed]
- Moellering, R.C., Jr. NDM-1—A cause for worldwide concern. N. Engl. J. Med. 2010, 363, 2377–2379. [Google Scholar] [CrossRef] [PubMed]
- Sreenivasan, P.; Sharma, B.; Kaur, S.; Rana, S.; Biswal, M.; Ray, P.; Angrup, A. In-vitro susceptibility testing methods for the combination of ceftazidime-avibactam with aztreonam in metallobeta-lactamase producing organisms: Role of combination drugs in antibiotic resistance era. J. Antibiot. 2022, 75, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Bassetti, M.; Righi, E.; Viscoli, C. Novel β-lactam antibiotics and inhibitor combinations. Expert Opin. Investig. Drugs 2008, 17, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.U.; Maryam, L.; Zarrilli, R. Structure, genetics and worldwide spread of New Delhi metallo-β-lactamase (NDM): A threat to public health. BMC Microbiol. 2017, 17, 101. [Google Scholar] [CrossRef]
- Bush, K.; Jacoby, G.A. Updated functional classification of β-lactamases. Antimicrob. Agents Chemother. 2010, 54, 969–976. [Google Scholar] [CrossRef]
- Helfand, M.S.; Bonomo, R.A. β-Lactamases: A survey of protein diversity. Curr. Drug Targets Infect. Disord. 2003, 3, 9–23. [Google Scholar] [CrossRef]
- Bebrone, C. Metallo-β-lactamases (classification, activity, genetic organization, structure, zinc coordination) and their superfamily. Biochem. Pharmacol. 2007, 74, 1686–1701. [Google Scholar] [CrossRef]
- Yong, D.; Toleman, M.A.; Giske, C.G.; Cho, H.S.; Sundman, K.; Lee, K.; Walsh, T.R. Characterization of a new metallo-β-lactamase gene, bla NDM-1, and a novel erythromycin esterase gene carried on a unique genetic structure in Klebsiella pneumoniae sequence type 14 from India. Antimicrob. Agents Chemother. 2009, 53, 5046–5054. [Google Scholar] [CrossRef] [PubMed]
- Quiñones, D.; Aung, M.S.; Carmona, Y.; González, M.K.; Pereda, N.; Hidalgo, M.; Rivero, M.; Zayas, A.; Del Campo, R.; Urushibara, N. High prevalence of CTX-M type extended-spectrum beta-lactamase genes and detection of NDM-1 carbapenemase gene in extraintestinal pathogenic Escherichia coli in Cuba. Pathogens 2020, 9, 65. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, X. Prevalence of metallo-β-lactamase genes among Pseudomonas aeruginosa isolated from various clinical samples in China. J. Lab. Med. 2020, 44, 197–203. [Google Scholar] [CrossRef]
- Cai, Y.; Chen, C.; Zhao, M.; Yu, X.; Lan, K.; Liao, K.; Guo, P.; Zhang, W.; Ma, X.; He, Y. High prevalence of metallo-β-lactamase-producing Enterobacter cloacae from three tertiary hospitals in China. Front. Microbiol. 2019, 10, 1610. [Google Scholar] [CrossRef] [PubMed]
- Thapa, A.; Upreti, M.K.; Bimali, N.K.; Shrestha, B.; Sah, A.K.; Nepal, K.; Dhungel, B.; Adhikari, S.; Adhikari, N.; Lekhak, B. Detection of NDM variants (blaNDM-1, blaNDM-2, blaNDM-3) from Carbapenem-resistant Escherichia coli and Klebsiella pneumoniae: First report from Nepal. Infect. Drug Resist. 2022, 15, 4419–4434. [Google Scholar] [CrossRef] [PubMed]
- Green, V.L.; Verma, A.; Owens, R.J.; Phillips, S.E.; Carr, S.B. Structure of New Delhi metallo-β-lactamase 1 (NDM-1). Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2011, 67, 1160–1164. [Google Scholar] [CrossRef] [PubMed]
- Zhang, g.; Hao, Q. Crystal structure of NDM-1 reveals a common β-lactam hydrolysis mechanism. FASEB J. 2011, 25, 2574–2582. [Google Scholar] [CrossRef] [PubMed]
- Poirel, L.; Lagrutta, E.; Taylor, P.; Pham, J.; Nordmann, P. Emergence of metallo-β-lactamase NDM-1-producing multidrug-resistant Escherichia coli in Australia. Antimicrob. Agents Chemother. 2010, 54, 4914–4916. [Google Scholar] [CrossRef]
- Wang, T.; Xu, K.; Zhao, L.; Tong, R.; Xiong, L.; Shi, J. Recent research and development of NDM-1 inhibitors. Eur. J. Med. Chem. 2021, 223, 113667. [Google Scholar] [CrossRef]
- Nordmann, P.; Naas, T.; Poirel, L. Global spread of carbapenemase-producing Enterobacteriaceae. Emerg. Infect. Dis. 2011, 17, 1791–1798. [Google Scholar] [CrossRef]
- Wu, W.; Feng, Y.; Tang, G.; Qiao, F.; McNally, A.; Zong, Z. NDM metallo-β-lactamases and their bacterial producers in health care settings. Clin. Microbiol. Rev. 2019, 32, e00115–e00118. [Google Scholar] [CrossRef] [PubMed]
- Buynak, J.D. β-Lactamase inhibitors: A review of the patent literature (2010–2013). Expert Opin. Ther. Pat. 2013, 23, 1469–1481. [Google Scholar] [CrossRef] [PubMed]
- Winkler, M.L.; Bonomo, R.A. SHV-129: A gateway to global suppressors in the SHV β-lactamase family? Mol. Biol. Evol. 2016, 33, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Bergstrom, A.; Katko, A.; Adkins, Z.; Hill, J.; Cheng, Z.; Burnett, M.; Yang, H.; Aitha, M.; Mehaffey, M.R.; Brodbelt, J.S. Probing the Interaction of Aspergillomarasmine A with Metallo-β-lactamases NDM-1, VIM-2, and IMP-7. ACS Infect. Dis. 2018, 4, 135–145. [Google Scholar] [CrossRef] [PubMed]
- King, A.M.; Reid-Yu, S.A.; Wang, W.; King, D.T.; De Pascale, G.; Strynadka, N.C.; Walsh, T.R.; Coombes, B.K.; Wright, G.D. Aspergillomarasmine A overcomes metallo-β-lactamase antibiotic resistance. Nature 2014, 510, 503–506. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.; Wang, S.; Wu, K.; Zhang, W.; Ahmad, A.; Hao, Q.; Lei, X.; Zhang, H. Structure-guided optimization of D-captopril for discovery of potent NDM-1 inhibitors. Bioorg. Med. Chem. 2021, 29, 115902. [Google Scholar] [CrossRef] [PubMed]
- Rotondo, C.M.; Wright, G.D. Inhibitors of metallo-β-lactamases. Curr. Opin. Microbiol. 2017, 39, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Brem, J.; van Berkel, S.S.; Zollman, D.; Lee, S.Y.; Gileadi, O.; McHugh, P.J.; Walsh, T.R.; McDonough, M.A.; Schofield, C.J. Structural basis of metallo-β-lactamase inhibition by captopril stereoisomers. Antimicrob. Agents Chemother. 2016, 60, 142–150. [Google Scholar] [CrossRef]
- Neveu, V.; Perez-Jiménez, J.; Vos, F.; Crespy, V.; du Chaffaut, L.; Mennen, L.; Knox, C.; Eisner, R.; Cruz, J.; Wishart, D. Phenol-Explorer: An online comprehensive database on polyphenol contents in foods. Database 2010, 2010, bap024. [Google Scholar] [CrossRef]
- Mohanraj, K.; Karthikeyan, B.S.; Vivek-Ananth, R.; Chand, R.; Aparna, S.; Mangalapandi, P.; Samal, A. IMPPAT: A curated database of Indian medicinal plants, phytochemistry and therapeutics. Sci. Rep. 2018, 8, 4329. [Google Scholar] [CrossRef]
- Dzotam, J.K.; Touani, F.K.; Kuete, V. Antibacterial activities of the methanol extracts of Canarium schweinfurthii and four other Cameroonian dietary plants against multi-drug resistant Gram-negative bacteria. Saudi J. Biol. Sci. 2016, 23, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Adelusi, T.I.; Oyedele, A.-Q.K.; Boyenle, I.D.; Ogunlana, A.T.; Adeyemi, R.O.; Ukachi, C.D.; Idris, M.O.; Olaoba, O.T.; Adedotun, I.O.; Kolawole, O.E. Molecular modeling in drug discovery. Inform. Med. Unlocked 2022, 29, 100880. [Google Scholar] [CrossRef]
- Salo-Ahen, O.M.; Alanko, I.; Bhadane, R.; Bonvin, A.M.; Honorato, R.V.; Hossain, S.; Juffer, A.H.; Kabedev, A.; Lahtela-Kakkonen, M.; Larsen, A.S. Molecular dynamics simulations in drug discovery and pharmaceutical development. Processes 2020, 9, 71. [Google Scholar] [CrossRef]
- Salari-Jazi, A.; Mahnam, K.; Sadeghi, P.; Damavandi, M.S.; Faghri, J. Discovery of potential inhibitors against New Delhi metallo-β-lactamase-1 from natural compounds: In silico-based methods. Sci. Rep. 2021, 11, 2390. [Google Scholar] [CrossRef] [PubMed]
- Pestana-Nobles, R.; Aranguren-Díaz, Y.; Machado-Sierra, E.; Yosa, J.; Galan-Freyle, N.J.; Sepulveda-Montaño, L.X.; Kuroda, D.G.; Pacheco-Londoño, L.C. Docking and molecular dynamic of microalgae compounds as potential inhibitors of beta-lactamase. Int. J. Mol. Sci. 2022, 23, 1630. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.; Khan, M.K.A. In silico based unraveling of New Delhi metallo-β-lactamase (NDM-1) inhibitors from natural compounds: A molecular docking and molecular dynamics simulation study. J. Biomol. Struct. Dyn. 2020, 38, 2093–2103. [Google Scholar] [CrossRef]
- Sharma, R.; Jade, D.; Mohan, S.; Chandel, R.; Sugumar, S. In-silico virtual screening for identification of potent inhibitor for L2-β-lactamase from Stenotrophomonas maltophilia through molecular docking, molecular dynamics analysis study. J. Biomol. Struct. Dyn. 2021, 39, 7123–7137. [Google Scholar] [CrossRef]
- Yusof, Y.; Tan, D.T.; Arjomandi, O.K.; Schenk, G.; McGeary, R.P. Captopril analogues as metallo-β-lactamase inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 1589–1593. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Zheng, M.; Chen, L.; Li, H. The development of New Delhi metallo-β-lactamase-1 inhibitors since 2018. Microbiol. Res. 2022, 261, 127079. [Google Scholar] [CrossRef] [PubMed]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef]
- Klingler, F.-M.; Wichelhaus, T.A.; Frank, D.; Cuesta-Bernal, J.; El-Delik, J.; Müller, H.F.; Sjuts, H.; Göttig, S.; Koenigs, A.; Pos, K.M. Approved drugs containing thiols as inhibitors of metallo-β-lactamases: Strategy to combat multidrug-resistant bacteria. J. Med. Chem. 2015, 58, 3626–3630. [Google Scholar] [CrossRef] [PubMed]
- Fullington, S.; Cheng, Z.; Thomas, C.; Miller, C.; Yang, K.; Ju, L.-C.; Bergstrom, A.; Shurina, B.A.; Bretz, S.L.; Page, R.C. An integrated biophysical approach to discovering mechanisms of NDM-1 inhibition for several thiol-containing drugs. J. Biol. Inorg. Chem. 2020, 25, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Wang, J.; Niu, G.; Shui, W.; Sun, Y.; Zhou, H.; Zhang, Y.; Yang, C.; Lou, Z.; Rao, Z. A structural view of the antibiotic degradation enzyme NDM-1 from a superbug. Protein Cell 2011, 2, 384–394. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Polli, J. Identification of inhibitor concentrations to efficiently screen and measure inhibition Ki values against solute carrier transporters. Eur. J. Pharm. Sci. 2010, 41, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Concha, N.O.; Janson, C.A.; Rowling, P.; Pearson, S.; Cheever, C.A.; Clarke, B.P.; Lewis, C.; Galleni, M.; Frere, J.-M.; Payne, D.J. Crystal structure of the IMP-1 metallo β-lactamase from Pseudomonas aeruginosa and its complex with a mercaptocarboxylate inhibitor: Binding determinants of a potent, broad-spectrum inhibitor. Biochemistry 2000, 39, 4288–4298. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Jin, W.; Matsunaga, K.; Ikemizu, S.; Yamagata, Y.; Wachino, J.-i.; Shibata, N.; Arakawa, Y.; Kurosaki, H. Crystallographic investigation of the inhibition mode of a VIM-2 metallo-β-lactamase from Pseudomonas aeruginosa by a mercaptocarboxylate inhibitor. J. Med. Chem. 2007, 50, 6647–6653. [Google Scholar] [CrossRef] [PubMed]
- Zhai, L.; Zhang, Y.-L.; Kang, J.S.; Oelschlaeger, P.; Xiao, L.; Nie, S.-S.; Yang, K.-W. Triazolylthioacetamide: A valid scaffold for the development of New Delhi metallo-β-lactmase-1 (NDM-1) inhibitors. ACS Med. Chem. Lett. 2016, 7, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hu, X.; Wu, Y.; Zhang, W.; Chen, X.; You, X.; Hu, L. Synthesis and structure-activity relationship of novel bisindole amidines active against MDR Gram-positive and Gram-negative bacteria. Eur. J. Med. Chem. 2018, 150, 771–782. [Google Scholar] [CrossRef]
- Yu, Z.-J.; Liu, S.; Zhou, S.; Li, H.; Yang, F.; Yang, L.-L.; Wu, Y.; Guo, L.; Li, G.-B. Virtual target screening reveals rosmarinic acid and salvianolic acid A inhibiting metallo-and serine-β-lactamases. Bioorg. Med. Chem. Lett. 2018, 28, 1037–1042. [Google Scholar] [CrossRef]
- Chen, A.Y.; Thomas, P.W.; Stewart, A.C.; Bergstrom, A.; Cheng, Z.; Miller, C.; Bethel, C.R.; Marshall, S.H.; Credille, C.V.; Riley, C.L. Dipicolinic acid derivatives as inhibitors of New Delhi metallo-β-lactamase-1. J. Med. Chem. 2017, 60, 7267–7283. [Google Scholar] [CrossRef]
- Wang, X.; Gao, Y.; Yu, Y.; Yang, Y.; Wang, G.; Sun, L.; Niu, X. Design of dipicolinic acid derivatives as New Delhi metallo-β-lactamase-1 inhibitors using a combined computational approach. J. Biomol. Struct. Dyn. 2020, 38, 3384–3395. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Xu, Y.; Xia, Q.; Bai, C.; Wang, T.; Wang, L.; He, D.; Xie, N.; Li, L.; Wang, J. Simplified captopril analogues as NDM-1 inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 386–389. [Google Scholar] [CrossRef]
- Cheng, Z.; Thomas, P.W.; Ju, L.; Bergstrom, A.; Mason, K.; Clayton, D.; Miller, C.; Bethel, C.R.; VanPelt, J.; Tierney, D.L. Evolution of New Delhi metallo-β-lactamase (NDM) in the clinic: Effects of NDM mutations on stability, zinc affinity, and mono-zinc activity. J. Biol. Chem. 2018, 293, 12606–12618. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhou, Y.; Niu, X.; Wang, T.; Li, J.; Liu, Z.; Wang, J.; Tang, S.; Wang, Y.; Deng, X. Magnolol restores the activity of meropenem against NDM-1-producing Escherichia coli by inhibiting the activity of metallo-beta-lactamase. Cell Death Discov. 2018, 20, 28. [Google Scholar] [CrossRef] [PubMed]
- Rivière, G.; Oueslati, S.; Gayral, M.; Créchet, J.-B.; Nhiri, N.; Jacquet, E.; Cintrat, J.-C.; Giraud, F.; Van Heijenoort, C.; Lescop, E. NMR characterization of the influence of Zinc (II) ions on the structural and dynamic behavior of the New Delhi Metallo-β-Lactamase-1 and on the binding with flavonols as inhibitors. ACS Omega 2020, 5, 10466–10480. [Google Scholar] [CrossRef] [PubMed]
- Linciano, P.; Cendron, L.; Gianquinto, E.; Spyrakis, F.; Tondi, D. Ten years with New Delhi metallo-β-lactamase-1 (NDM-1): From structural insights to inhibitor design. ACS Infect. Dis. 2019, 5, 9–34. [Google Scholar] [CrossRef]
- Sohail, S.; Rajiv, L.; Praneeta, K.; Tandon, G. Beta lactamase inhibitors from indigenous herbs and spices. Res. J. Pharm. Biol. Chem. Sci. 2014, 5, 275–285. [Google Scholar]
- Zhang, J.; Wang, S.; Wei, Q.; Guo, Q.; Bai, Y.; Yang, S.; Song, F.; Zhang, L.; Lei, X. Synthesis and biological evaluation of Aspergillomarasmine A derivatives as novel NDM-1 inhibitor to overcome antibiotics resistance. Bioorg. Med. Chem. 2017, 25, 5133–5141. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.P.; Rees, S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef]
- Tabassum, N.; Lee, J.-H.; Yim, S.-H.; Batkhuu, G.J.; Jung, D.-W.; Williams, D.R. Isolation of 4, 5-O-dicaffeoylquinic acid as a pigmentation inhibitor occurring in Artemisia capillaris Thunberg and its validation in vivo. Evid.-Based Complement. Altern. Med. 2016, 2016, 7823541. [Google Scholar] [CrossRef]
- Koolaji, N.; Shammugasamy, B.; Schindeler, A.; Dong, Q.; Dehghani, F.; Valtchev, P. Citrus peel flavonoids as potential cancer prevention agents. Curr. Dev. Nutr. 2020, 4, nzaa025. [Google Scholar] [CrossRef] [PubMed]
- Kar, B.; Kundu, C.N.; Pati, S.; Bhattacharya, D. Discovery of phyto-compounds as novel inhibitors against NDM-1 and VIM-1 protein through virtual screening and molecular modelling. J. Biomol. Struct. Dyn. 2023, 41, 1267–1280. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, Y.; Gao, Y.; Niu, X. Discovery of the novel inhibitor against New Delhi Metallo-β-Lactamase based on virtual screening and molecular modelling. Int. J. Mol. Sci. 2020, 21, 3567. [Google Scholar] [CrossRef] [PubMed]
- Jin, B.-R.; Chung, K.-S.; Cheon, S.-Y.; Lee, M.; Hwang, S.; Noh Hwang, S.; Rhee, K.-J.; An, H.-J. Rosmarinic acid suppresses colonic inflammation in dextran sulphate sodium (DSS)-induced mice via dual inhibition of NF-κB and STAT3 activation. Sci. Rep. 2017, 7, 46252. [Google Scholar] [CrossRef] [PubMed]
- Javidanpour, S.; Dianat, M.; Badavi, M.; Mard, S.A. The cardioprotective effect of rosmarinic acid on acute myocardial infarction and genes involved in Ca2+ homeostasis. Free Radic. Res. 2017, 51, 911–923. [Google Scholar] [CrossRef] [PubMed]
- Lou, K.; Yang, M.; Duan, E.; Zhao, J.; Yu, C.; Zhang, R.; Zhang, L.; Zhang, M.; Xiao, Z.; Hu, W. Rosmarinic acid stimulates liver regeneration through the mTOR pathway. Phytomedicine 2016, 23, 1574–1582. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-Y.; Wang, Y.-Y.; Chen, W.-Y.; Liao, S.-L.; Chou, S.-T.; Yang, C.-P.; Chen, C.-J. Hepatoprotective activities of rosmarinic acid against extrahepatic cholestasis in rats. Food Chem. Toxicol. 2017, 108, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhao, S.; Zhao, Q.; Chen, Y.; Jia, S.; Xiang, R.; Zhang, J.; Sun, J.; Xu, Y.; Zhao, M. Butein, a potential drug for the treatment of bone cancer pain through bioinformatic and network pharmacology. Toxicol. Appl. Pharmacol. 2023, 472, 116570. [Google Scholar] [CrossRef]
- Sulaiman, S.; Arafat, K.; Al-Azawi, A.M.; AlMarzooqi, N.A.; Lootah, S.N.A.H.; Attoub, S. Butein and frondoside-A combination exhibits additive anti-cancer effects on tumor cell viability, colony growth, and invasion and synergism on endothelial cell migration. Int. J. Mol. Sci. 2021, 23, 431. [Google Scholar] [CrossRef]
- Kapoor, N.; Ghorai, S.M.; Khuswaha, P.K.; Bandichhor, R.; Brogi, S. Butein as a potential binder of human ACE2 receptor for interfering with SARS-CoV-2 entry: A computer-aided analysis. J. Mol. Model. 2022, 28, 270. [Google Scholar] [CrossRef]
- Xie, X.-Q.S. Exploiting PubChem for virtual screening. Expert Opin. Drug Discov. 2010, 5, 1205–1220. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Sterling, T.; Irwin, J.J. ZINC 15–ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Norgan, A.P.; Coffman, P.K.; Kocher, J.-P.A.; Katzmann, D.J.; Sosa, C.P. Multilevel parallelization of AutoDock 4.2. J. Cheminform. 2011, 3, 12. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; MacKerell, A.D., Jr. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; MacKerell, A.D., Jr. Automation of the CHARMM General Force Field (CGenFF) I: Bond perception and atom typing. J. Chem. Inf. Model. 2012, 52, 3144–3154. [Google Scholar] [CrossRef]
- Ruder, S. An overview of gradient descent optimization algorithms. arXiv 2016, arXiv:1609.04747. [Google Scholar]
- Bajda, M.; Więckowska, A.; Hebda, M.; Guzior, N.; Sotriffer, C.A.; Malawska, B. Structure-based search for new inhibitors of cholinesterases. Int. J. Mol. Sci. 2013, 14, 5608–5632. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Compound Name | Binding Energy (kcal/mol) | Ki (µM) | LogS 1 | LogP 2 | Ames 3 | Carcinogenicity | Toxicophores | Synthetic Accessibility | Lipinski | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| Butein | −9.1 | 2.04 | −2.99 | 2.63 | 0.826 | 0.509 | 3 | 2.22 | Accepted | This Study |
| Monodemethylcurcumin | −9.3 | 3.33 | −3.33 | 2.57 | 0.289 | 0.599 | 3 | 2.49 | Accepted | |
| Rosmarinic acid | −8.9 | 7.35 | −2.43 | 1.77 | 0.035 | 0.275 | 3 | 2.90 | Accepted | |
| Aspergillomarasmine | −7.36 | 508.94 | −2.15 | −5.94 | 0.013 | 0.067 | 3 | 3.42 | Rejected | [25] |
| Tiopronin | −7.94 | 586.05 | −0.51 | −0.47 | 0.015 | 0.03 | 3 | 2.92 | Accepted | [41] |
| Thiorphan | −7.04 | 1.25 | −1.73 | 1.23 | 0.603 | 0.117 | 3 | 2.44 | Accepted | [41] |
| Dimercaprol | −3.1 | 510.9 | −0.54 | 0.59 | 0.814 | 0.931 | 2 | 4.39 | Accepted | [42] |
| D-captopril | −6.63 | 600.3 | −0.61 | 0.27 | 0.01 | 0.029 | 3 | 3.03 | Accepted | [43] |
| Names of Compounds | Average Energy Interactions kJ.mol−1 (Coul-SR) | Average Energy Interactions kJ.mol−1 (LJ-SR) |
|---|---|---|
| Butein | −77.533 | −61.658 |
| Monodemethylcurcumin | −46.918 | −98.748 |
| Rosmarinic acid | −50.221 | −101.427 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bibi, Z.; Asghar, I.; Ashraf, N.M.; Zeb, I.; Rashid, U.; Hamid, A.; Ali, M.K.; Hatamleh, A.A.; Al-Dosary, M.A.; Ahmad, R.; et al. Prediction of Phytochemicals for Their Potential to Inhibit New Delhi Metallo β-Lactamase (NDM-1). Pharmaceuticals 2023, 16, 1404. https://doi.org/10.3390/ph16101404
Bibi Z, Asghar I, Ashraf NM, Zeb I, Rashid U, Hamid A, Ali MK, Hatamleh AA, Al-Dosary MA, Ahmad R, et al. Prediction of Phytochemicals for Their Potential to Inhibit New Delhi Metallo β-Lactamase (NDM-1). Pharmaceuticals. 2023; 16(10):1404. https://doi.org/10.3390/ph16101404
Chicago/Turabian StyleBibi, Zainab, Irfa Asghar, Naeem Mahmood Ashraf, Iftikhar Zeb, Umer Rashid, Arslan Hamid, Maria Kanwal Ali, Ashraf Atef Hatamleh, Munirah Abdullah Al-Dosary, Raza Ahmad, and et al. 2023. "Prediction of Phytochemicals for Their Potential to Inhibit New Delhi Metallo β-Lactamase (NDM-1)" Pharmaceuticals 16, no. 10: 1404. https://doi.org/10.3390/ph16101404
APA StyleBibi, Z., Asghar, I., Ashraf, N. M., Zeb, I., Rashid, U., Hamid, A., Ali, M. K., Hatamleh, A. A., Al-Dosary, M. A., Ahmad, R., & Ali, M. (2023). Prediction of Phytochemicals for Their Potential to Inhibit New Delhi Metallo β-Lactamase (NDM-1). Pharmaceuticals, 16(10), 1404. https://doi.org/10.3390/ph16101404