
1-Piperidine Propionic Acid as an Allosteric Inhibitor of Protease Activated Receptor-2

 , , , , , and
, , , , , and 
Abstract
:1. Introduction
2. Results and Discussion
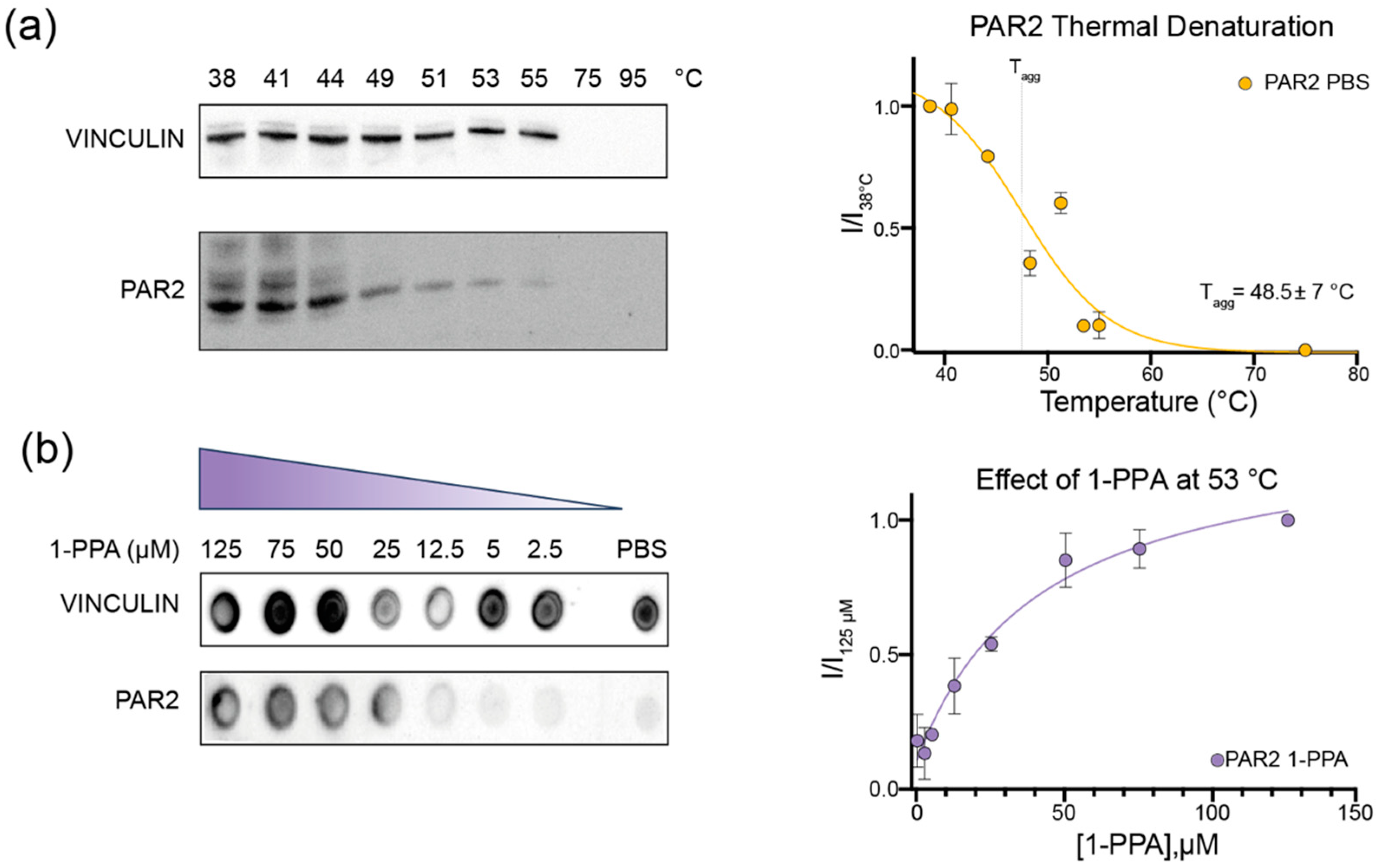
2.1. CETSA Assay Suggests That 1-PPA Has a Stabilizing Effect on PAR2
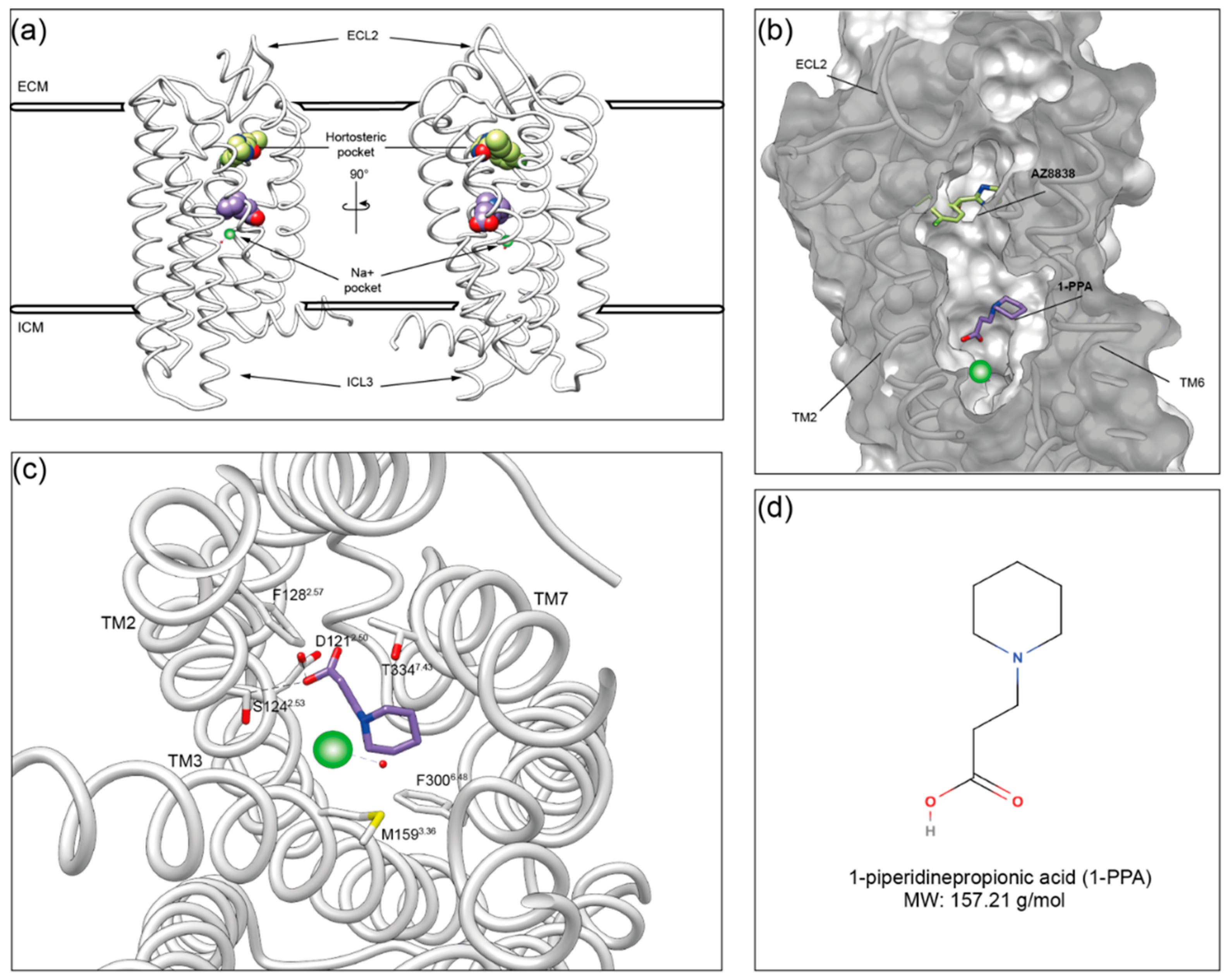
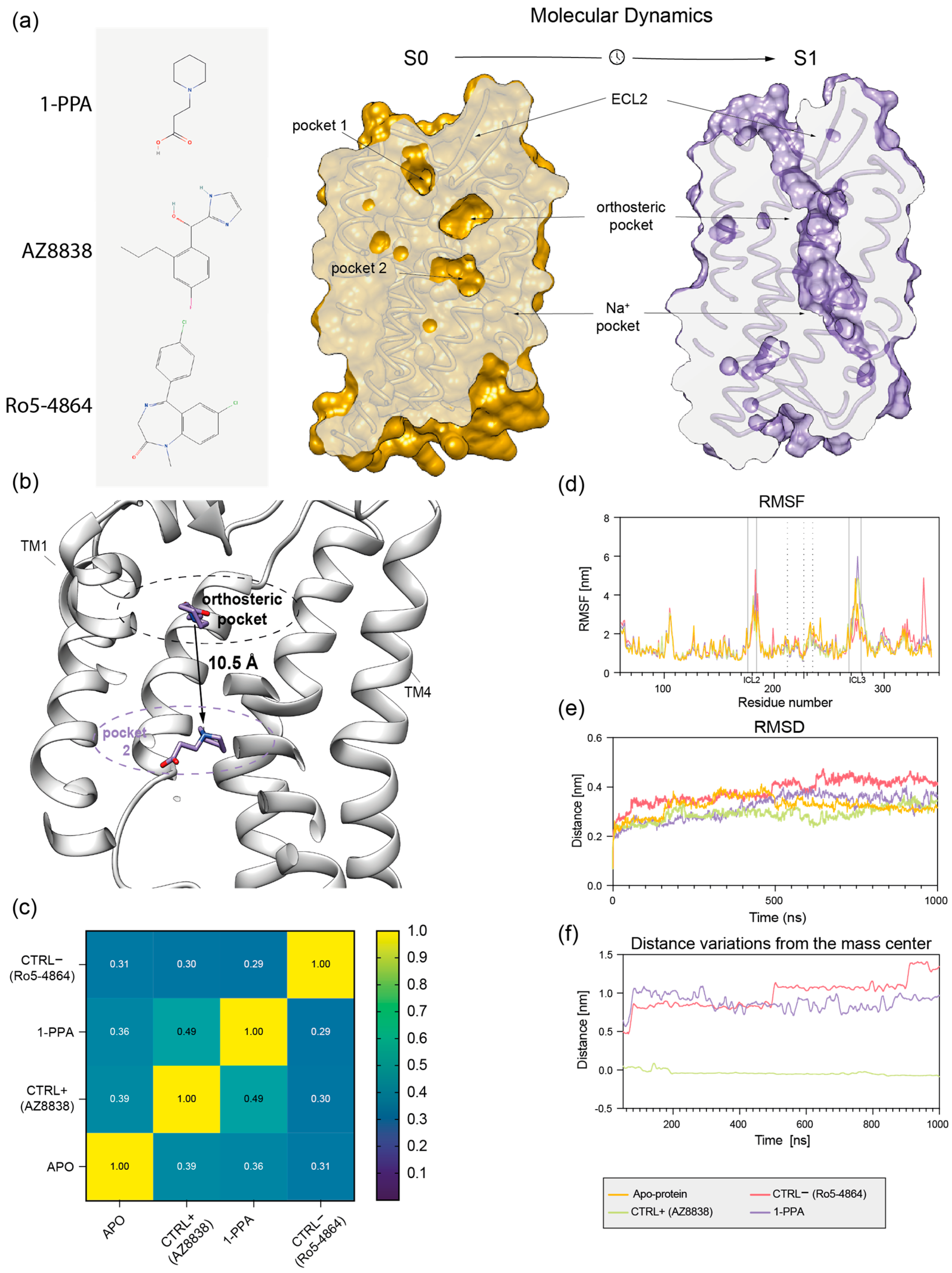
2.2. Molecular Dynamics Simulations Suggest That 1-PPA Occupies an Allosteric Pocket Buried within the PAR2 Receptor
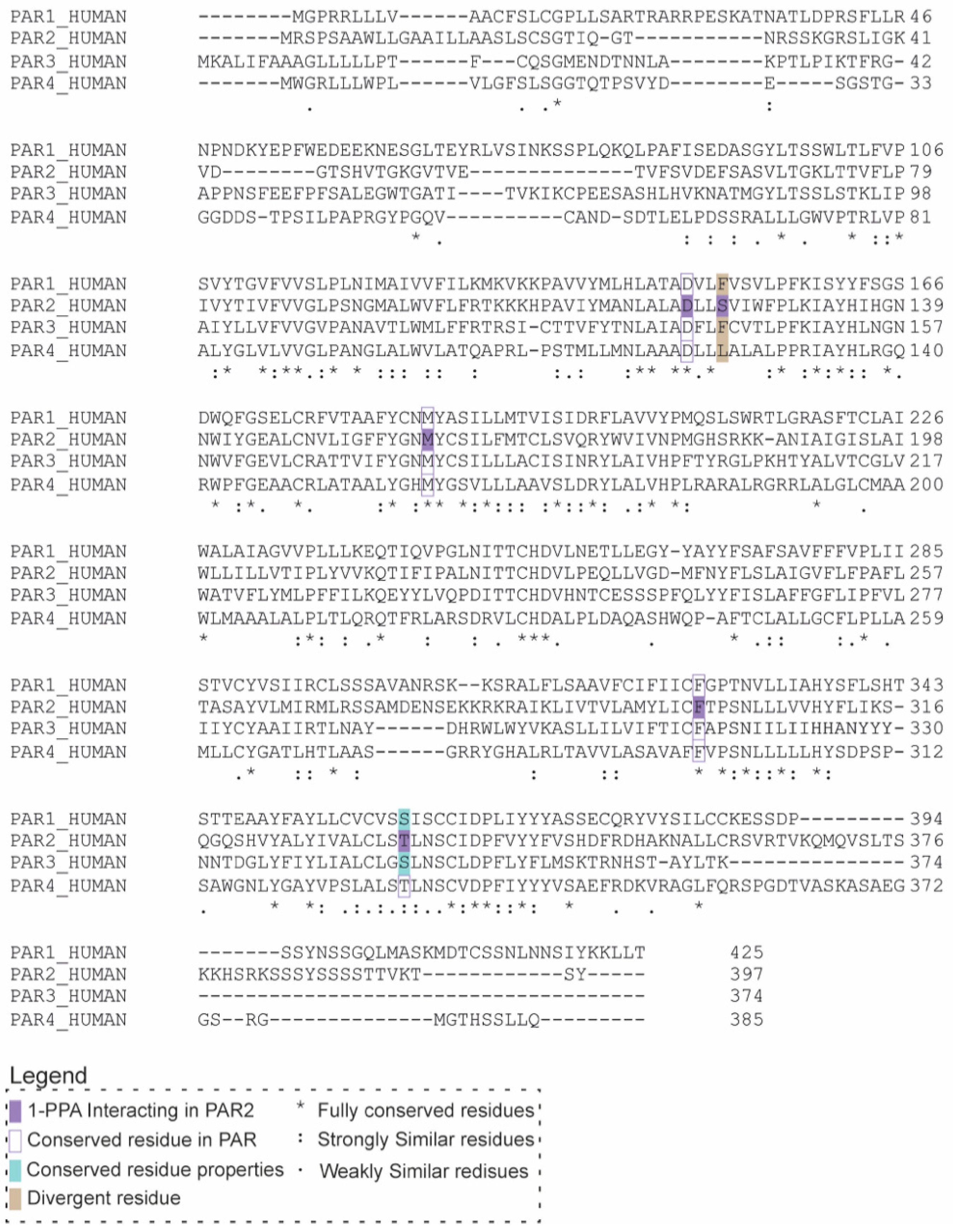
2.3. 1-PPA as a Putative Pan-PAR Ligand
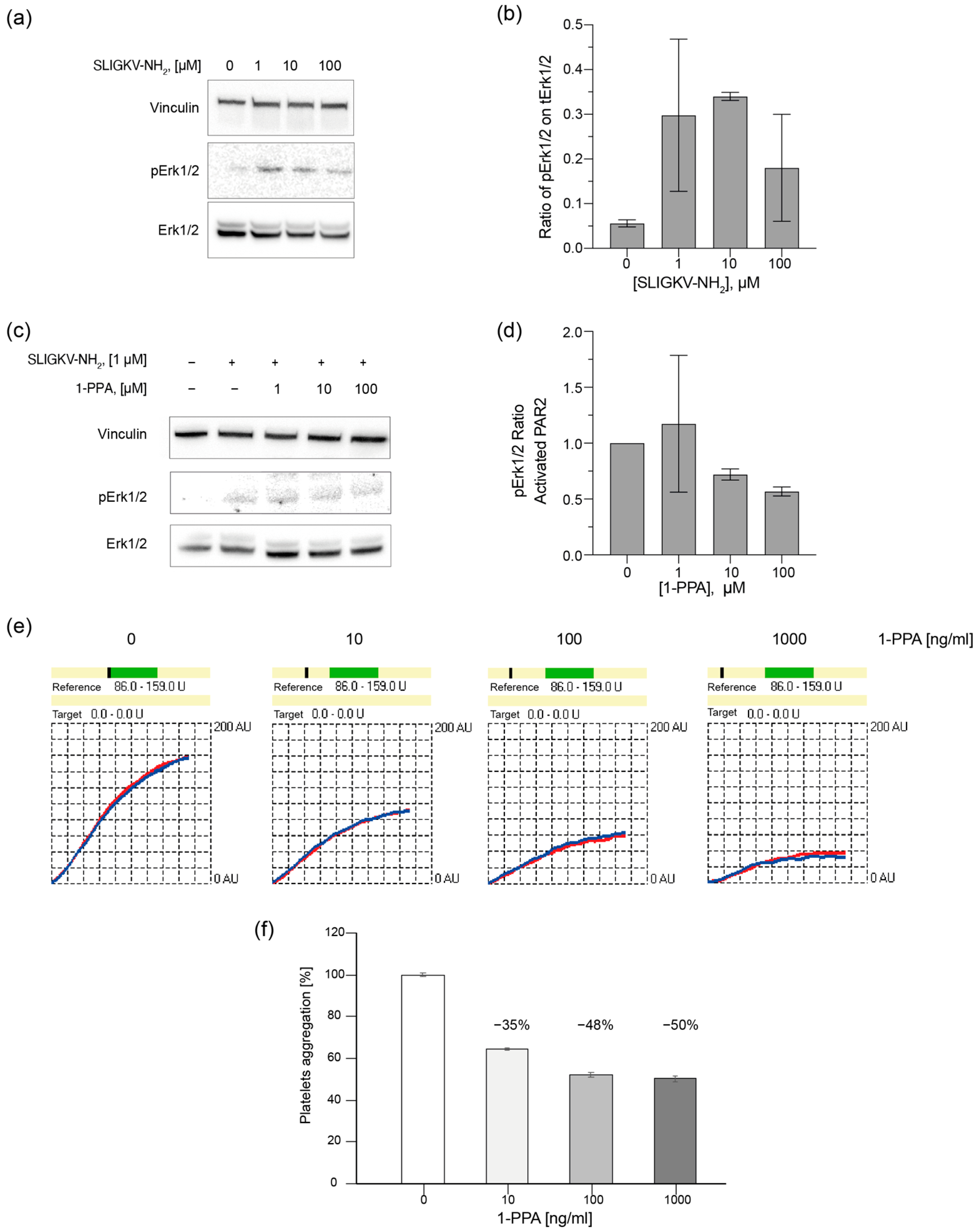
2.4. 1-PPA Antagonizes Cell Signaling and Platelet Aggregation
3. Materials and Methods
3.1. Chemicals
3.2. Suspension Cell Culture and Transfection
3.3. Adhesion Cell Culture
3.4. Cellular Thermal Shift Assay
3.5. Mechanistic Study
3.6. Erk1/2 Phoshorylation
3.7. Gel Electrophoresis and Western Blot
3.8. Immunoblotting
3.9. Protein Modelling and Docking
3.10. Molecular Dynamics
3.11. TRAPtest
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hilger, D.; Masureel, M.; Kobilka, B.K. Structure and Dynamics of GPCR Signaling Complexes. Nat. Struct. Mol. Biol. 2018, 25, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Tesmer, J.J.G. Hitchhiking on the Heptahelical Highway: Structure and Function of 7TM Receptor Complexes. Nat. Rev. Mol. Cell Biol. 2016, 17, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Song, G.; de Graaf, C.; Stevens, R.C. Structure and Function of Peptide-Binding G Protein-Coupled Receptors. J. Mol. Biol. 2017, 429, 2726–2745. [Google Scholar] [CrossRef]
- Hedderich, J.B.; Persechino, M.; Becker, K.; Heydenreich, F.M.; Gutermuth, T.; Bouvier, M.; Bünemann, M.; Kolb, P. The Pocketome of G-Protein-Coupled Receptors Reveals Previously Untargeted Allosteric Sites. Nat. Commun. 2022, 13, 2567. [Google Scholar] [CrossRef] [PubMed]
- Latorraca, N.R.; Venkatakrishnan, A.J.; Dror, R.O. GPCR Dynamics: Structures in Motion. Chem. Rev. 2017, 117, 139–155. [Google Scholar] [CrossRef] [PubMed]
- Miller-Gallacher, J.L.; Nehmé, R.; Warne, T.; Edwards, P.C.; Schertler, G.F.X.; Leslie, A.G.W.; Tate, C.G.; Van Veen, H.W. The 2.1 Å Resolution Structure of Cyanopindolol-Bound Β1-Adrenoceptor Identifies an Intramembrane Na+ Ion That Stabilises the Ligand-Free Receptor. PLoS ONE 2014, 9, e92727. [Google Scholar] [CrossRef]
- Manglik, A.; Kobilka, B. The Role of Protein Dynamics in GPCR Function: Insights from the Β2AR and Rhodopsin. Curr. Opin. Cell Biol. 2014, 27, 136–143. [Google Scholar] [CrossRef]
- Chandrabalan, A.; Ramachandran, R.; Ramachandran, C.R. Molecular Mechanisms Regulating Proteinase-Activated Receptors (PARs). FEBS J. 2021, 288, 2697–2726. [Google Scholar] [CrossRef]
- Heuberger, D.M.; Schuepbach, R.A. Protease-Activated Receptors (PARs): Mechanisms of Action and Potential Therapeutic Modulators in PAR-Driven Inflammatory Diseases. Thromb. J. 2019, 17, 22. [Google Scholar] [CrossRef]
- Rasmussen, U.B.; Vouret-Craviari, V.; Jallat, S.; Schlesinger, Y.; Pages, G.; Pavirani, A.; Lecocq, J.-P.; Pouyssegur, J.; Van Obberghen-Schilling, E. DNA Cloning and Expression of a Hamster A-Thrombin Receptor Coupled to Ca2+ Mobilization. FEBS Lett. 1991, 288, 123–128. [Google Scholar] [CrossRef]
- Kanke, T.; Takizawa, T.; Kabeya, M.; Kawabata, A. Physiology and Pathophysiology of Proteinase-Activated Receptors (PARs): PAR-2 as a Potential Therapeutic Target. J. Pharmacol. Sci. 2005, 97, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Suhaj, P.; Olejar, T.; Matej, R.; Suhaj, P. PAR2: The Cornerstone of Pancreatic Diseases. Physiol. Res. 2022, 71, 583–596. [Google Scholar] [CrossRef]
- Cheng, R.K.; Fiez-Vandal, C.; Schlenker, O.; Edman, K.; Aggeler, B.; Brown, D.G.; Brown, G.A.; Cooke, R.; Dumelin, C.E.; Doré, A.S.; et al. Structural Insight into Allosteric Modulation of Protease-Activated Receptor 2. Nature 2017, 545, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.J.; Sundström, L.; Geschwindner, S.; Poon, E.K.Y.; Jiang, Y.; Chen, R.; Cooke, R.; Johnstone, S.; Madin, A.; Lim, J.; et al. Protease-Activated Receptor-2 Ligands Reveal Orthosteric and Allosteric Mechanisms of Receptor Inhibition. Commun. Biol. 2020, 3, 782. [Google Scholar] [CrossRef] [PubMed]
- Pontisso, P.; Biasiolo, A.; Martini, A.; Quarta, S.; Ruvoletto, M.; Turato, C.; Villano, G. The Serpinb3 Inhibitor Piperidinpropionic Acid for Tumor Treatment. U.S. Patent US20220265626A1, 25 August 2022. [Google Scholar]
- Pontisso, P.; Biasiolo, A.; Cappon, A.; Martini, A.; Quarta, S.; Ruvoletto, M.; Turato, C.; Villano, G. 1-Piperidinepropionic Acid for Treating a Fibrosing Disease. U.S. Patent US11628163B2, 18 April 2022. [Google Scholar]
- Yau, M.K.; Lim, J.; Liu, L.; Fairlie, D.P. Protease Activated Receptor 2 (PAR2) Modulators: A Patent Review (2010–2015). Expert Opin. Ther. Pat. 2016, 26, 471–483. [Google Scholar] [CrossRef]
- Kawatkar, A.; Schefter, M.; Hermansson, N.-O.; Snijder, A.; Dekker, N.; Brown, D.G.; Lundbäck, T.; Zhang, A.X.; Castaldi, M.P. CETSA beyond Soluble Targets: A Broad Application to Multipass Transmembrane Proteins. ACS Chem. Biol. 2019, 14, 1913–1920. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.M.; Glukhova, A.; Sexton, P.M.; Christopoulos, A. Structural Insights into G-Protein-Coupled Receptor Allostery. Nature 2018, 559, 45–53. [Google Scholar] [CrossRef]
- Ballesteros, J.A.; Weinstein, H. Integrated Methods for the Construction of Three-Dimensional Models and Computational Probing of Structure-Function Relations in G Protein-Coupled Receptors. In Methods in Neurosciences; Academic Press: Cambridge, MA, USA, 1995; Volume 25, pp. 366–428. [Google Scholar]
- Hollenberg, M.D.; Mihara, K.; Polley, D.; Suen, J.Y.; Han, A.; Fairlie, D.P.; Ramachandran, R.; Hollenberg, M.D. Biased Signalling and Proteinase-Activated Receptors (PARs): Targeting Inflammatory Disease. Br. J. Pharmacol. 2014, 171, 1180–1194. [Google Scholar] [CrossRef] [PubMed]
- Barry, G.D.; Suen, J.Y.; Le, G.T.; Cotterell, A.; Reid, R.C.; Fairlie, D.P. Novel Agonists and Antagonists for Human Protease Activated Receptor 2. J. Med. Chem. 2010, 53, 7428–7440. [Google Scholar] [CrossRef]
- Jiang, Y.; Yau, M.-K.; Lim, J.; Wu, K.-C.; Xu, W.; Suen, J.Y.; Fairlie, D.P. A Potent Antagonist of Protease-Activated Receptor 2 That Inhibits Multiple Signaling Functions in Human Cancer Cells. J. Pharmacol. Exp. Ther. 2018, 364, 246–257. [Google Scholar] [CrossRef]
- Camerer, E.; Qazi, A.A.; Duong, D.N.; Cornelissen, I.; Advincula, R.; Coughlin, S.R. Platelets, Protease-Activated Receptors, and Fibrinogen in Hematogenous Metastasis. Blood 2004, 104, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Schrödinger, LLC. The {PyMOL} Molecular Graphics System; Version 1.8; Schrödinger, LLC: New York, NY, USA, 2015. [Google Scholar]
- Šali, A.; Blundell, T.L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Grosdidier, A.; Zoete, V.; Michielin, O. Fast Docking Using the CHARMM Force Field with EADock DSS. J. Comput. Chem. 2011, 32, 2149–2159. [Google Scholar] [CrossRef] [PubMed]
- Lien Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock, a Protein-Small Molecule Docking Web Service Based on EADock DSS. Nucleic Acids Res. 2021, 39, W270–W277. [Google Scholar] [CrossRef]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A Fast Force Field Generation Tool for Small Organic Molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef]
- Haberthur, U.; Caflisch, A. FACTS: Fast Analytical Continuum Treatment of Solvation. J. Comput. Chem. 2008, 29, 701–715. [Google Scholar] [CrossRef]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed Atlas of Surface Topography of Proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A Message-Passing Parallel Molecular Dynamics Implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM General Force Field: A Force Field for Drug-like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed]
- Soteras Gutiérrez, I.; Lin, F.-Y.; Vanommeslaeghe, K.; Lemkul, J.A.; Armacost, K.A.; Brooks, C.L.; MacKerell, A.D. Parametrization of Halogen Bonds in the CHARMM General Force Field: Improved Treatment of Ligand–Protein Interactions. Bioorg. Med. Chem. 2016, 24, 4812–4825. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; MacKerell, A.D. Automation of the CHARMM General Force Field (CGenFF) I: Bond Perception and Atom Typing. J. Chem. Inf. Model. 2012, 52, 3144–3154. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Raman, E.P.; MacKerell, A.D. Automation of the CHARMM General Force Field (CGenFF) II: Assignment of Bonded Parameters and Partial Atomic Charges. J. Chem. Inf. Model. 2012, 52, 3155–3168. [Google Scholar] [CrossRef]
- Zorzan, M.; Castellan, M.; Gasparotto, M.; de Melo, G.D.; Zecchin, B.; Leopardi, S.; Chen, A.; Rosato, A.; Angelini, A.; Bourhy, H.; et al. Antiviral Mechanisms of Two Broad-Spectrum Monoclonal Antibodies for Rabies Prophylaxis and Therapy. Front Immunol 2023, 14, 1186063. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Bernetti, M.; Bussi, G. Pressure Control Using Stochastic Cell Rescaling. J. Chem. Phys. 2020, 153, 114107. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N·log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Braun, S.; Jawansky, S.; Vogt, W.; Mehilli, J.; Schömig, A.; Kastrati, A.; vonBeckerath, N.; DirkSibbing, D. Assessment of ADP-Induced Platelet aggregation with light Transmission Aggregometry and Multiple Electrode platelet Aggregometry before and After clopidogrel Treatment Platelets and Blood Cells. Thromb. Haemost. 2008, 99, 121–126. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Dilution | Code | Producer |
|---|---|---|---|
| hPAR2 (HA tag) | 1:5000 | 2-2.2.14 | Invitrogen, Waltham, MA, USA |
| PAR2 | 1:1500 | ||
| Vinculin | 1:5000 | HL1964 | GeneTex, Irvine, CA, USA |
| Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) | 1:1000 | 9101 | Cell Signaling Danvers, MA, USA |
| Total p44/42 MAPK (Erk1/2) | 1:100 | Sc-514302 | Santa Cruz Biotechnology, Dallas, TX, USA |
| Mouse IgG | 1:10,000 | A16066 | Invitrogen, Waltham, MA, USA |
| Rabbit IgG | 1:10,000 | SSA004 | SinoBiological, Beijing, China |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chinellato, M.; Gasparotto, M.; Quarta, S.; Ruvoletto, M.; Biasiolo, A.; Filippini, F.; Spiezia, L.; Cendron, L.; Pontisso, P. 1-Piperidine Propionic Acid as an Allosteric Inhibitor of Protease Activated Receptor-2. Pharmaceuticals 2023, 16, 1486. https://doi.org/10.3390/ph16101486
Chinellato M, Gasparotto M, Quarta S, Ruvoletto M, Biasiolo A, Filippini F, Spiezia L, Cendron L, Pontisso P. 1-Piperidine Propionic Acid as an Allosteric Inhibitor of Protease Activated Receptor-2. Pharmaceuticals. 2023; 16(10):1486. https://doi.org/10.3390/ph16101486
Chicago/Turabian StyleChinellato, Monica, Matteo Gasparotto, Santina Quarta, Mariagrazia Ruvoletto, Alessandra Biasiolo, Francesco Filippini, Luca Spiezia, Laura Cendron, and Patrizia Pontisso. 2023. "1-Piperidine Propionic Acid as an Allosteric Inhibitor of Protease Activated Receptor-2" Pharmaceuticals 16, no. 10: 1486. https://doi.org/10.3390/ph16101486
APA StyleChinellato, M., Gasparotto, M., Quarta, S., Ruvoletto, M., Biasiolo, A., Filippini, F., Spiezia, L., Cendron, L., & Pontisso, P. (2023). 1-Piperidine Propionic Acid as an Allosteric Inhibitor of Protease Activated Receptor-2. Pharmaceuticals, 16(10), 1486. https://doi.org/10.3390/ph16101486