
Umbelliferone and Its Synthetic Derivatives as Suitable Molecules for the Development of Agents with Biological Activities: A Review of Their Pharmacological and Therapeutic Potential



Abstract
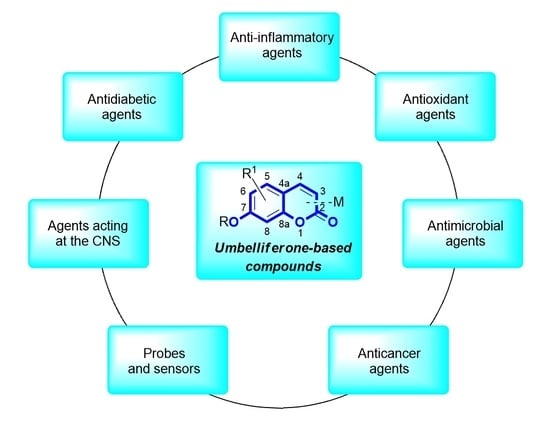
:
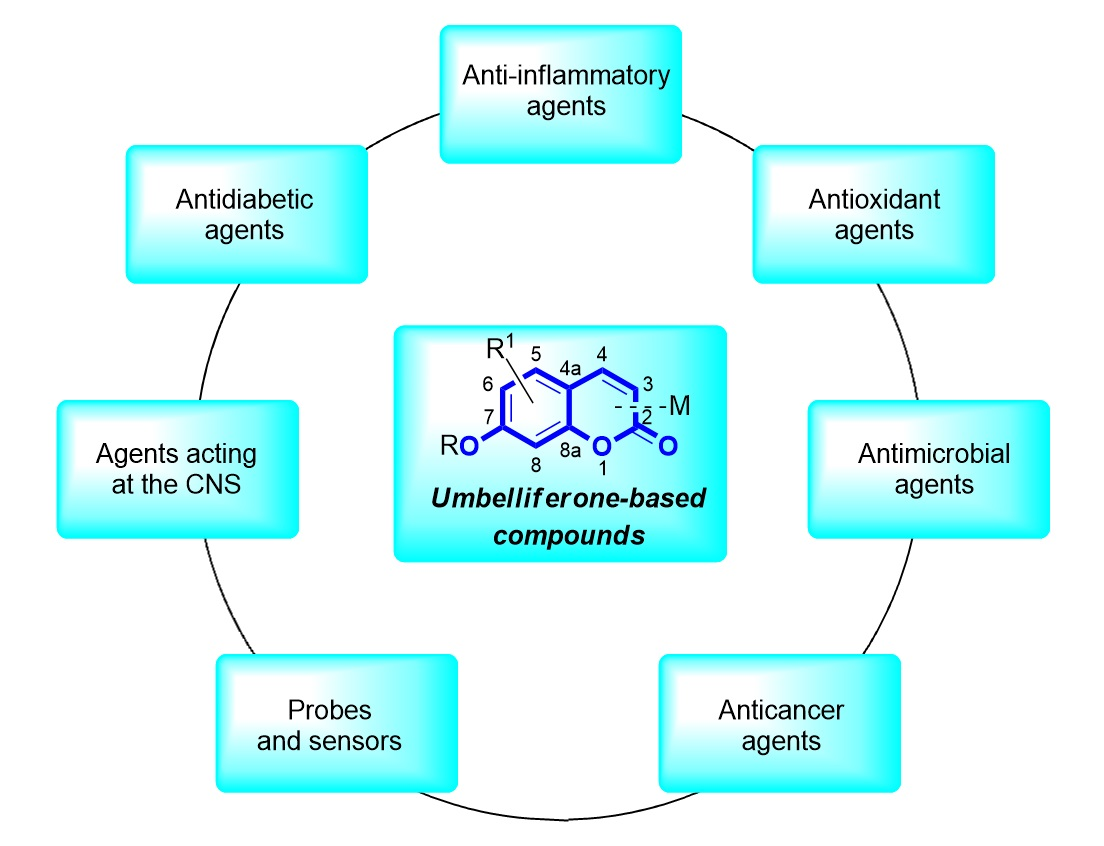
1. Introduction
2. Anti-Inflammatory Activity
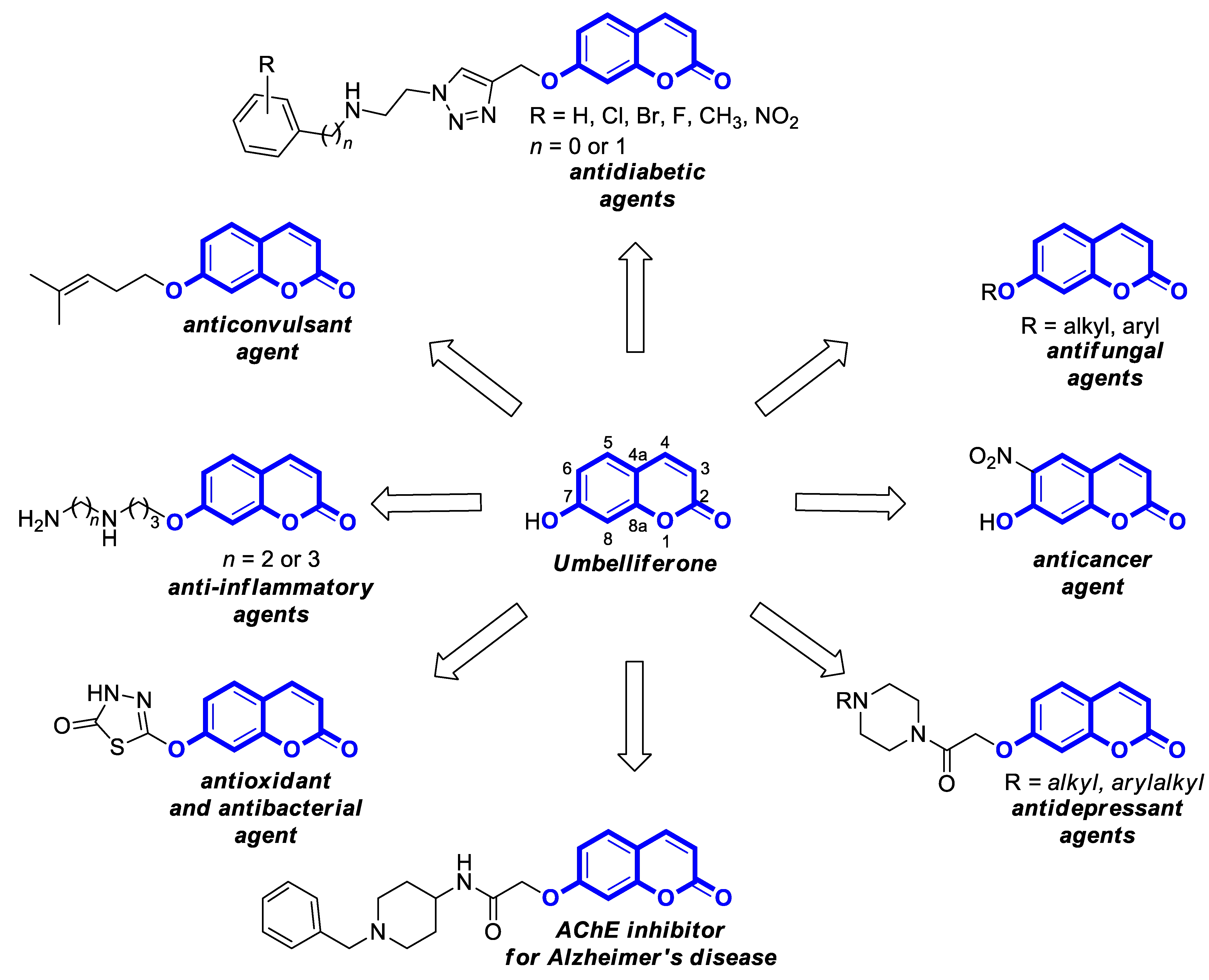
2.1. Anti-Inflammatory Properties of Umbelliferone
2.2. Synthetic 7-Hydroxycoumarin-Based Compounds as Anti-Inflammatory Agents
3. Antioxidant Activity
3.1. Antioxidant Properties of Umbelliferone
3.2. Synthetic 7-Hydroxycoumarin-Based Compounds as Antioxidant Agents
3.3. Metal Complexes with 7-Hydroxycoumarin-Based Compounds as Antioxidant Agents
4. Umbelliferone and 7-Hydroxycoumarin-Based Compounds Acting in the Central Nervous System (CNS)
4.1. Neurodegenerative Disorders
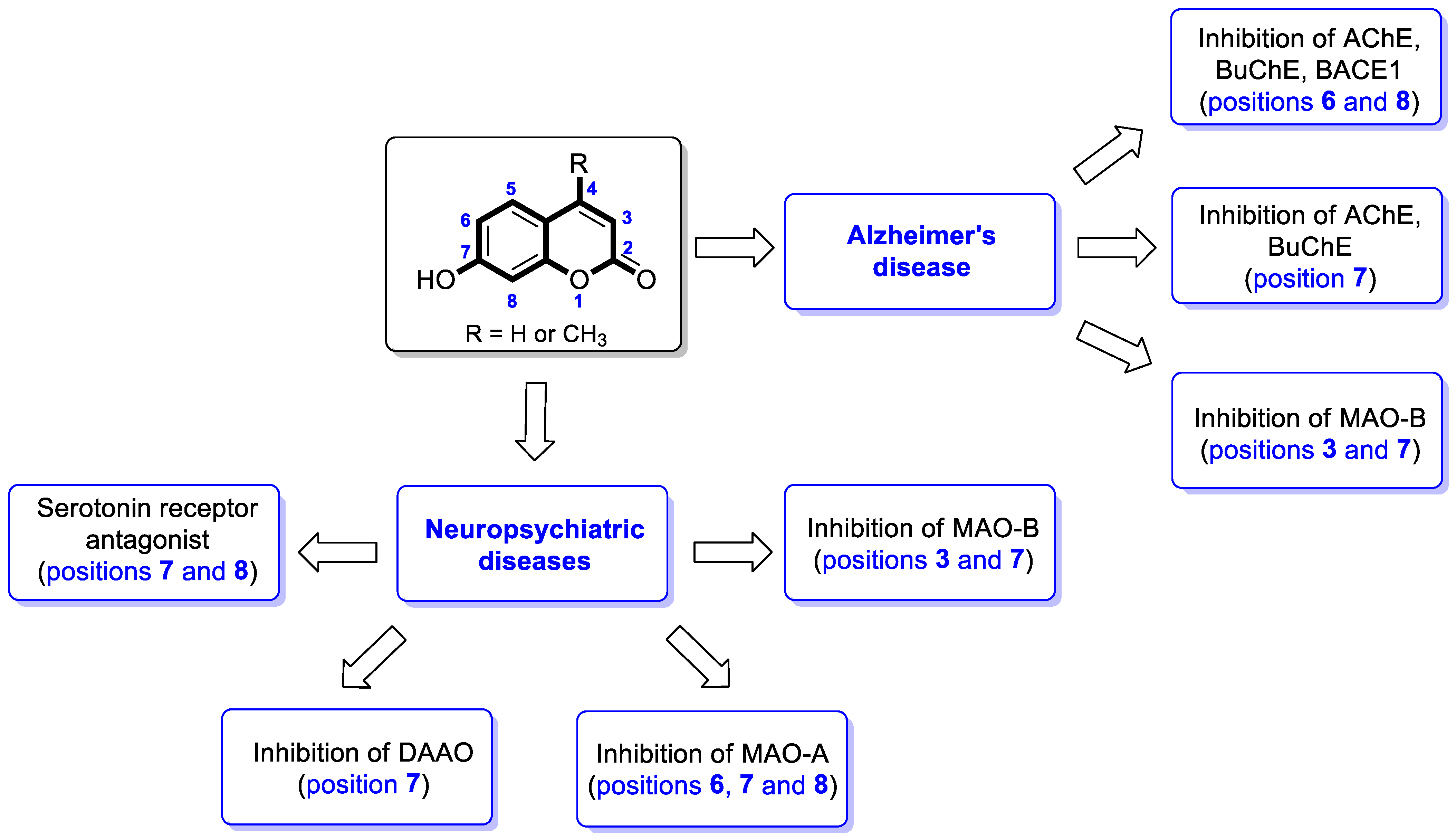
4.2. Neuropsychiatric Diseases
4.2.1. Synthetic 7-Hydroxycoumarin-Based Compounds Targeting Monoamine Oxidase (MAO) and D-Amino Acid Oxidase (DAAO)
4.2.2. Synthetic 7-Hydroxycoumarin-Based Compounds Targeting Serotonin Receptors
4.3. Antiepileptic Agents
5. Umbelliferone and 7-Hydroxycoumarin-Based Compounds as Antidiabetic Agents
6. Chemotherapeutic Activity
6.1. Antimicrobial Properties of Umbelliferone and 7-Hydroxycoumarin-Based Compounds
6.1.1. Synthetic 7-Hydroxycoumarin-Based Compounds as Antibacterial and Antifungal Agents
6.1.2. Metal Complexes of 7-Hydroxycoumarin-Based Compounds as Antibacterial and Antifungal Agents
6.2. Synthetic 7-Hydroxycoumarin-Based Compounds as Antituberculosis Agents
6.3. Synthetic 7-Hydroxycoumarin-Based Compounds as Antimalarial Agents
6.4. Umbelliferone and 7-Hydroxycoumarin-Based Compounds as Antiviral Agents
6.5. Umbelliferone and 7-Hydroxycoumarin-Based Compounds as Anticancer Agents
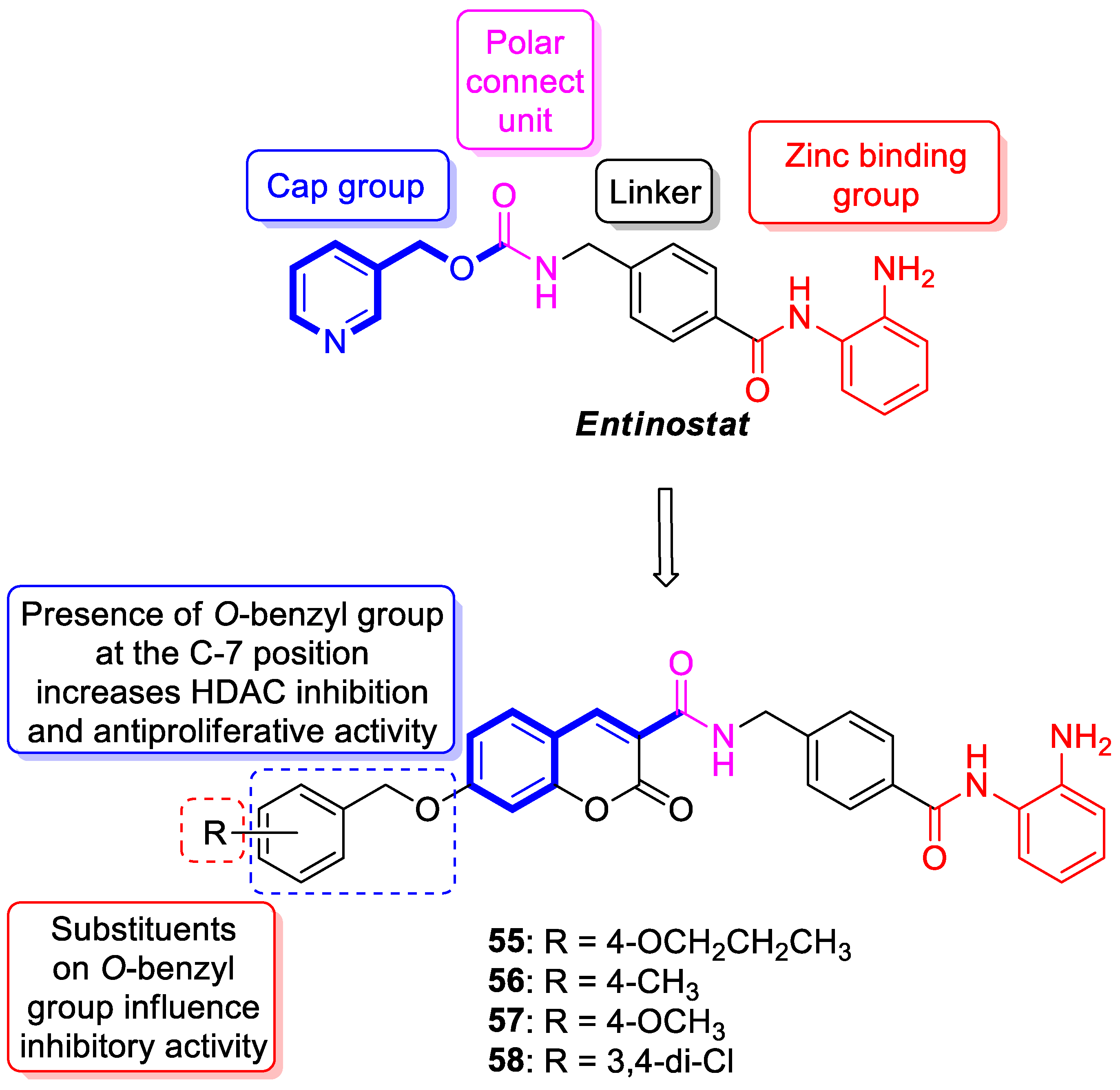
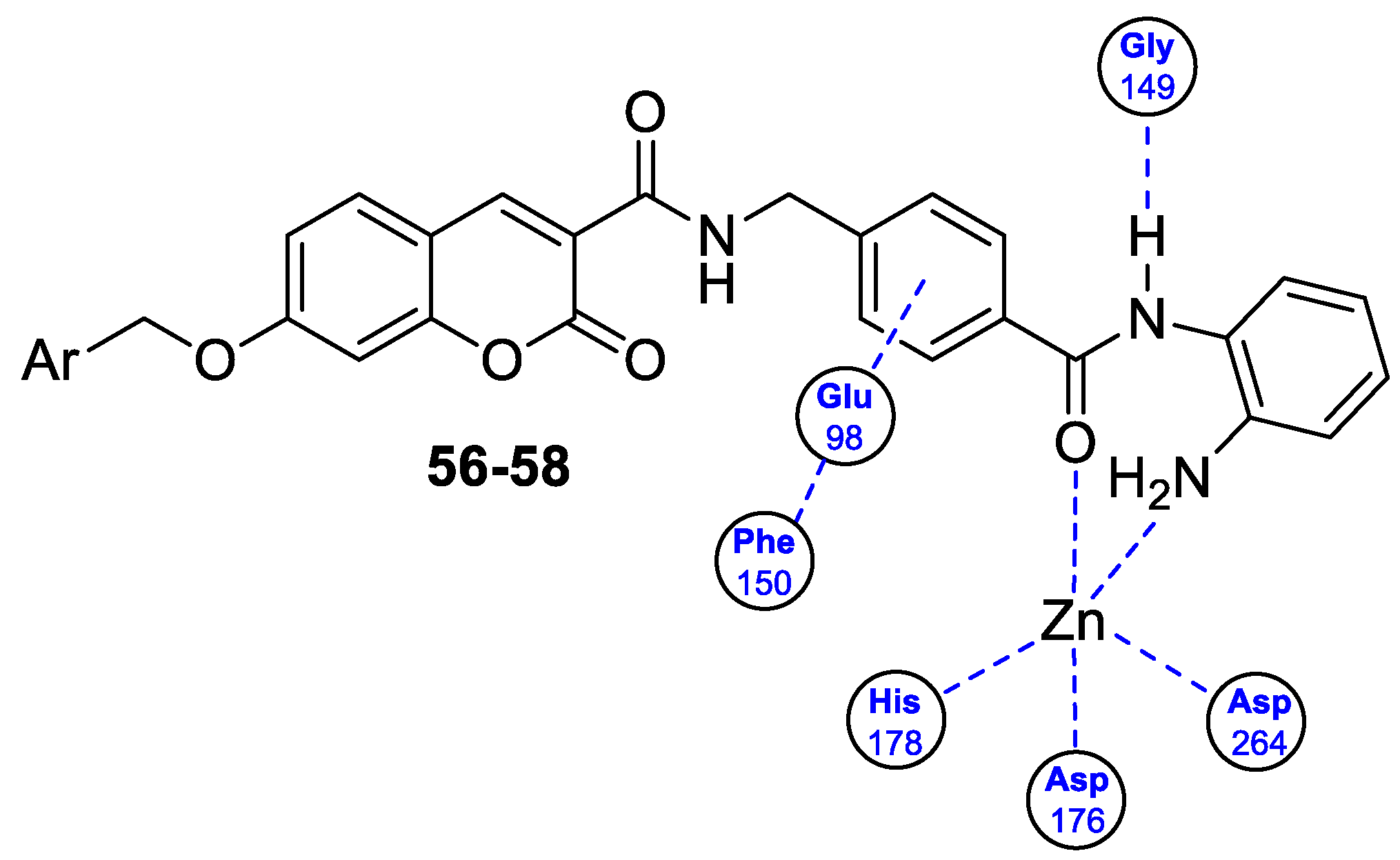
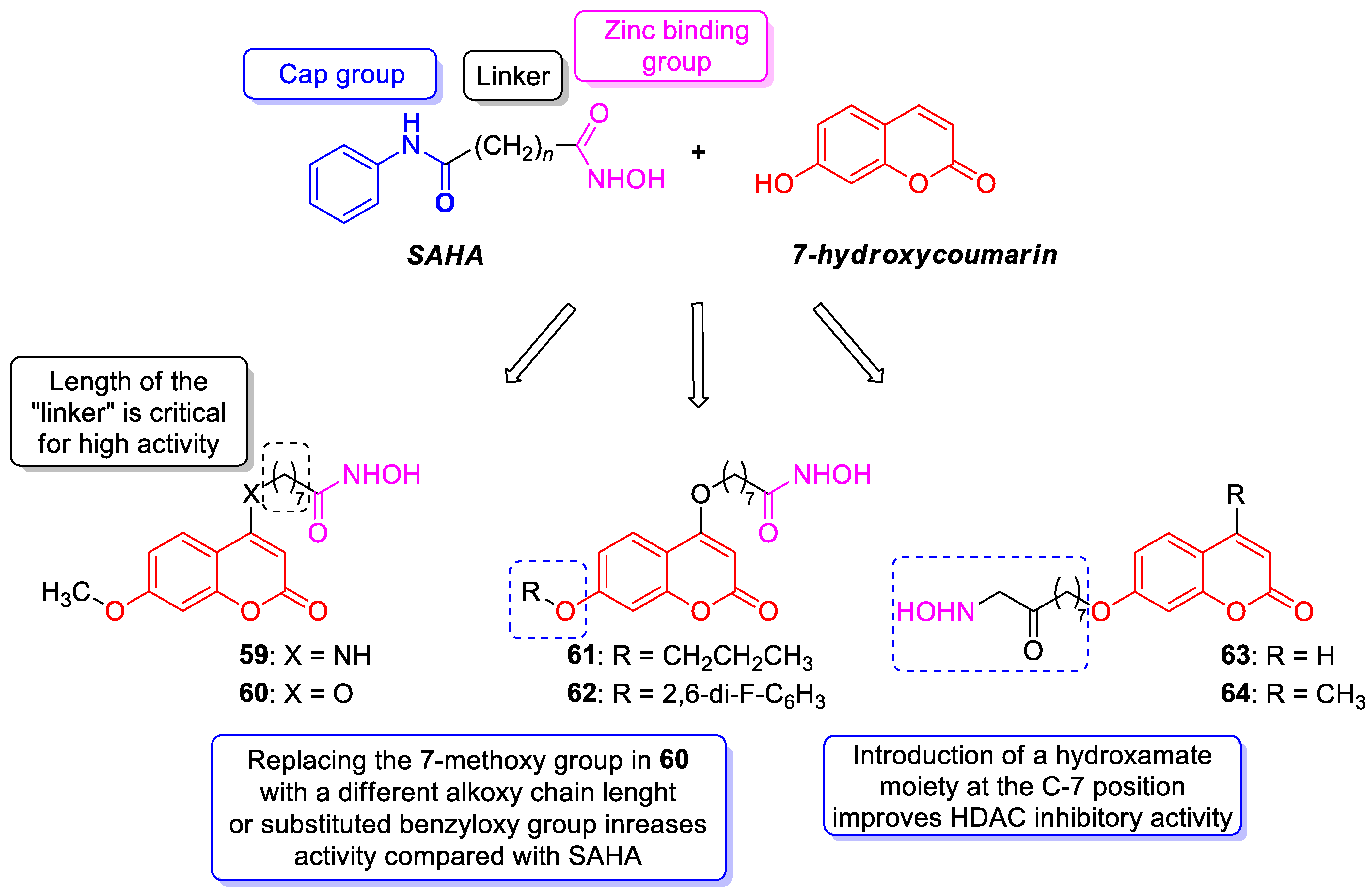
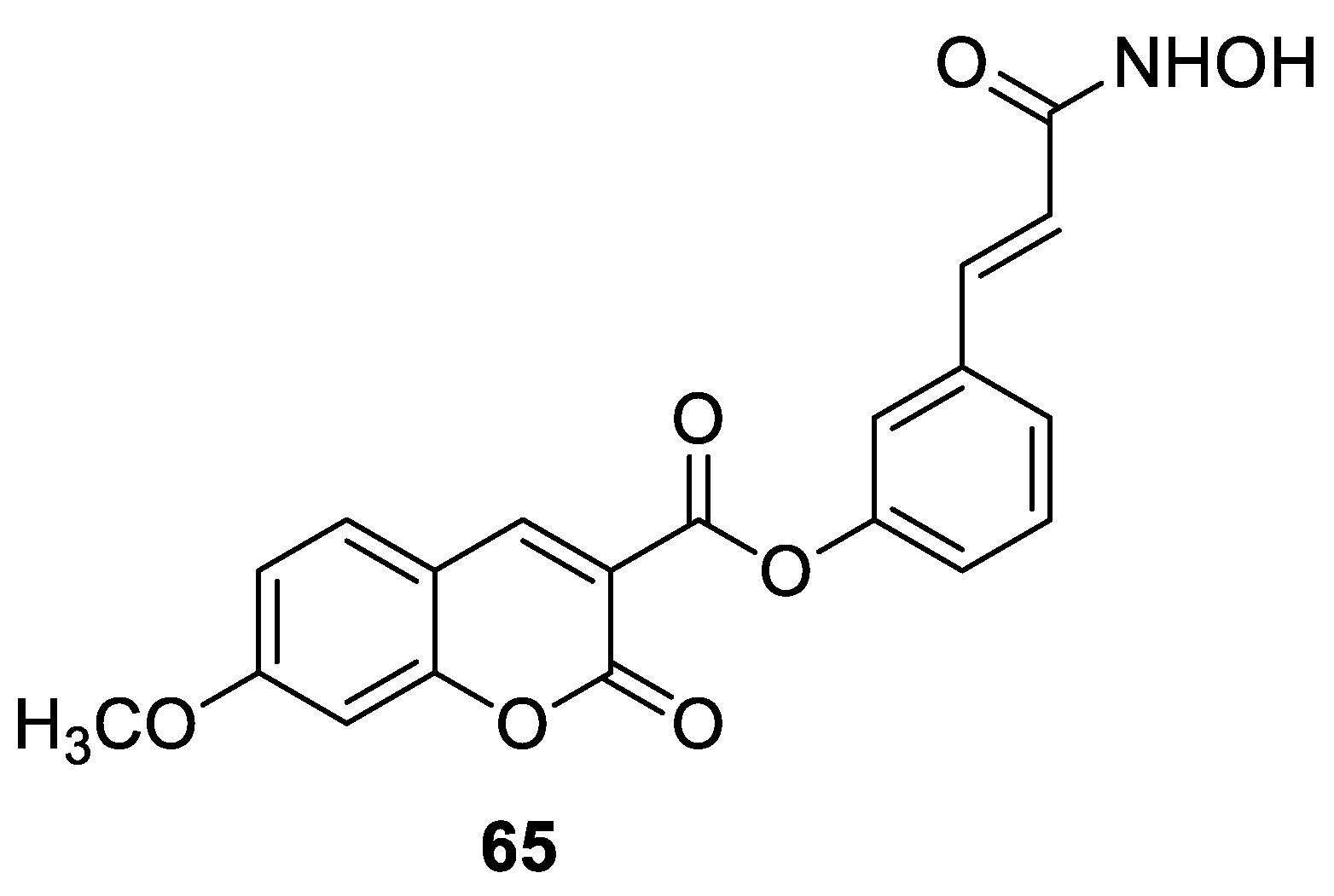
6.5.1. Synthetic 7-Hydroxycoumarin-Based Compounds as Histone Deacetylase (HDAC) Inhibitors
6.5.2. Synthetic 7-Hydroxycoumarin-Based Compounds as Androgen Receptor (AR) Antagonists
6.5.3. Synthetic 7-Hydroxycoumarin-Based Compounds as Inhibitors of the PIK3/Akt Signaling Pathway
6.5.4. Monoterpene-Coumarin Hybrids as Tyrosyl-DNA Phosphodiesterase 1 (Tdp1) Inhibitors
6.5.5. Synthetic 7-Hydroxycoumarin-Based Compounds as Carbonic Anhydrase (CA) Inhibitors
6.5.6. Synthetic 7-Hydroxycoumarin-Based Compounds as Cyclooxygenase-2 (COX-2) and 5-Lipoxygenas (5-LOX) Inhibitors
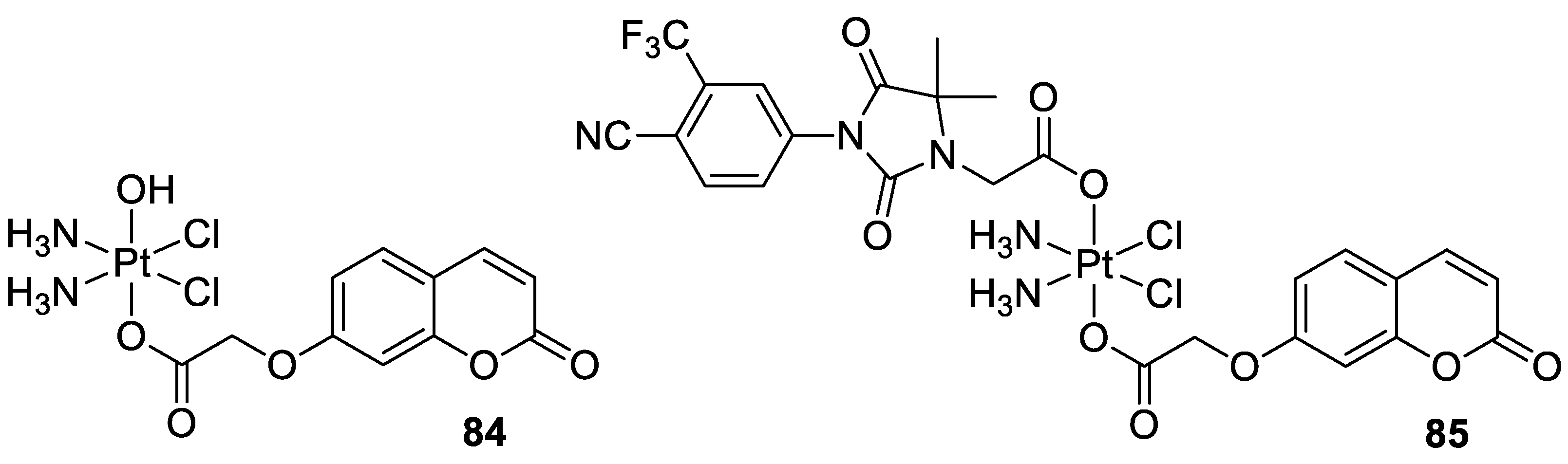
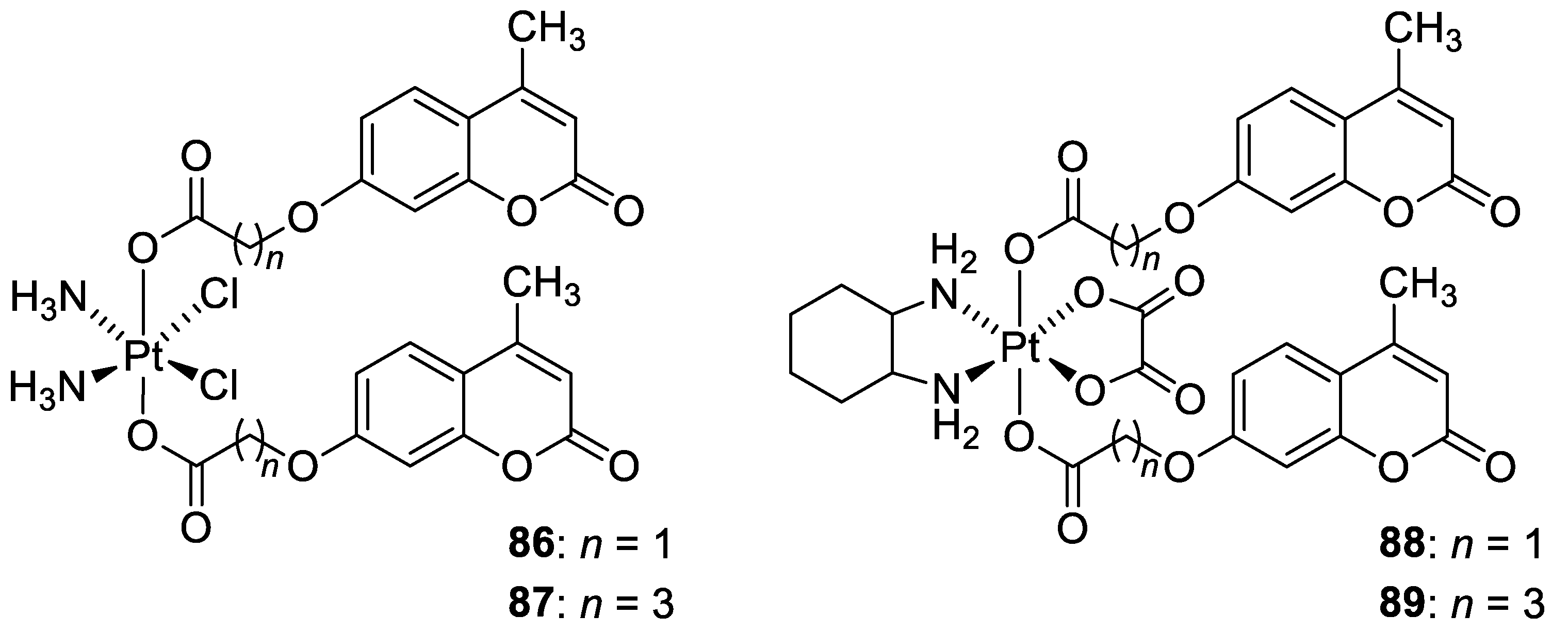
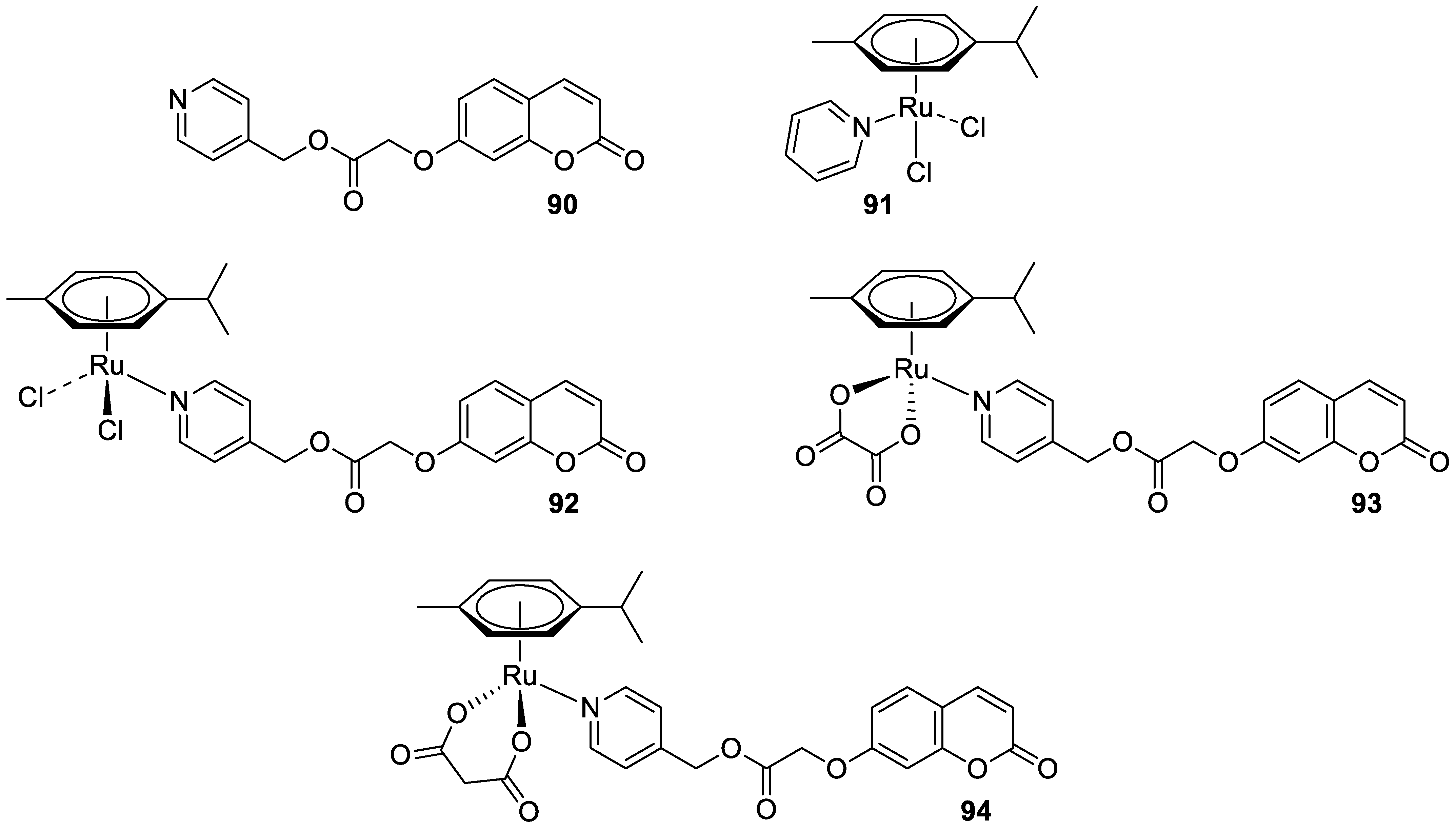
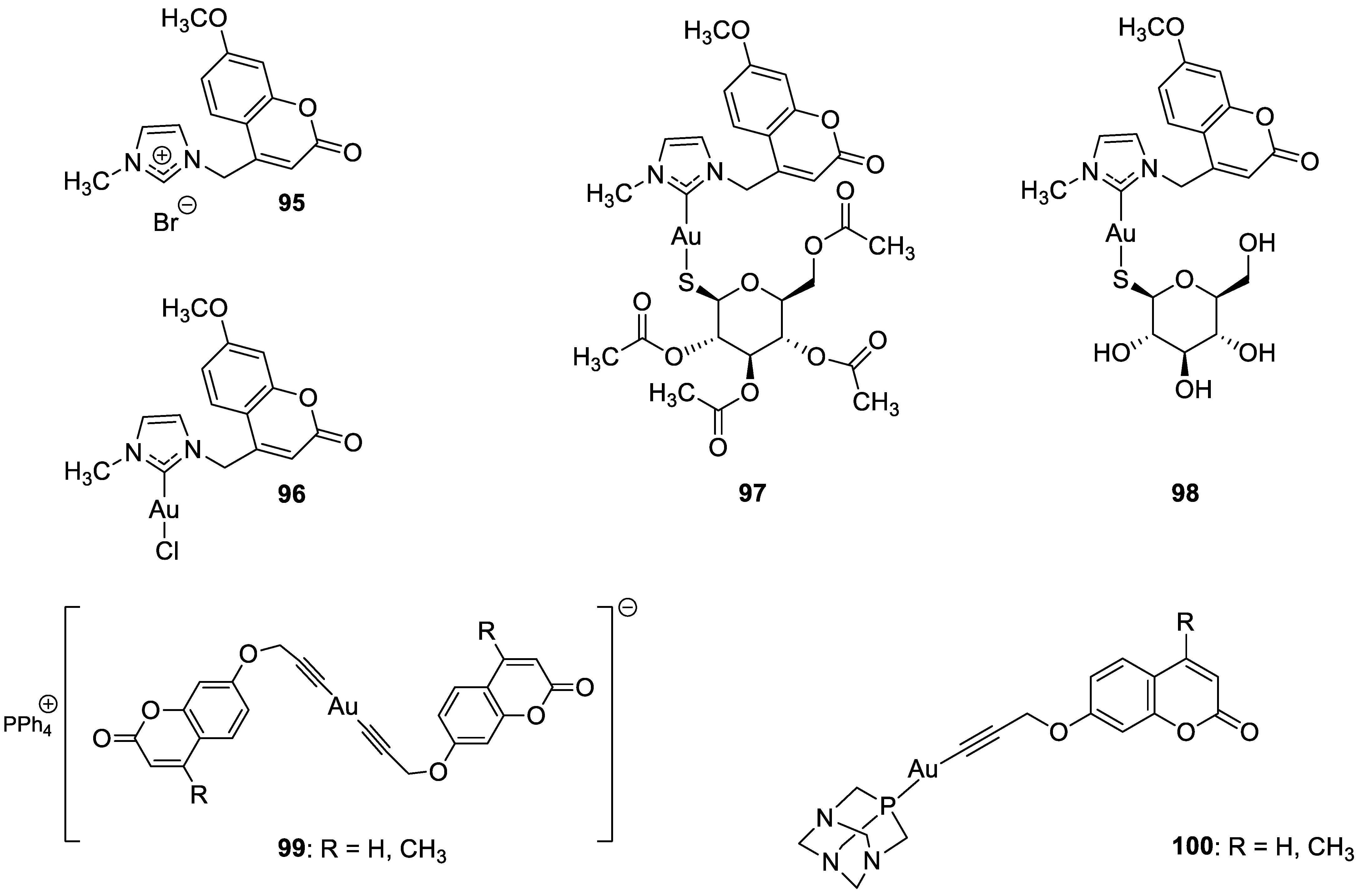
6.5.7. Metal Complexes of 7-Hydroxycoumarin-Based Compounds as Anticancer Agents
7. Umbelliferone and 7-Hydroxycoumarin-Based Compounds as Probes and Sensors
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sharifi-Rad, J.; Cruz-Martins, N.; López-Jornet, P.; Pons-Fuster Lopez, E.; Harun, N.; Yeskaliyeva, B.; Beyatli, A.; Sytar, O.; Shaheen, S.; Sharopov, F.; et al. Natural coumarins: Exploring the pharmacological complexity and underlying molecular mechanisms. Oxid. Med. Cell. Longev. 2021, 2021, 6492346. [Google Scholar] [CrossRef]
- Dawidowicz, A.L.; Bernacik, K.; Typek, R. Umbelliferone instability during an analysis involving its extraction process. Monatsh. Chem. 2018, 149, 1327–1340. [Google Scholar] [CrossRef]
- Mazimba, O. Umbelliferone: Sources, chemistry and bioactivities review. Bull. Fac. Pharm. Cairo Univ. 2017, 55, 223–232. [Google Scholar] [CrossRef]
- Radha, G.V.; Sadhana, B.; Trideva Sastri, K.; Ganapaty, S. Biooactive umbelliferone and its derivatives: An update. J. Pharmacogn. Phytochem. 2019, 8, 59–66. [Google Scholar]
- Lin, Z.; Zheng, H. Umbelliferon: A review of its pharmacology, toxicity and pharmacokinetics. Inflammopharmacology 2023, 31, 1731–1750. [Google Scholar] [CrossRef]
- Fylaktakidou, K.C.; Hadjipavlou-Litina, D.J.; Litinas, K.E.; Nicolaides, D.N. Natural and synthetic coumarin derivatives with anti-inflammatory/antioxidant activities. Curr. Pharm. Des. 2004, 10, 3813–3833. [Google Scholar] [CrossRef] [PubMed]
- Emami, S.; Dadashpour, S. Current developments of coumarin-based anti-cancer agents in medical chemistry. Eur. J. Med. Chem. 2015, 102, 611–630. [Google Scholar] [CrossRef]
- Pan, Y.; Liu, T.; Wang, X.; Sun, J. Research progress of coumarins and their derivatives in the treatment of diabetes. J. Enzyme Inhib. Med. Chem. 2022, 37, 616–628. [Google Scholar] [CrossRef] [PubMed]
- Genovese, S.; Epifano, F.; Curini, M.; Dudra-Jastrzebska, M.; Luszczki, J.J. Prenyloxyphenylpropanoids as a novel class of anticonvulsive agents. Bioorg. Med. Chem. Lett. 2009, 19, 5419–5422. [Google Scholar] [CrossRef] [PubMed]
- Alipour, M.; Khoobi, M.; Moradi, A.; Nadri, H.; Moghadam, F.H.; Emami, S.; Hasanpour, Z.; Foroumadi, A.; Shafiee, A. Synthesis and anticholinesterase activity of new 7-hydroxycoumarin derivatives. Eur. J. Med. Chem. 2014, 82, 536–544. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, H.; Wang, X.; Lei, K.; Wang, S. Design, synthesis, and in vivo and in silico evaluation of coumarin derivatives with potential antidepressant effects. Molecules 2021, 26, 5556. [Google Scholar] [CrossRef]
- Vasconcelos, J.F.; Teixeira, M.M.; Barbosa-Filho, J.M.; Agra, M.F.; Nunes, X.P.; Giulietti, A.M.; Ribeiro-dos-Santos, R.; Soares, M.B.P. Effects of umbelliferone in a murine model of allergic airway inflammation. Eur. J. Pharmacol. 2009, 609, 126–131. [Google Scholar] [CrossRef]
- Zinovieva, M.L.; Zhminko, P.G. Single and repeat dose toxicity study of 7-hydroxycoumarin, ethanol, and their mixture in rats. J. Pharm. Pharmacol. 2017, 5, 237–244. [Google Scholar]
- Cruz, L.F.; de Figueiredo, G.F.; Pedro, L.P.; Amorin, Y.M.; Andrade, J.T.; Passos, T.F.; Rodrigues, F.F.; Souza, I.L.A.R.; Gonçalves, T.P.R.; Dos Santos Lima, L.A.R.; et al. Umbelliferone (7-hydroxycoumarin): A non-toxic antidiarrheal and antiulcerogenic coumarin. Biomed. Pharmacother. 2020, 129, 110432. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef] [PubMed]
- Kishore, N.; Kumar, P.; Shanker, K.; Kumar Verma, A. Human disorders associated with inflammation and the evolving role of natural products to overcome. Eur. J. Med. Chem. 2019, 179, 272–309. [Google Scholar] [CrossRef] [PubMed]
- Grover, J.; Jachak, S.M. Coumarins as privileged scaffold for antiinflammatory drug development. RSC Adv. 2015, 5, 38892–38905. [Google Scholar] [CrossRef]
- Rostom, B.; Karaky, R.; Kassab, I.; Veitía, M.S.-I. Coumarins derivatives and inflammation: Review of their effects on the inflammatory signaling pathways. Eur. J. Pharmacol. 2022, 922, 174867. [Google Scholar] [CrossRef] [PubMed]
- Di Stasi, L.C. Natural coumarin derivatives activating Nrf2 signaling pathway as lead compounds for the design and synthesis of intestinal anti-inflammatory drugs. Pharmaceuticals 2023, 16, 511. [Google Scholar] [CrossRef]
- Lee, J.H.; Cho, S.H. Korean red ginseng extract ameliorates skin lesions in NC/ Nga mice: An atopic dermatitis model. J. Ethnopharmacol. 2011, 133, 810–817. [Google Scholar] [CrossRef]
- Akdis, C.A.; Akdis, M. Mechanisms and treatment of allergic disease in the big picture of regulatory T cells. J. Allergy Clin. Immunol. 2009, 123, 735–746. [Google Scholar] [CrossRef]
- Huang, Y.; Li, W.; Su, Z.; Kong, A.T. The complexity of the Nrf2 pathway: Beyond the antioxidant response. J. Nutr. Biochem. 2015, 26, 1401–1413. [Google Scholar] [CrossRef]
- Saho, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An overview of Nrf2 signaling pathway and its role in inflammation. Molecules 2020, 25, 5474. [Google Scholar] [CrossRef] [PubMed]
- Younas; Khan, A.; Shehzad, O.; Seo, E.K.; Onder, A.; Khan, S. Anti-allergic activities of umbelliferone against histamine- and picryl chloride- induced ear edema by targeting Nrf2/iNOS signaling in mice. BMC Complement. Med. 2021, 21, 215. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, B.A.; Alkahtani, S.A.; Alqahtani, A.A.; Hassanein, E.H.M. Umbelliferone ameliorates ulcerative colitis induced by acetic acid via modulation of TLR4/NF-κB-p65/iNOS and SIRT1/PPARγ signaling pathways in rats. Environ. Sci. Pollut. Res. 2022, 29, 37644–37659. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zhou, W.; Huang, Y.; Tian, Y.; Wong, S.Y.; Lam, W.K.; Ying, K.Y.; Zhang, J.; Chen, H. Umbelliferone and scopoletin target tyrosine kinases on fibroblast-like synoviocytes to block NF-κB signaling to combat rheumatoid arthritis. Front. Pharmacol. 2022, 13, 946210. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Zong, P.; Zhou, M.-Y.; Liu, F.Y.; Meng, B.; Liu, M.-M.; Li, Z.; Li, R. 7-Hydroxycoumarin mitigates the severity of collagen-induced arthritis in rats by inhibiting proliferation and inducing apoptosis of fibroblast-like synoviocytes via suppression of Wnt/β-catenin signaling pathway. Phytomedicine 2022, 94, 153841. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Lakshmanan, L. Dose-dependent efficacy of umbelliferone and gelatin-coated ZnO/ZnS core-shell nanoparticles: A novel arthritis agent for severe knee arthritis. Oxid. Med. Cell. Longev. 2022, 2022, 7795602. [Google Scholar] [CrossRef]
- Ji-ye, L.; Jim-Hyun, L.; Dong-Hyun, L.; Jeong-Heon, L.; Dea-Ki, K. Umbelliferone reduces the expression of inflammatory chemokines in HaCaT cells and DNCB/DFE-induced atopic dermatitis symptoms in mice. Int. Immunopharmacol. 2019, 75, 105830. [Google Scholar]
- Telange, D.R.; Nirgulkar, S.B.; Umekar, M.J.; Patil, A.T.; Petheb, A.M.; Bali, N.R. Enhanced transdermal permeation and anti-inflammatory potential of phospholipids complex-loaded matrix film of umbelliferone: Formulation development, physico-chemical and functional characterization. Eur. J. Pharm. Sci. 2019, 131, 23–38. [Google Scholar] [CrossRef]
- Bansal, Y.; Sethi, P.; Bansal, G. Coumarin: A potential nucleus for anti-inflammatory molecules. Med. Chem. Res. 2013, 22, 3049–3060. [Google Scholar] [CrossRef]
- Zhang, H.-J.; Li, Y.-F.; Cao, Q.; Tian, Y.-S.; Quan, Z.-S. Pharmacological evaluation of 9,10-dihydrochromeno[8,7-e][1,3]oxazin-2(8H)-one derivatives as potent anti-inflammatory agent. Pharmacol. Rep. 2017, 69, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T.; Willoughby, D.A.; Gilroy, D.W. Anti-inflammatory lipid mediators and insights into the resolution of inflammation. Nat. Rev. Immunol. 2002, 2, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Mu, C.; Wu, M.; Li, Z. Anti-inflammatory effect of novel 7-substituted coumarin derivatives through inhibition of NF-κB signaling pathway. Chem. Biodivers. 2019, 16, e1800559. [Google Scholar] [CrossRef]
- Gao, F.; Tao, D.; Ju, C.; Yang, B.-B.; Bao, X.-Q.; Zhang, D.; Zhang, T.-T.; Li, L. Regioselectivity of aminomethylation in 3-acetyl-7-hydroxycoumarins: Mannich bases and Betti bases. New J. Chem. 2021, 45, 9864–9871. [Google Scholar] [CrossRef]
- Sharifi-Rad, M.; Kumar, N.V.A.; Zucca, P.; Varoni, E.M.; Dini, L.; Panzarini, E.; Rajkovic, J.; Fokou, P.V.T.; Azzini, E.; Peluso, I.; et al. Lifestyle, oxidative stress and antioxidants: Back and forth in the pathophysiology of chronic diseases. Front. Physiol. 2020, 11, 694. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations od antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689. [Google Scholar] [CrossRef]
- Jing, T.; Chen, C. Umbelliferone delays the progression of diabetic nephropathy by inhibiting ferroptosis through activation of the Nff-2/OH-1 pathway. Food Chem. Toxicol. 2022, 163, 112892. [Google Scholar]
- Al-Majedy, Y.K.; Al-Amiery, A.; Kadhum, A.A.H.; Mohamad, A.B. Antioxidant activities of 4-methylumbelliferone derivatives. PLoS ONE 2016, 11, e0156625. [Google Scholar] [CrossRef]
- Al-Majedy, Y.K.; Al-Duhaidahawi, D.; Al-Azawi, K.; Al-Amiery, A.A.; Kadhum, A.A.H.; Mohamad, A.B. Coumarins as potential antioxidant agents complemented with suggested mechanisms and approved by molecular modeling studies. Molecules 2016, 21, 135. [Google Scholar] [CrossRef]
- Kurt, B.Z.; Gazioglu, I.; Kandas, N.O.; Sonmez, F. Synthesis, anticholinesterase, antioxidant, and anti-aflatoxigenic activity of novel coumarin carbamates. ChemistrySelect 2018, 3, 3978–3983. [Google Scholar] [CrossRef]
- Joy, M.N.; Bodke, Y.D.; Telkar, S.; Bakulev, V.A. Synthesis of coumarins linked with 1,2,3-triazoles under microwave irradiation and evaluation of their antimicrobial and antioxidant activity. J. Mex. Chem. Soc. 2020, 64, 53–73. [Google Scholar]
- Kaushik, C.P.; Chahal, M. Synthesis, antimalarial and antioxidant activity of coumarin appended 1,4-disubstituted 1,2,3-triazoles. Mon. Chem. Chem. Mon. 2021, 152, 1001–1012. [Google Scholar] [CrossRef]
- Kecel-Gunduz, S.; Budama-Kilinic, Y.; Bicak, B.; Gok, B.; Belmen, B.; Aydogan, F.; Yolacan, C. New coumarin derivative with potential antioxidant activity: Synthesis, DNA binding and in silico studies (Docking, MD, ADMET). Arab. J. Chem. 2023, 16, 104440. [Google Scholar] [CrossRef]
- Balewski, Ł.; Szulta, S.; Jalińska, A.; Kornicka, A. A mini-review: Recent advances in coumarin-metal complexes with biological properties. Front. Chem. 2021, 9, 781779. [Google Scholar] [CrossRef]
- Todorov, L.; Saso, L.; Kostova, I. Antioxidant activity of coumarins and their metal complexes. Pharmaceuticals 2023, 16, 651. [Google Scholar] [CrossRef]
- Kalaiarasi, G.; Rajkumar, S.R.J.; Dharani, S.; Małecki, J.G.; Prabhakaran, R. An investigation on 3-acetyl-7-methoxy-coumarin Shiff bases and their Ru(II) metallates with potent antiproliferative activity and enhanced LDH and NO release. RSC Adv. 2018, 8, 1539–1561. [Google Scholar] [CrossRef]
- Özdemir, M.; Köksoy, B.; Yalçin, B.; Taşkin, T.; Selçuki, N.A.; Salan, Ü.; Durmuş, M.; Bulut, M. Novel lutetium(III) phthalocyanine-coumarin dyades; synthesis, characterization, photochemical, theoretical and antioxidant activity. Inorg. Chem. Acta 2020, 517, 120145. [Google Scholar] [CrossRef]
- Li, S.; Li, A.J.; Travers, J.; Xu, T.; Sakamuru, S.; Klumpp-Thomas, C.; Huang, R.; Xia, M. Identification of compounds for butyrylcholinesterase inhibition. SLAS Discov. 2021, 26, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Osswald, H.L. BACE1 (β-secretase) inhibitors for the treatment of Alzheimer’s disease. Chem. Soc. Rev. 2014, 43, 6765–6813. [Google Scholar] [CrossRef] [PubMed]
- Moussa-Pacha, N.M.; Abdin, S.M.; Hany, A.; Omar, H.A.; Alniss, H.; Al-Tel, T.H. BACE1 inhibitors: Current status and future directions in treating Alzheimer’s disease. Med. Res. Rev. 2020, 40, 339–384. [Google Scholar] [CrossRef]
- Ali, M.Y.; Jannat, S.; Jung, H.A.; Choi, R.J.; Roy, A.; Choi, J.S. Anti-Alzheimer’s disease potential of coumarins from Angelica decursiva and Artemisia capillaris and structure-activity analysis. Asian Pac. J. Trop. Med. 2016, 9, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.Y.; Seong, S.H.; Reddy, M.R.; Seo, S.Y.; Choi, J.S.; Jung, H.A. Kinetics and molecular docking studies of 6-formyl umbelliferone isolated from Angelica decursiva as an inhibitor of cholinesterase and BACE1. Molecules 2017, 22, 1604. [Google Scholar] [CrossRef]
- Karakaya, S.; Koca, M.; Sytar, O.; Duman, H. The natural phenolic compounds and their antioxidant and anticholinesterase potential of herb Leiotulus dasyanthus (K. Koch) Pimenov & Ostr. Nat. Prod. Res. 2020, 1303–1305. [Google Scholar]
- Hindam, M.O.; Sayed, R.H.; Skalicka-Woźniak, K.; Barbara Budzyńska, B.; EL Sayed, N.S. Xanthotoxin and umbelliferone attenuate cognitive dysfunction in a streptozotocin-induced rat model of sporadic Alzheimer’s disease: The role of JAK2/STAT3 and Nrf2/HO-1 signalling pathway modulation. Phytother. Res. 2020, 34, 2351–2365. [Google Scholar] [CrossRef]
- Kurach, Ł.; Kulczycka-Mamona, S.; Kowalczyk, J.; Skalicka-Woźniak, K.; Boguszewska-Czubara, A.; El Sayed, N.; Osmani, M.; Iwaniak, K.; Budzyńska, B. Mechanisms of the procognitive effects of xanthotoxin and umbelliferone on LPS-induced amnesia in mice. Int. J. Mol. Sci. 2021, 22, 1779. [Google Scholar] [CrossRef]
- Decker, M. Hybrid Molecules for Drug Development; Elsevier Ltd.: New York, NY, USA, 2017; ISBN 9780081011188. [Google Scholar]
- Decker, M. Hybrid molecules incorporating natural products: Applications in cancer therapy, neurodegenerative disorders and beyond. Curr. Med. Chem. 2011, 18, 1464–1475. [Google Scholar] [CrossRef] [PubMed]
- Spilovska, K.; Korabecny, J.; Sepsova, V.; Jun, D.; Hrabinova, M.; Jost, P.; Muckova, L.; Soukup, O.; Janockova, J.; Kucera, T.; et al. Novel tacrine-scutellarin hybrids as multipotent anti-Alzheimer’s agents: Design, synthesis and biological evaluation. Molecules 2017, 22, 1006. [Google Scholar] [CrossRef] [PubMed]
- Hirbod, K.; Jalili-Baleh, L.; Nadri, H.; Ebrahimi, S.E.S.; Moradi, A.; Pakseresht, B.; Foroumadi, A.; Shafiee, A.; Khoob, M. Coumarin derivatives bearing benzoheterocycle moiety: Synthesis, cholinesterase inhibitory, and docking simulation study. Iran. J. Basic. Med. Sci. 2017, 20, 631–638. [Google Scholar] [PubMed]
- Wang, Y.; Sun, Y.; Guo, Y.; Wang, Z.; Huang, L.; Li, X. Dual functional cholinesterase and MAO inhibitors for the treatment of Alzheimer’s disease: Synthesis, pharmacological analysis and molecular modeling of homoisoflavonoid derivatives J. Enzyme Inhibit. Med. Chem. 2016, 31, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Mateev, E.; Kondeva-Burdina, M.; Georgieva, M.; Zlatkov, A. Repurposing of FDA-approved drugs as dual-acting MAO-B and AChE inhibitors against Alzheimer’s disease: An in silico and in vitro study. J. Mol. Graph. Model. 2023, 122, 108471. [Google Scholar] [CrossRef]
- Mzezewa, S.C.; Omoruyib, S.I.; Zondagha, L.S.; Malana, S.F.; Ekpoband, O.E.; Joubert, J.J. Design, synthesis, and evaluation of 3,7-substituted coumarin derivatives asmultifunctional Alzheimer’s disease agents. Enzyme Inhib. Med. Chem. 2021, 36, 1606–1620. [Google Scholar] [CrossRef]
- Venugopala, K.N.; Rashmi, V.; Odhav, B. Review on natural coumarin lead compounds for their pharmacological activity. Biomed. Res. Int. 2013, 2013, 963248. [Google Scholar] [CrossRef]
- Seong, S.H.; Ali, M.Y.; Jung, H.A.; Cho, J.S. Umbelliferone derivatives exert neuroprotective effects by inhibiting monoamine oxidase A, self-amyloidβ aggregation, and lipid peroxidation. Bioorg. Chem. 2019, 92, 103293. [Google Scholar] [CrossRef]
- Dhiman, P.; Malik, N.; Khatkar, A. Exploration of umbelliferone based derivatives as potent MAO inhibitors: Dry vs. wet lab evaluation. Curr. Top. Med. Chem. 2018, 18, 1857–1871. [Google Scholar] [CrossRef]
- Fradley, R.; Goetghebeur, P.; Miller, D.; Burley, R.; Almond, S.; Massó, A.G.; García, J.M.D.; Zhu, B.; Howley, E.; Neill, J.C.; et al. Luvadaxistat: A novel potent and selective D-amino acid oxidase inhibitor improves cognitive and social deficits in rodent models for schizophrenia. Neurochem. Res. 2023, 48, 3027–3041. [Google Scholar] [CrossRef] [PubMed]
- Tsapakis, E.M.T.; Diakaki, K.; Miliaras, A.; Fountoulakis, K.N. Novel compounds in the treatment of schizophrenia-a selective review. Brain Sci. 2023, 13, 1193. [Google Scholar] [CrossRef] [PubMed]
- Bester, E.; Petzer, A.; Petzer, J.P. Coumarin derivatives as inhibitors of D-amino acid oxidase and monoamine oxidase. Bioorg. Chem. 2022, 123, 105791. [Google Scholar] [CrossRef] [PubMed]
- Ostrowska, K.; Leśniak, A.; Czarnocka, Z.; Chmiel, J.; Bujalska-Zadrożny, M.; Trzaskowski, B. Design, synthesis, and biological evaluation of a series of 5- and 7-hydroxycoumarin derivatives as 5-HT1A serotonin receptor antagonists. Pharmaceuticals 2021, 14, 179. [Google Scholar] [CrossRef] [PubMed]
- Bryda, J.; Zagaja, M.; Szewczyk, A.; Andres-Mach, M. Coumarins as potential supportive medication for the treatment of epilepsy. Acta Neurobiol. Exp. 2019, 79, 126–132. [Google Scholar] [CrossRef]
- Zagaja, M.; Anna Zagaja, A.; Szala-Rycaj, J.; Szewczyk, A.; Lemieszek, M.K.; Raszewski, G.; Andres-Mach, M. Influence of umbelliferone on the anticonvulsant and neuroprotective activity of selected antiepileptic drugs: An in vivo and in vitro study. Int. J. Mol. Sci. 2022, 23, 3492. [Google Scholar] [CrossRef] [PubMed]
- Yakovleva, E.E.; Myznikov, L.V.; Shabanov, P.D. Comparison of the anticonvulsant activities of substituted hydroxycoumarins and 4-[(3-nitro-2-oxo-2H-chromen-yl)amino]butanoic acid. Pharm. Chem. J. 2020, 54, 904–908. [Google Scholar] [CrossRef]
- Ramu, R.; Shirahatti, P.S.; Swamy, S.N.; Zameer, F.; Dhananjaya, B.L.; Prasad, M.N.N. Assessment of in vivo antidiabetic properties of umbelliferone and lupeol constituents of banana (Musa sp. var. Nanjangud Rasa Bale) flower in hyperglycaemic rodent model. PLoS ONE 2016, 11, e0151135. [Google Scholar] [CrossRef] [PubMed]
- Al Mouslem, A.K.; Khalil, H.E.; Emeka, P.M.; Alotaibi, G. Investigation of the chemical composition, antihyperglycemic and antilipidemic effects of Bassia eriophora and its derived constituent, umbelliferone on high-fat diet and streptozotocin-induced diabetic rats. Molecules 2022, 27, 6941. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Tao, W.; Wang, H.; Chen, Y.; Huang, H.; Chen, G. Umbelliferone attenuates unpredictable chronic mild stress induced-insulin resistance in rats. IUBMB Life 2016, 68, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Khadrawy, S.M.; El Sayed, R.A. Umbelliferone attenuates diabetic cardiomyopathy by suppression of JAK/STAT signaling pathway through amelioration of oxidative stress and inflammation in rats. J. Biochem. Mol. Toxicol. 2023, 37, e23296. [Google Scholar] [CrossRef]
- Ali, M.Y.; Zamponi, G.W.; Seong, S.H.; Jung, H.A.; Choi, J.S. 6-Formyl umbelliferone, a furanocoumarin from Angelica decursiva L., inhibits key diabetes-related enzymes and advanced glycation end-product formation. Molecules 2022, 27, 5720. [Google Scholar] [CrossRef]
- Wang, G.; Wang, J.; He, D.; Li, X.; Li, J.; Peng, Z. Synthesis, in vitro evaluation and molecular docking studies of novel coumarin-isatin derivatives as α-glucosidase inhibitors. Chem. Biol. Drug Des. 2017, 89, 456–463. [Google Scholar] [CrossRef]
- Ojala, T.; Remes, S.; Haansuu, P.; Vuorela, H.; Hiltunen, R.; Haahtela, K.; Vuorela, P. Antimicrobial activity of some coumarin containing herbal plants growing in Finland. J. Ethnopharmacol. 2000, 73, 299–305. [Google Scholar] [CrossRef]
- Dadak, V.; Hodak, K. Some relations between the structure and the antibacterial activity of natural coumarins. Experientia 1966, 22, 38–39. [Google Scholar] [CrossRef] [PubMed]
- Jurd, L.; King, A.D., Jr.; Mihara, K. Antimicrobial properties of umbelliferone derivatives. Phytochemistry 1971, 10, 2965–2970. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, Y.G.; Cho, H.S.; Ryu, S.Y.; Cho, M.H.; Lee, J. Coumarins reduce biofilm formation and the virulence of Escherichia coli O157:H7. Phytomedicine 2014, 21, 1037–1042. [Google Scholar] [CrossRef] [PubMed]
- Swetha, K.T.; Pooranachthra, M.; Subramenium, G.A.; Divya, V.; Balamurugan, K.; Pandian, S.K. Umbelliferone impedes biofilm formation and virulence of methicillin-resistant Staphylococcus epidermis via impairment of initial attachment and intercellular adhesion. Front. Cell. Infect. Microbiol. 2019, 9, 357. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, R.; Santhakumari, S.; Poonguzhali, P.; Geetha, M.; Dyavaiah, M.; Xiangmin, L. Bacterial biofilm inhibition: A focused review on recent therapeutic strategies for combating the biofilm mediated infections. Front. Microbiol. 2021, 12, 676458. [Google Scholar] [CrossRef] [PubMed]
- Kasthuri, T.; Barath, S.; Nandhakumar, M.; Karutha Pandian, S. Proteomic profiling spotlights the molecular targets and the impact of the natural antivirulent umbelliferone on stress response, virulence factors, and the quorum sensing network of Pseudomonas aeruginosa. Front. Cell. Infect. Microbiol. 2022, 12, 998540. [Google Scholar] [CrossRef] [PubMed]
- Giovannuzzi, S.; Hewitt, C.S.; Nocentini, A.; Capasso, C.; Flaherty, D.P.; Supuran, C.T. Coumarins effectively inhibit bacterial α-carbonic anhydrases. J. Enzyme Inhib. Med. Chem. 2022, 37, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Darla, M.M.; Krishna, B.S.; Umamaheswara Rao, K.; Reddy, N.B.; Srivash, M.K.; Adeppa, K.; Sundar, C.S.; Reddy, C.S.; Misra, K. Synthesis and bio-evaluation of novel 7-hydroxy coumarin derivatives via Knoevenagel reaction. Res. Chem. Intermed. 2015, 41, 1115–1133. [Google Scholar] [CrossRef]
- Sokol, I.; Toma, M.; Krnić, M.; Macan, A.M.; Drenjančević, D.; Liekens, S.; Raić-Malić, S.; Gazivoda Kraljević, T. Transition metal-catalyzed synthesis of new 3-substituted coumarin derivatives as antibacterial and cytostatic agents. Future Med. Chem. 2021, 13, 1865–1884. [Google Scholar] [CrossRef] [PubMed]
- Jund, L.; Corse, J.; King, A.S.; Bayne, H.; Mihrag, K. Antimicrobial properties of 6,7-dihydroxy-7,8-dihydroxy-, 6-hydroxy- and 8-hydroxycoumarins. Phytochemistry 1971, 10, 2971–2974. [Google Scholar]
- Farshori, N.N.; Banday, M.R.; Ahmad, A.; Khan, A.U.; Rauf, A. 7-Hydroxy-coumarin derivatives: Synthesis, characterization and preliminary antimicrobial activities. Med. Chem. Res. 2010, 20, 535–541. [Google Scholar] [CrossRef]
- Soares, V.; Marini, M.B.; de Paula, L.A.; Gabry, P.S.; Amaral, A.C.F.; Malafaia, C.A.; Leal, I.C.R. Umbelliferone esters with antibacterial activity produced by lipase-mediated biocatalytic pathway. Biotechnol. Lett. 2020, 43, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.-L.; Ke, X.; Liu, M. Coumarin–triazole hybrids and their biological activities. J. Heterocycl. Chem. 2018, 55, 791–802. [Google Scholar] [CrossRef]
- Shi, Y.; Zhou, C.H. Synthesis and evaluation of a class of new coumarin triazole derivatives as potential antimicrobial agents. Bioorg. Med. Chem. Lett. 2011, 21, 956–961. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, M.H.; Subhedar, D.D.; Shingate, B.B.; Kalam Khan, F.A.; Sangshetti, J.N.; Khedkar, V.M.; Nawale, L.; Sarkar, D.; Navale, G.R.; Shinde, S.S. Synthesis, biological evaluation and molecular docking of novel coumarin incorporated triazoles as antitubercular, antioxidant and antimicrobial agents. Med. Chem. Res. 2016, 25, 790–804. [Google Scholar] [CrossRef]
- Gazivoda Kraljević, T.; Harej, A.; Sedić, M.; Kraljević Pavelić, S.; Stepanić, V.; Drenjančević, D.; Talapko, J.; Raić-Malić, S. Synthesis, in vitro anticancer and antibacterial activities and in silico studies of new 4-substituted 1,2,3-triazole-coumarin hybrids. Eur. J. Med. Chem. 2016, 124, 794–808. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Shen, Y.; Wu, X.; Tu, X.; Wang, G.-X. Synthesis and biological evaluation of coumarin derivatives containing imidazole skeleton as potential antibacterial agents. Eur. J. Med. Chem. 2018, 143, 958–969. [Google Scholar] [CrossRef] [PubMed]
- El-Sherief, H.A.; Abuo-Rahma, G.E.-D.A.; Shoman, M.E.; Beshr, E.A.; Abdel-baky, R.M. Design and synthesis of new coumarin–chalcone/NO hybrids of potential biological activity. Med. Chem. Res. 2017, 26, 3077–3090. [Google Scholar] [CrossRef]
- Şahin Gül, D.; Ogutcu, H.; Hayvalı, Z. Investigation of photophysical behaviours and antimicrobial activity of novel benzo-15-crown-5 substituted coumarin and chromone derivatives. J. Mol. Struct. 2020, 1204, 127569. [Google Scholar] [CrossRef]
- Nath, M.; Jairath, R.; Eng, G.; Song, X.; Kumar, A. Triorganotin(IV) derivatives of umbelliferone (7-hydroxycoumarin) and their adducts with 1,10-phenanthroline: Synthesis, structural and biological studies. J. Organomet. Chem. 2005, 690, 134–144. [Google Scholar] [CrossRef]
- Yernule, N.G.; Bennikallu Hire Mathada, M. Preparation of octahedral Cu(II), Co(II), Ni(II) and Zn(II) complexes derived from 8-formyl-7-hydroxy-4-methylcoumarin: Synthesis, characterization and biological study. J. Mol. Struct. 2020, 1220, 128659. [Google Scholar] [CrossRef]
- Klepka, M.T.; Drzewiecka-Antonik, A.; Wolska, A.; Rejmak, P.; Ostrowska, K.; Hejchman, E.; Kruszewska, H.; Czajkowska, A.; Młynarczuk-Biały, I.; Ferenc, W. Synthesis, structural studies and biological activity of new Cu(II) complexes with acetyl derivatives of 7-hydroxy-4-methylcoumarin. J. Inorg. Biochem. 2015, 145, 94–100. [Google Scholar] [CrossRef]
- El-Attar, M.S.; Sadeek, S.A.; Abd El-Hamid, S.M.; Elshafie, H.S. Spectroscopic analyses and antimicrobial activity of novel ciprofloxacin and 7-hydroxy-4-methylcoumarin, the plant-based natural benzopyrone derivative. Int. J. Mol. Sci. 2022, 23, 8019. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.-C.; Cheng, M.-J.; Peng, C.-F.; Huang, H.-Y.; Chen, I.-S. A novel dimeric coumarin analog and antimycobacterial constituents from Fatoua Pilosa. Chem. Biodivers. 2010, 7, 1728–1736. [Google Scholar] [CrossRef] [PubMed]
- Siqueira-Neto, J.L.; Wicht, K.J.; Chibale, K.; Burrows, J.N.; Fidock, D.; Winzeler, E.A. Antimalarial drug discovery: Progress and approaches. Nat. Rev. Drug Discov. 2023, 22, 807–826. [Google Scholar] [CrossRef] [PubMed]
- Batra, N.; Rajendran, V.; Wadi, I.; Ghosh, P.C.; Nath, M. Synthesis and antimalarial activity of sulfonamide-attached coumarin-[1,2,3]-triazoles. Indian J. Chem. 2020, 59B, 1545–1555. [Google Scholar]
- Paget, J.; Spreeuwenberg, P.; Charu, V.; Taylor, R.J.; Iuliano, A.D.; Bresee, J.; Simonsen, L.; Viboud, C. Global mortality associated with seasonal influenza epidemics: New burden estimates and predictors from the GLaMOR Project. J. Glob. Health 2019, 9, 020421. [Google Scholar] [CrossRef]
- Kanazawa, R.; Morimoto, R.; Horio, Y.; Sumitani, H.; Isegawa, Y. Inhibition of influenza virus replication by Apiaceae plants, with special reference to Peucedanum japonicum (Sacna) constituents. J. Ethnopharmacol. 2022, 292, 115243. [Google Scholar] [CrossRef]
- Cheng, F.-J.; Ho, C.-Y.; Li, T.-S.; Chen, Y.; Yeh, Y.-L.; Wei, Y.-L.; Huynh, T.K.; Chen, B.-R.; Ko, H.-Y.; Hsueh, C.-S.; et al. Umbelliferone and eriodictyol suppress the cellular entry of SARS-CoV-2. Cell Biosci. 2023, 13, 118. [Google Scholar] [CrossRef]
- Khomenko, T.M.; Zarubaev, V.V.; Orshanskaya, I.R.; Kadyrova, R.A.; Sannikova, V.A.; Korchagina, D.V.; Volcho, K.P.; Salakhutdinov, N.F. Anti-influenza activity of monoterpene-containing substituted coumarins. Bioorg. Med. Chem. Lett. 2017, 27, 2920–2925. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, W.; Shen, Y.; Zhu, B.; Wang, G.-X. Synthesis and antiviral activity of coumarin derivatives against infectious hematopoietic necrosis virus. Bioorg. Med. Chem. Lett. 2019, 29, 1749–1755. [Google Scholar] [CrossRef]
- He, R.; Zhang, Y.; Wu, L.; Nie, H.; Huang, Y.; Liu, B.; Deng, S.; Yang, R.; Huang, S.; Nong, Z.; et al. Benzofuran glycosides and coumarins from the bark of Streblus indicus (Bur.). Corner. Phytochem. 2017, 138, 170–177. [Google Scholar] [CrossRef]
- Chu, L.L.; Pandey, R.P.; Lim, H.N.; Jung, H.J.; Thuan, N.H.; Kim, T.-S.; Sohng, J.K. Synthesis of umbelliferone derivatives in Escherichia coli and their biological activities. J. Biol. Eng. 2017, 11, 15. [Google Scholar] [CrossRef]
- Ben Salem, S.; Jabrane, A.; Harzallah-Skhiri, F.; Ben Jannet, H. New bioactive dihydrofuranocoumarins from the roots of the Tunisian Ferula lutea (Poir.) Maire. Bioorg. Med. Chem. Lett. 2013, 23, 4248–4252. [Google Scholar] [CrossRef]
- Vijayalakshmi, A.; Sindhu, G. Umbelliferone arrest cell cycle at G0/G1 phase and induces apoptosis in human oral carcinoma (KB) cells possibly via oxidative DNA damage. Biomed. Pharmacother. 2017, 92, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.-M.; Hu, D.-H.; Zhang, J.-J. Umbelliferone exhibits anticancer activity via the induction of apoptosis and cell cycle arrest in HepG2 hepatocellular carcinoma cells. Mol. Med. Rep. 2015, 12, 3869–3873. [Google Scholar] [CrossRef]
- Khunluck, T.; Kukongviriyapan, V.; Senggunprai, L.; Duangarsong, W.; Prawan, A. The inhibition kinetics and potential antimigration activity of NQO1 inhibitory coumarins on cholangiocarcinoma cells. Integr. Cancer. Ther. 2019, 18. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Gonzalez, J.S.; Prado-Garcia, H.; Aguilar-Cazares, D.; Molina-Guarneros, J.A.; MoralesFuentes, J.; Mandoki, J.J. Apoptosis and cell cycle disturbances induced by coumarin and 7-hydroxycoumarin on human lung carcinoma cell lines. Lung Cancer 2004, 43, 275–283. [Google Scholar] [CrossRef]
- Jiménez-Orozco, F.A.; López-González, J.S.; Nieto-Rodriguez, A.; Velasco-Velázquez, M.A.; Molina-Guarneros, J.A.; Mendoza-Patiño, N.; García-Mondragón, M.J.; Elizalde-Galvan, P.; LeónCedeño, F.; Mandoki, J.J. Decrease of cyclin D1 in the human lung adenocarcinoma cell line A-427 by 7-hydroxycoumarin. Lung Cancer 2001, 34, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Elinos-Baez, C.M.; Leon, F.; Santos, E. Effects of coumarin and 7OH-coumarin on Bcl-2 and Bax expression in two human lung cancer cell lines in vitro. Cell Biol. Int. 2005, 29, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Huang, S.; Xin, X.; Ren, Y.; Weng, G.; Wang, P. The antitumor activity of umbelliferone in human renal cell carcinoma via regulation of the p110γ catalytic subunit of PI3Kγ. Acta Pharm. 2019, 69, 111–119. [Google Scholar] [CrossRef]
- Shen, J.Q.; Zhang, Z.X.; Shen, C.F.; Liao, J.Z. Anticarcinogenic effect of umbelliferone in human prostate carcinoma: An in vitro study. J. BUON 2017, 22, 94–101. [Google Scholar] [PubMed]
- Kim, H.-J.; Jin, B.-R.; An, H.-J. Umbelliferone ameliorates benign prostatic hyperplasia by inhibiting cell proliferation and G1/S phase cell cycle progression through regulation of STAT3/E2F1 axis. Int. J. Mol. Sci. 2021, 22, 9019. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, Y. Anticancer effect of 7-hydroxycoumarin in cisplatin-resistant ovarian cancer cell is mediated via apoptosis induction, caspase activation and cell cycle arrest at G2M phase. Trop. J. Pharm. Res. 2021, 20, 281–286. [Google Scholar] [CrossRef]
- Rashmi, R.; Prakash, N.; Narayana Swamy, H.D.; Narayana Swamy, M.; Rathnamma, D.; Suguna Rao, A.; Sahadev, A.; Santhosh, C.R.; Sunilchandra, U.; Naveen Kumar, S.; et al. Evaluation of anticancer efficacy of umbelliferone with or without piperine. J. Entomol. Zool. Stud. 2020, 8, 225–229. [Google Scholar]
- Sumorek-Wiadro, J.; Zając, A.; Bądziul, D.; Langner, E.; Skalicka-Woźniak, K.; Maciejczyk, A.; Wertel, I.; Rzeski, W.; Jakubowicz-Gil, J. Coumarins modulate the anti-glioma properties of temozolomide. Eur. J. Pharmacol. 2020, 881, 173207. [Google Scholar] [CrossRef]
- Sumorek-Wiadro, J.; Zając, A.; Langner, E.; Skalicka-Woźniak, K.; Maciejczyk, A.; Rzeski, W.; Jakubowicz-Gil, J. Antiglioma potential of coumarins combined with Sorafenib. Molecules 2020, 25, 5192. [Google Scholar] [CrossRef] [PubMed]
- Kundu, M.; Chatterjee, S.; Ghosh, N.; Manna, P.; Das, J.; Sil, P.C. Tumor targeted delivery of umbelliferone via a smart mesoporous silica nanoparticles controlled-release drug delivery system for increased anticancer efficiency. Mater. Sci. Eng. C Mater. Biol. Appl. 2020, 116, 111239. [Google Scholar] [CrossRef]
- Verdone, L.; Agricola, E.; Caserta, M.; Di Mauro, E. Histone acetylation in gene regulation. Brief. Funct. Genom. Proteom. 2006, 5, 209–221. [Google Scholar] [CrossRef]
- Pramanik, S.D.; Kumar Halder, A.; Mukherjee, U.; Kumar, D.; Dey, Y.N. Potential of histone deacetylase inhibitors in the control and regulation of prostate, breast and ovarian cancer. Front. Chem. 2022, 10, 847. [Google Scholar] [CrossRef]
- Abdizadeh, T.; Kalani, M.R.; Abnous, K.; Tayarani-Najaran, Z.; Khashyarmanesh, B.Z.; Abdizadeh, R.; Hadizadeh, F. Design, synthesis and biological evaluation of novel coumarin-based benzamides as potent histone deacetylase inhibitors and anticancer agents. Eur. J. Med. Chem. 2017, 132, 42–62. [Google Scholar] [CrossRef]
- Yang, F.; Zhao, N.; Song, J.; Zhu, K.; Jiang, C.; Shan, P.; Zhang, H. Design, synthesis and biological evaluation of novel coumarin-based hydroxamate derivatives as histone deacetylase (Hdac) inhibitors with antitumor activities. Molecules 2019, 24, 2569. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Yang, F.; Han, L.; Yuhua, Q.; Ge, D.; Zhang, H. Development of coumarin-based hydroxamates as histone deacetylase inhibitors with antitumor activities. Molecules 2020, 25, 717. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Liu, J.; Zhang, Z.; Guo, J.; Cheng, M.; Wan, Y.; Wang, R.; Fang, Y.; Guan, Z.; Jin, Y.; et al. Design, synthesis and biological evaluation of coumarin-based N-hydroxycinnamide derivatives as novel histone deacetylase inhibitors with anticancer activities. Bioorg. Chem. 2020, 101, 104023. [Google Scholar] [CrossRef]
- Chang, C.; Lee, S.O.; Yeh, S.; Chang, T.M. Androgen receptor (AR) differential roles in hormone-related tumors including prostate, bladder, kidney, lung, breast and liver. Oncogene 2014, 33, 3225–3234. [Google Scholar] [CrossRef]
- Voet, A.; Helsen, C.; Zhang, K.Y.J.; Claessens, F. The discovery of novel human androgen receptor antagonist chemotypes using a combined pharmacophore screening procedure. ChemMedChem 2013, 8, 644–651. [Google Scholar] [CrossRef]
- Makkonen, H.; Kauhanen, M.; Jääskeläinen, T.; Palvimo, J.J. Androgen receptor amplification is reflected in the transcriptional responses of vertebral-cancer of the prostate cells. Mol. Cell. Endocrinol. 2011, 331, 57–65. [Google Scholar] [CrossRef]
- Kandil, S.; Westwell, A.D.; McGuigan, C. 7-Substituted umbelliferone derivatives as androgen receptor antagonists for the potential treatment of prostate and breast cancer. Bioorg. Med. Chem. Lett. 2016, 26, 2000–2004. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Jain, V.K.; Rizwanullah, M.; Ahmad, J.; Jain, K. PI3K/AKT/mTOR pathway inhibitors in triple-negative breast cancer: A review on drug discovery and future challenges. Drug Discov. Today 2019, 24, 2181–2191. [Google Scholar] [CrossRef]
- O’Donnell, J.; Massi, D.; Teng, M.W.; Mandala, M. PI3K-AKT-mTOR inhibition in cancer immunotherapy, redux. Semin. Cancer Biol. 2018, 48, 91–103. [Google Scholar] [CrossRef]
- Miricescu, D.; Totan, A.; Stanescu-Spinu, I.-I.; Badoiu, S.C.; Stefani, C.; Greabu, M. PI3K/AKT/mTOR signaling pathway in breast cancer: From molecular landscape to clinical aspects. Int. J. Mol. Sci. 2020, 22, 173. [Google Scholar] [CrossRef]
- Miller, T.W.; Rexer, B.N.; Garrett, J.T.; Arteaga, C.L. Mutations in the phosphatidylinositol 3-kinase pathway: Role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 2011, 13, 224. [Google Scholar] [CrossRef]
- Xue, G.; Zippelius, A.; Wicki, A.; Mandalà, M.; Tang, F.; Massi, D.; Hemmings, B.A. Integrated Akt/PKB signaling in immunomodulation and its potential role in cancer immunotherapy. J. Natl. Cancer Inst. 2015, 107, djv171. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.-C.; Liu, Z.-P. Design and synthesis of coumarin derivatives as novel PI3K inhibitors. Anti-Cancer Agents Med. Chem. 2017, 17, 395–403. [Google Scholar] [CrossRef]
- Abdelnaby, R.M.; Rateb, H.S.; Ali, O.; Saad, A.S.; Nadeem, R.I.; Abou-Seri, S.M.; Amin, K.M.; Younis, N.S.; Abdelhady, R. Dual PI3K/Akt inhibitors bearing coumarin-thiazolidine pharmacophores as potential apoptosis inducers in MCF-7 cells. Pharmaceuticals 2022, 15, 428. [Google Scholar] [CrossRef]
- Laev, S.; Salakhutdinov, N.; Lavrik, O. Tyrosyl–DNA phosphodiesterase inhibitors: Progress and potential. Bioorg. Med. Chem. 2016, 24, 5017–5027. [Google Scholar] [CrossRef] [PubMed]
- Zakharenko, A.; Dyrkheeva, N.; Lavrik, O. Dual DNA topoisomerase 1 and tyrosyl-DNA phosphodiesterase 1 inhibition for improved anticancer activity. Med. Res. Rev. 2019, 39, 1427–1441. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhou, S.; Begum, S.; Sidransky, D.; Westra, W.H.; Brock, M.; Califano, J.A. Increased expression and activity of repair genes TDP1 and XPF in non-small cell lung cancer. Lung Cancer 2007, 55, 303–311. [Google Scholar] [CrossRef]
- Fam, H.K.; Walton, C.; Mitra, S.A.; Chowdhury, M.; Osborne, N.; Choi, K.; Sun, G.; Wong, P.C.; O’Sullivan, M.J.; Turashvili, G.; et al. TDP1 and PARP1 deficiency are cytotoxic to rhabdomyosarcoma cells. Mol. Cancer Res. 2013, 11, 1179–1192. [Google Scholar] [CrossRef]
- Keil, A.; Frese-Schaper, M.; Steiner, S.K.; Korner, M.; Schmid, R.A.; Frese, S. The topoisomerase I inhibitor irinotecan and the tyrosyl-DNA phosphodiesterase 1 inhibitor furamidine synergistically suppress murine lupus nephritis. Arthritis Rheumatol. 2015, 67, 1858–1867. [Google Scholar] [CrossRef]
- Interthal, H.; Chen, H.J.; Kehl-Fie, T.E.; Zotzmann, J.; Leppard, J.B.; Champoux, J.J. SCAN1 mutant Tdp1 accumulates the enzyme-DNA intermediate and causes camptothecin hypersensitivity. EMBO J. 2005, 24, 2224–2233. [Google Scholar] [CrossRef]
- Khomenko, T.; Zakharenko, A.; Odarchenko, T.; Arabshahi, H.J.; Sannikova, V.; Zakharova, O.; Korchagina, D.; Reynisson, J.; Volcho, K.; Salakhutdinov, N.; et al. New inhibitors of tyrosyl-DNA phosphodiesterase I (Tdp 1) combining 7-hydroxycoumarin and monoterpenoid moieties. Bioorg. Med. Chem. 2016, 24, 5573–5581. [Google Scholar] [CrossRef] [PubMed]
- Khomenko, T.M.; Zakharenko, A.L.; Chepanova, A.A.; Ilina, E.S.; Zakharova, O.D.; Kaledin, V.I.; Nikolin, V.P.; Popova, N.A.; Korchagina, D.V.; Reynisson, J.; et al. Promising new inhibitors of tyrosyl-DNA phosphodiesterase I (Tdp 1) combining 4-arylcoumarin and monoterpenoid moieties as components of complex antitumor therapy. Int. J. Mol. Sci. 2020, 21, 126. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrases as drug targets-an overview. Curr. Top. Med. Chem. 2007, 7, 825–833. [Google Scholar] [CrossRef] [PubMed]
- De Luca, L.; Mancuso, F.; Ferro, S.; Buemi, M.R.; Angeli, A.; Del Prete, S.; Capasso, C.; Supuran, C.T.; Gitto, R. Inhibitory effects and structural insights for a novel series of coumarin-based compounds that selectively target human CA IX and CA XII carbonic anhydrases. Eur. J. Med. Chem. 2018, 143, 276–282. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbonic anhydrase inhibitors: An update on experimental agents for the treatment and imaging of hypoxic tumors. Expert. Opin. Investig. Drugs 2021, 30, 1197–1208. [Google Scholar] [CrossRef] [PubMed]
- McDonald, P.C.; Chafe, S.C.; Supuran, C.T.; Dedhar, S. Cancer therapeutic targeting of hypoxia induced carbonic anhydrase IX: From bench to bedside. Cancers 2022, 14, 3297. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Coumarin carbonic anhydrase inhibitors from natural sources. J. Enzyme Inhib. Med. Chem. 2020, 35, 1462–1470. [Google Scholar] [CrossRef]
- Maresca, A.; Supuran, C.T. Coumarins incorporating hydroxy- and chloro-moieties selectively inhibit the transmembrane, tumor-associated carbonic anhydrase isoforms IX and XII over the cytosolic ones I and II. Bioorg. Med. Chem. Lett. 2010, 20, 4511–4514. [Google Scholar] [CrossRef]
- Nocentini, A.; Carta, F.; Ceruso, M.; Bartolucci, G.; Supuran, C.T. Click-tailed coumarins with potent and selective inhibitory action against the tumor-associated carbonic anhydrases IX and XII. Bioorg. Med. Chem. 2015, 23, 6955–6966. [Google Scholar] [CrossRef]
- Kurt, B.Z.; Sonmez, F.; Ozturk, D.; Akdemir, A.; Angeli, A.; Supuran, C.T. Synthesis of coumarin-sulfonamide derivatives and determination of their cytotoxicity, carbonic anhydrase inhibitory and molecular docking studies. Eur. J. Med. Chem. 2019, 183, 111702. [Google Scholar] [CrossRef]
- Thacker, P.S.; Alvala, M.; Arifuddin, M.; Angeli, A.; Supuran, C.T. Design, synthesis and biological evaluation of coumarin-3-carboxamides as selective carbonic anhydrase IX and XII inhibitors. Bioorg. Chem. 2019, 86, 386–392. [Google Scholar] [CrossRef]
- Thacker, P.S.; Goud, N.S.; Argulwar, O.S.; Soman, J.; Angeli, A.; Alvala, M.; Arifuddin, M.; Supuran, C.T. Synthesis and biological evaluation of some coumarin hybrids as selective carbonic anhydrase IX and XII inhibitors. Bioorg. Chem. 2020, 104, 104272. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, B.L.; Bornaghi, L.F.; Houston, T.A.; Innocenti, A.; Supuran, C.T.; Poulsen, S.A. A novel class of carbonic anhydrase inhibitors: Glycoconjuate benzene sulfonamides prepared by “click-tailing”. J. Med. Chem. 2006, 49, 6539–6548. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, B.L.; Bornaghi, L.F.; Houston, T.A.; Innocenti, A.; Vullo, D.; Supuran, C.T.; Poulsen, S.A. Inhibition of membrane-associated carbonic anhydrase isozymes IX, XII and XIV with a library of glycoconjugate benzenesulfonamides. Bioorg. Med. Chem. Lett. 2007, 17, 987–992. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, B.L.; Bornaghi, L.F.; Houston, T.A.; Innocenti, A.; Vullo, D.; Supuran, C.T.; Poulsen, S.A. Carbonic anhydrase inhibitors: Inhibition of isozymes I, II, and IX with triazole-linked O-glycosides of benzene sulfonamides. J. Med. Chem. 2007, 50, 1651–1657. [Google Scholar] [CrossRef] [PubMed]
- Chu, N.; Wang, Y.; Jia, H.; Han, J.; Wang, X.; Hou, Z. Design, synthesis and biological evaluation of new carbohydrate-based coumarin derivatives as selective carbonic anhydrase IX inhibitors via “click” reaction. Molecules 2022, 27, 5464. [Google Scholar] [CrossRef] [PubMed]
- Claus, S.; Pozzi, A. Cyclooxygenases and lipoxygenases in cancer. Cancer Metastasis Rev. 2011, 30, 277–294. [Google Scholar]
- Kennedy, B.M.; Harris, R.E. Cyclooxygenase and lipoxygenase gene expression in the inflammogenesis of colorectal cancer: Correlated expression of EGFR, JAK STAT and Src genes, and a natural antisense transcript, RP11-C67.2.2. Cancers 2023, 15, 2380. [Google Scholar] [CrossRef]
- Harris, R.E. Cyclooxygenase-2 (COX-2) and the inflammogenesis of cancer. Subcell. Biochem. 2007, 42, 93–126. [Google Scholar]
- Wang, D.; DuBois, R.N. The role of COX-2 in intestinal inflammation and colorectal cancer. Oncogene 2010, 29, 781–788. [Google Scholar] [CrossRef]
- Romano, M.; Clària, J. Cyclooxygenase-2 and 5-lipoxygenase converging functions on cell proliferation and tumor angiogenesis: Implications for cancer therapy. FASEB J. 2003, 17, 1986–1995. [Google Scholar] [CrossRef]
- Chang, J.; Tang, N.; Fang, Q.; Zhu, K.; Liu, L.; Xiong, X.; Zhu, Z.; Zhang, B.; Zhang, M.; Tao, J. Inhibition of COX-2 and 5-LOX regulates the progression of colorectal cancer by promoting PTEN and suppressing PI3K/AKT pathway. Biochem. Biophys. Res. Commun. 2019, 517, 1–7. [Google Scholar] [CrossRef]
- Shen, F.-Q.; Wang, Z.-C.; Wu, S.-Y.; Ren, S.-Z.; Man, R.-J.; Wang, B.-Z.; Zhu, H.-L. Synthesis of novel hybrids of pyrazole and coumarin as dual inhibitors of COX-2 and 5-LOX. Bioorg. Med. Chem. Lett. 2017, 27, 3653–3660. [Google Scholar] [CrossRef]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef]
- Hua, W.; Zhao, J.; Hu, W.; Gou, S. Combination of 7-hydroxycoumarin in a platinum(IV) complex derived from cisplatin enhanced cytotoxicity with multiple mechanisms of action. J. Inorg. Biochem. 2018, 186, 17–23. [Google Scholar] [CrossRef]
- Qin, X.; Fang, L.; Zhao, J.; Gou, S. Theranostic Pt(IV) conjugate with target selectivity for androgen receptor. Inorg. Chem. 2018, 57, 5019–5029. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chen, Y.; Li, G.; Liu, Z.; Ma, J.; Liu, M.; Li, D.; Han, J.; Wang, B. Synthesis and evaluation of bi-functional 7-hydroxycoumarin platinum(IV) complexes as antitumor agents. Bioorg. Med. Chem. 2019, 27, 2112–2121. [Google Scholar] [CrossRef] [PubMed]
- Guichard, S.M.; Else, R.; Reid, E.; Zeitlin, B.; Aird, R.; Muir, M.; Dodds, M.; Fiebig, H.; Sadler, P.J.; Jodrell, D.I. Anti-tumour activity in non-small cell lung cancer models and toxicity profiles for novel ruthenium(II) based organo-metallic compounds. Biochem. Pharmacol. 2006, 71, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Scolaro, C.; Bergamo, A.; Brescacin, L.; Delfino, R.; Cocchietto, M.; Laurenczy, G.; Geldbach, T.J.; Sava, G.; Dyson, P.J. In vitro and in vivo evaluation of ruthenium(II)−arene PTA complexes. J. Med. Chem. 2005, 48, 4161–4171. [Google Scholar] [CrossRef] [PubMed]
- Renfrew, A.K.; Phillips, A.D.; Tapavicza, E.; Scopelliti, R.; Rothlisberger, U.; Dyson, P.J. Tuning the efficacy of ruthenium(II)-arene (RAPTA) antitumor compounds with fluorinated arene ligands. Organometallics 2009, 28, 5061–5071. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, D.; Hua, W.; Li, W.; Xu, G.; Gou, S. Anticancer activity of bifunctional organometallic Ru(II) arene complexes containing a 7-hydroxycoumarin group. Organometallics 2018, 37, 441–447. [Google Scholar] [CrossRef]
- Schuh, E.; Pflüger, C.; Citta, A.; Folda, A.; Rigobello, M.P.; Bindoli, A.; Casini, A.; Mohr, F. Gold(I) carbene complexes causing thioredoxin 1 and thioredoxin 2 oxidation as potential anticancer agents. J. Med. Chem. 2012, 55, 5518–5528. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, B.; de Almeida, A.; van der Burgt, E.P.M.; Picquet, M.; Citta, A.; Folda, A.; Rigobello, M.P.; Le Gendre, P.; Bodio, E.; Casini, A. New gold(I) organometallic compounds with biological activity in cancer cells. Eur. J. Inorg. Chem. 2014, 27, 4532–4536. [Google Scholar] [CrossRef]
- Arcau, J.; Andermark, V.; Aguiló, E.; Gandioso, A.; Moro, A.; Cetina, M.; Lima, J.C.; Rissanen, K.; Ott, I.; Rodríguez, L. Luminescent alkynyl-gold(I) coumarin derivatives and their biological activity. Dalton Trans. 2014, 43, 4426–4436. [Google Scholar] [CrossRef]
- Raunio, H.; Pentikäinen, O.; Juvonen, R.O. Coumarin-based profluorescent and fluorescent substrates for determining xenobiotic-metabolizing enzyme activities in vitro. Int. J. Mol. Sci. 2020, 21, 4708. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Li, M.; Zheng, S.; Wang, B. Rational design of a fluorescent hydrogen peroxide probe based on the umbelliferone fluorophore. Tetrahedron Lett. 2008, 49, 3045–3048. [Google Scholar] [CrossRef]
- Levin, P.P.; Liubimov, A.V.; Shashkov, A.S.; Mardaleishvili, I.R.; Venidiktova, O.V.; Shienok, A.L.; Koltsova, L.S.; Astafiev, A.A.; Barachevsky, V.A.; Zaichenko, N.L. Multiple fluorescence of tetraarylimidazole and azomethinocoumarin dyad with dual excited-state intramolecular proton transfer. Dyes Pigm. 2020, 183, 108716. [Google Scholar] [CrossRef]
- Xiao, Z.; Chen, D.; Song, S.; Vlag, R.; Wouden, P.; Merkerk, R.; Cool, R.H.; Hirsch, A.K.H.; Melgert, B.N.; Quax, W.J.; et al. 7-Hydroxycoumarins are affinity-based fluorescent probes for competitive binding studies of macrophage migration inhibitory factor. J. Med. Chem. 2020, 63, 11920–11933. [Google Scholar] [CrossRef]
- Shi, B.; Zhang, Z.; Jin, Q.; Wang, Z.; Tang, J.; Xu, G.; Zhu, T.; Gong, X.; Tang, X.; Zhao, C. Selective tracking of ovarian-cancer-specific γ-glutamyltranspeptidase using a ratiometric two-photon fluorescent probe. J. Mater. Chem. B 2018, 6, 7439. [Google Scholar] [CrossRef]
- Li, S.; Kan, W.; Zhao, B.; Liu, T.; Fang, Y.; Bai, L.; Wang, L. A fluorescent pH probe for an aqueous solution composed of 7-hydroxycoumarin, Schiff base and phenanthro[9,10-d]imidazole moieties (PICO). Heterocycl. Commun. 2018, 24, 93–97. [Google Scholar] [CrossRef]
- Shukla, L.; Moodie, L.W.K.; Kindahl, T.; Hedberg, C. Synthesis and spectroscopic properties of fluorinated coumarin lysine derivatives. J. Org. Chem. 2018, 83, 4792–4799. [Google Scholar] [CrossRef] [PubMed]
- Gleason, P.R.; Kelly, P.I.; Grisingher, D.W.; Mills, J.H. An intrinsic FRET sensor of protein-ligand interactions. Org. Biomol. Chem. 2020, 18, 4079–4084. [Google Scholar] [CrossRef] [PubMed]
- Gleason, P.R.; Kolbaba-Kartchner, B.; Henderson, J.N.; Stahl, E.P.; Simmons, C.R.; Mills, J.H. Structural origins of altered spectroscopic properties upon ligand binding in proteins containing a fluorescent noncanonical amino acid. Biochemistry 2021, 60, 2577–2585. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Li, Y.; Liu, A.Y.; Tan, Y.H.; Ling, J.; Ding, Z.T.; Cao, Q.E. Highly selective visual sensing of copper based on fluorescence enhanced glutathione-Au nanoclusters. Spectrochim. Acta Part. A Mol. Biomol. Spectrosc. 2020, 224, 117472. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Yu, Y.; Lin, B.; Cai, Y.; Cao, Y.; Guo, M.; Zhu, D. Copper nanoclusters reversible switches based on ions-triggered for detection of inorganic pyrophosphatase activity. Sens. Actuators B Chem. 2019, 284, 36–44. [Google Scholar] [CrossRef]
- You, J.G.; Lu, C.-Y.; Kumar, A.S.K.; Tseng, W.-L. Cerium(III)-directed assembly of glutathione-capped gold nanoclusters for sensing and imaging of alkaline phosphatase-mediated hydrolysis of adenosine triphosphate. Nanoscale 2018, 10, 17691–17698. [Google Scholar] [CrossRef] [PubMed]
- Mei, H.; Wang, J.; Zhu, X.; Sun, J.; Shi, W.; Wang, H.; Qu, S.; Wang, X. Ce3+ and Fe2+ co-enhanced ratiometric fluorescence probe utilizing copper nanoclusters and coumarin for sensitive assay of hydrogen peroxide and glucose. Ecotoxicol. Environ. Saf. 2022, 245, 114117. [Google Scholar] [CrossRef] [PubMed]
- Ohguri, N.; Nosaka, A.Y.; Nosaka, Y. Detection of OH radicals as the effect of Pt particles in the membrane of polymer electrolyte fuel cells. J. Power Sources 2010, 195, 4647–4652. [Google Scholar] [CrossRef]
- Hirano, K.; Kobayashi, T. Coumarin fluorometry to quantitatively detectable OH radicals in ultrasound aqueous medium. Ultrason. Sonochem. 2016, 30, 18–27. [Google Scholar] [CrossRef]
- Wang, K.; Yao, T.; Xue, J.; Guo, Y.; Xu, X. A novel fluorescent probe for the detection of hydrogen peroxide. Biosensors 2023, 13, 658. [Google Scholar] [CrossRef]
- Zhu, G.; Huang, Y.; Wang, C.; Lu, L.; Sun, T.; Wang, M.; Tang, Y.; Shan, D.; Wen, S.; Zhu, J. A novel coumarin-based fluorescence chemosensor for Al3+ and its application in cell imaging. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2019, 210, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Duan, Q.; Yu, Y.; Wang, K.; Zhu, H.; Zhang, X.; Liu, C.; Jia, P.; Li, Z.; Sheng, W.; et al. A coumarin-based fluorescent probe for Hg2+ and its application in living cells and zebrafish. Luminescence 2020, 35, 941–946. [Google Scholar] [CrossRef]
- Rojas-Montoyaa, S.M.; Vonlanthena, M.; Huerta-Roldána, J.M.; Aguilar-Ortíza, E.; Burillob, G.; Morales-Espinoza, E.G.; Rivera, E. Incorporation of photoluminescent 7-hydroxycoumarin units onto a polyethylene matrix by means of gamma radiation. Radiat. Phys. Chem. 2019, 163, 52–57. [Google Scholar] [CrossRef]
- Stefanachi, A.; Leonetti, F.; Pisani, L.; Catto, M.; Carotti, A. Coumarin: A natural, privileged and versatile scaffold for bioactive compounds. Molecules 2018, 23, 250. [Google Scholar] [CrossRef] [PubMed]
- Flores-Morales, V.; Villasana-Ruiz, A.P.; Garza-Veloz, I.; González-Delgado, S. Therapeutic effects of coumarins with different substitution patterns. Molecules 2023, 28, 2413. [Google Scholar] [CrossRef]












































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Biological Activity | Molecular Target | Name/ Number | Ref. |
|---|---|---|---|---|
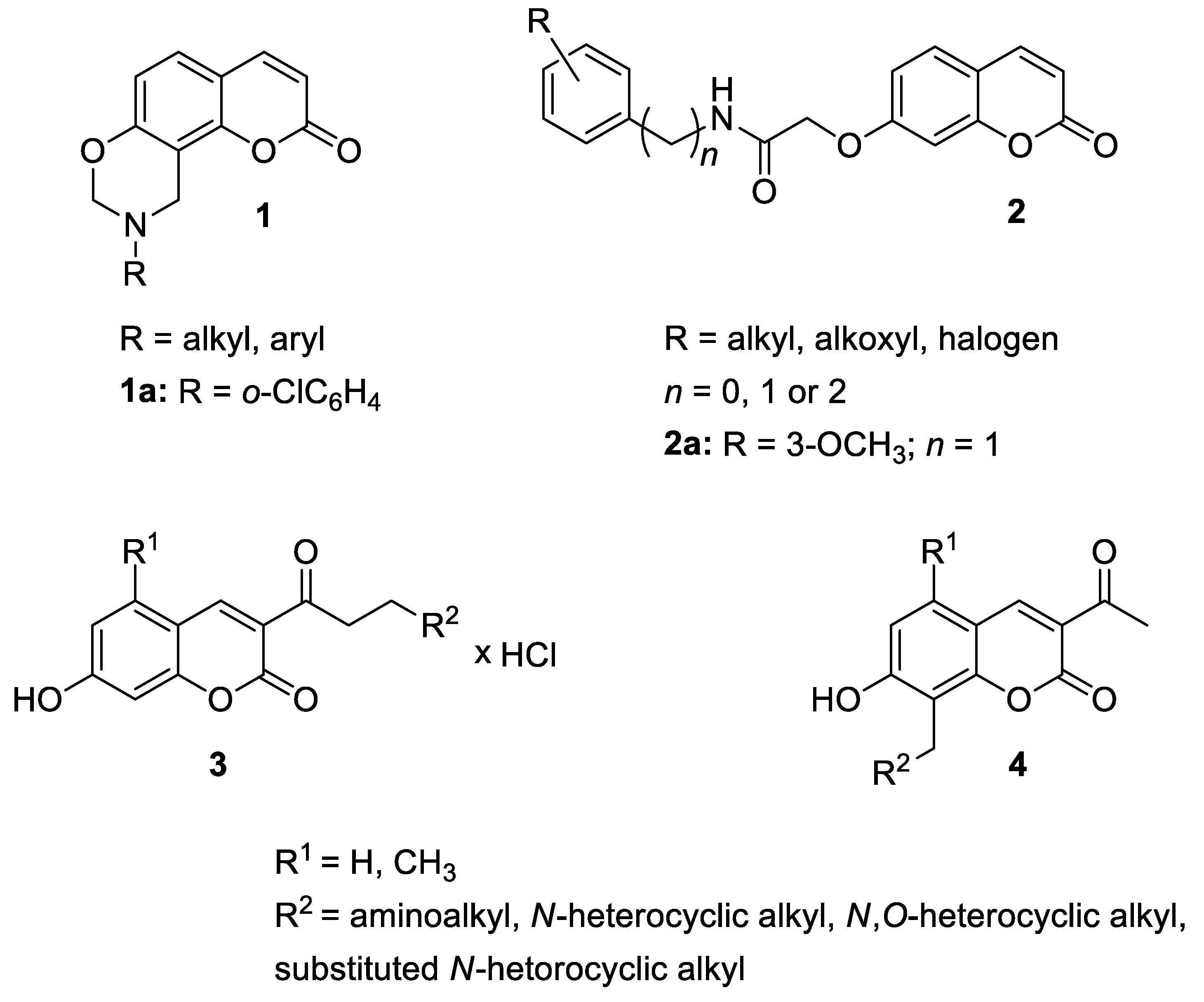
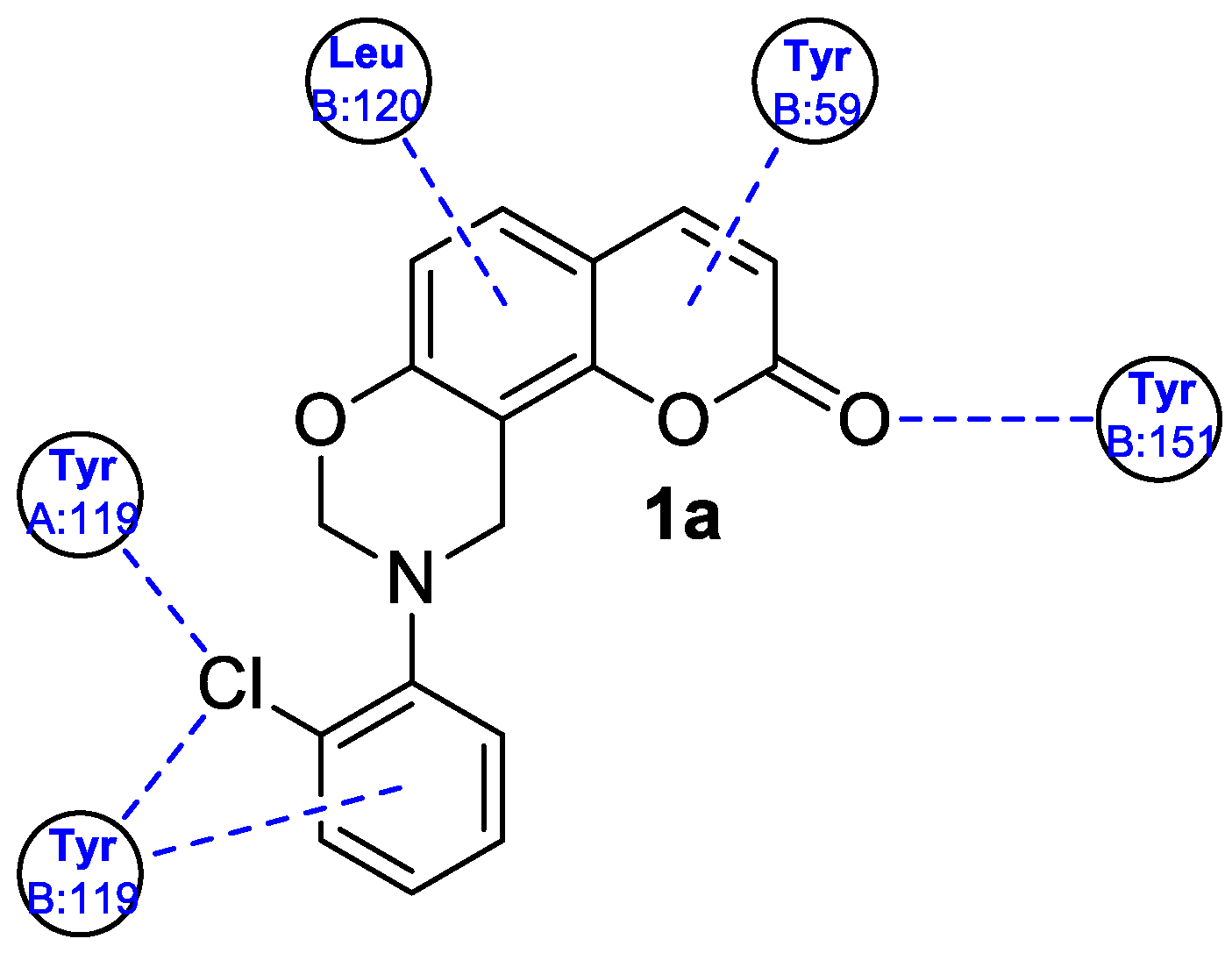
 | Anti-inflammatory | MAPK and NK-κB | 1a | [32] |
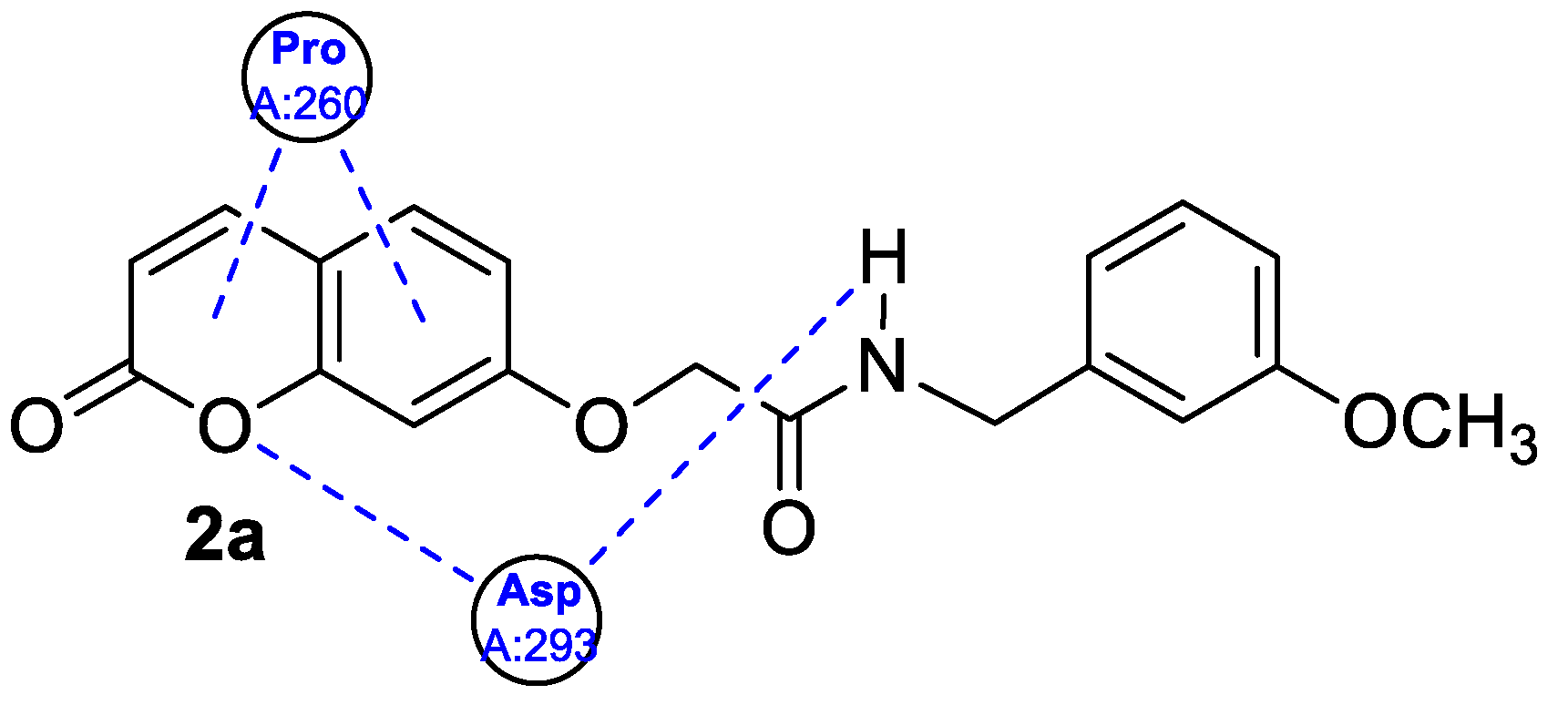
 | Anti-inflammatory | NK-κB p65 | 2a | [34] |
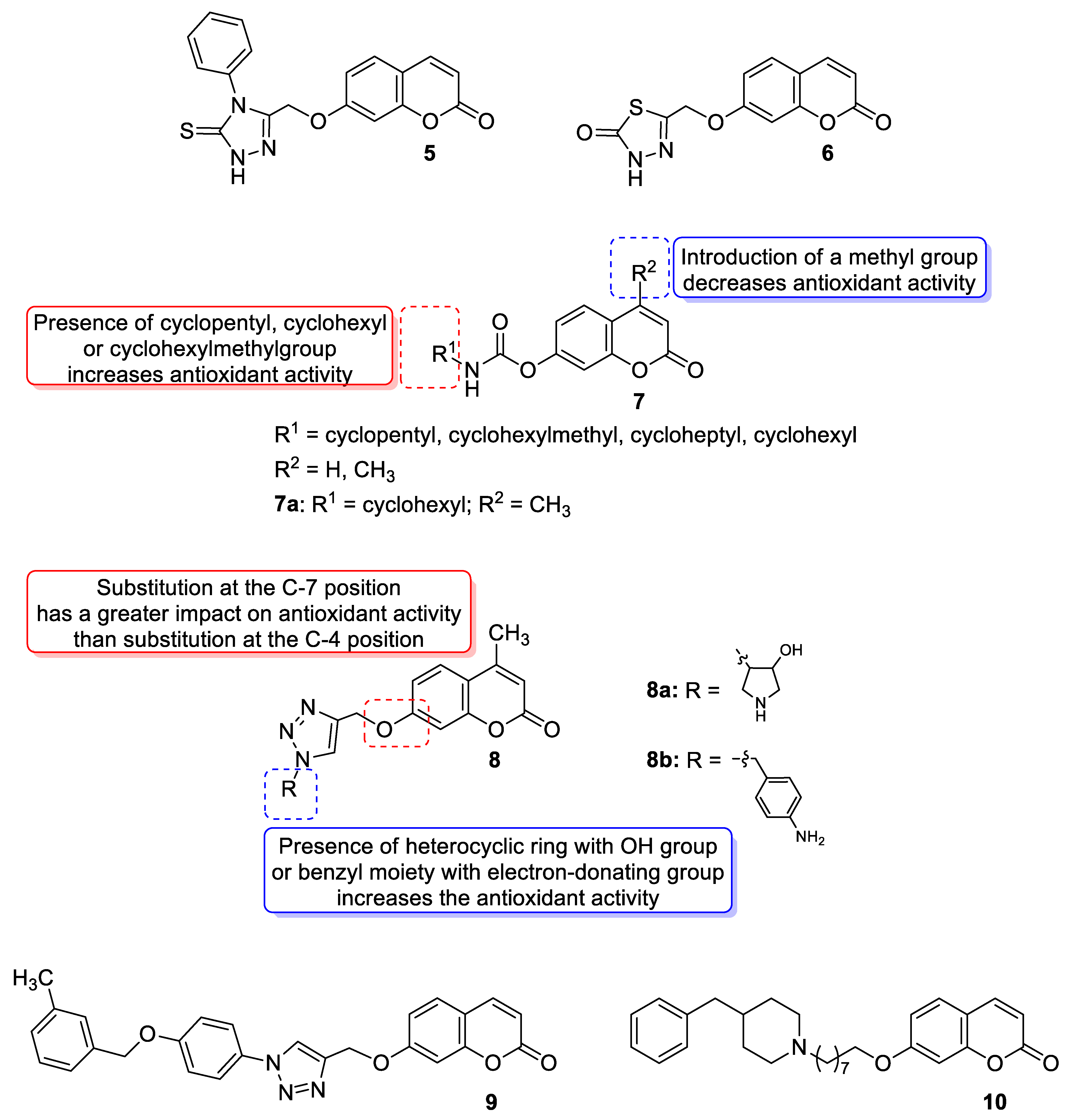
 | Antioxidant and anti-Alzheimer’s disease (anti-AD) | Free radicals and BuChE | 7 | [41] |
 | Antioxidant | Free radicals | 8a | [42] |
 | Antioxidant | Free radicals | 8b | [42] |
 | Antioxidant | Free radicals | 10 | [44] |
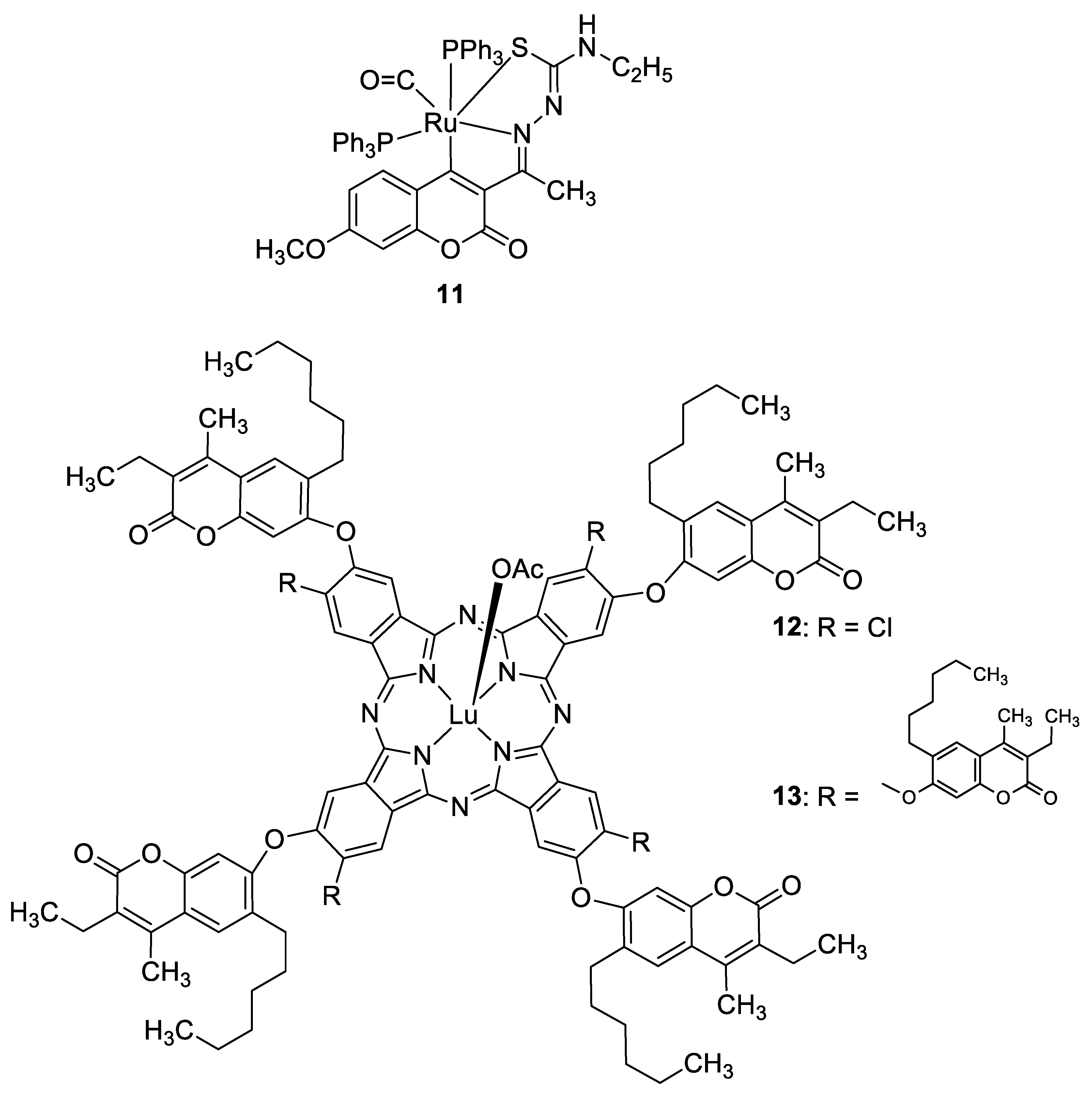
 | Antioxidant | Free radicals | 11 | [47] |
 | Anti-Alzheimer’s disease (anti-AD) | AChE and BuChE | 17 | [60] |
 | Anti-Alzheimer’s disease (anti-AD) and neuroprotective | MAO-B | 18 | [63] |
 | Anti-Alzheimer’s disease (anti-AD) and neuroprotective | MAO-B | 19 | [63] |
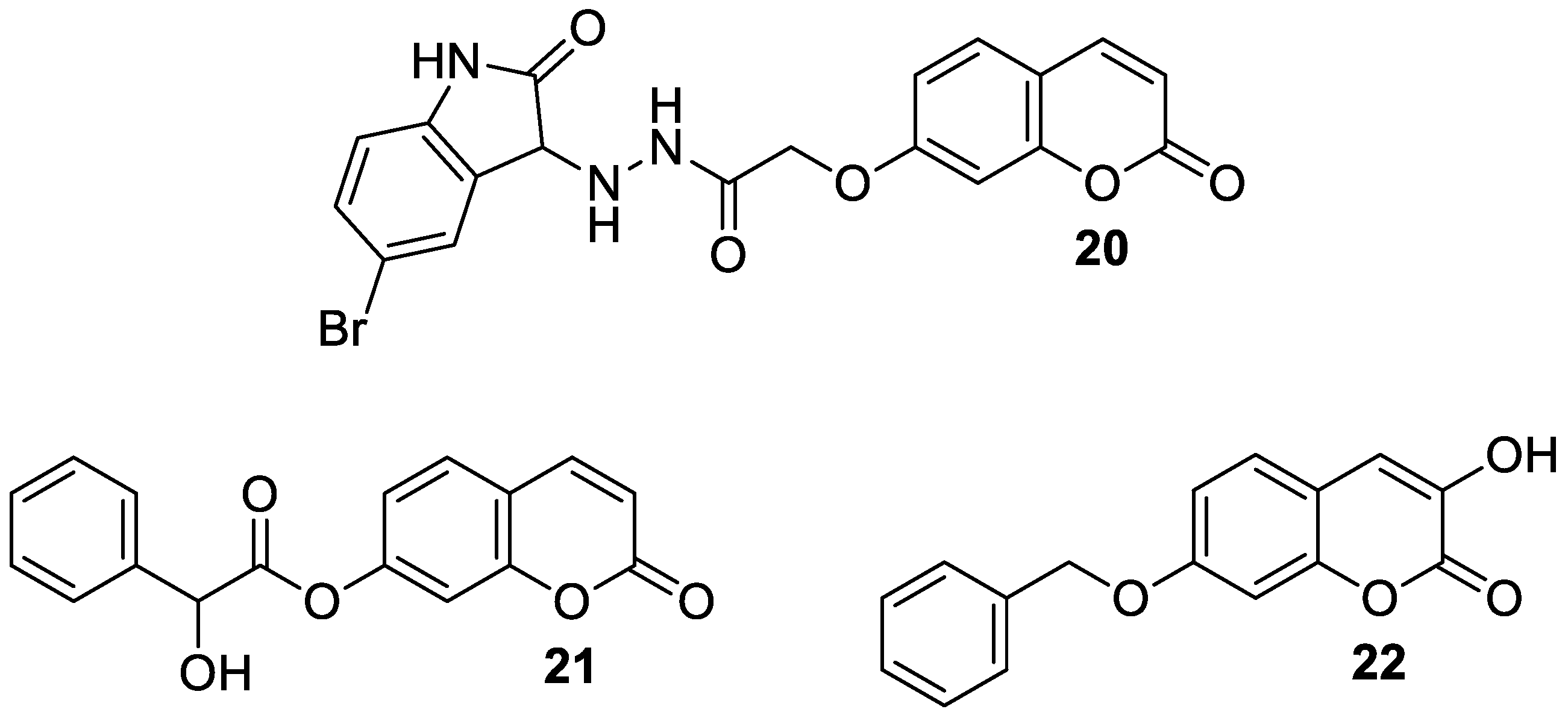
 | Treatment of neuropsychiatric diseases (schizophrenia) | DAAO | 22 | [69] |
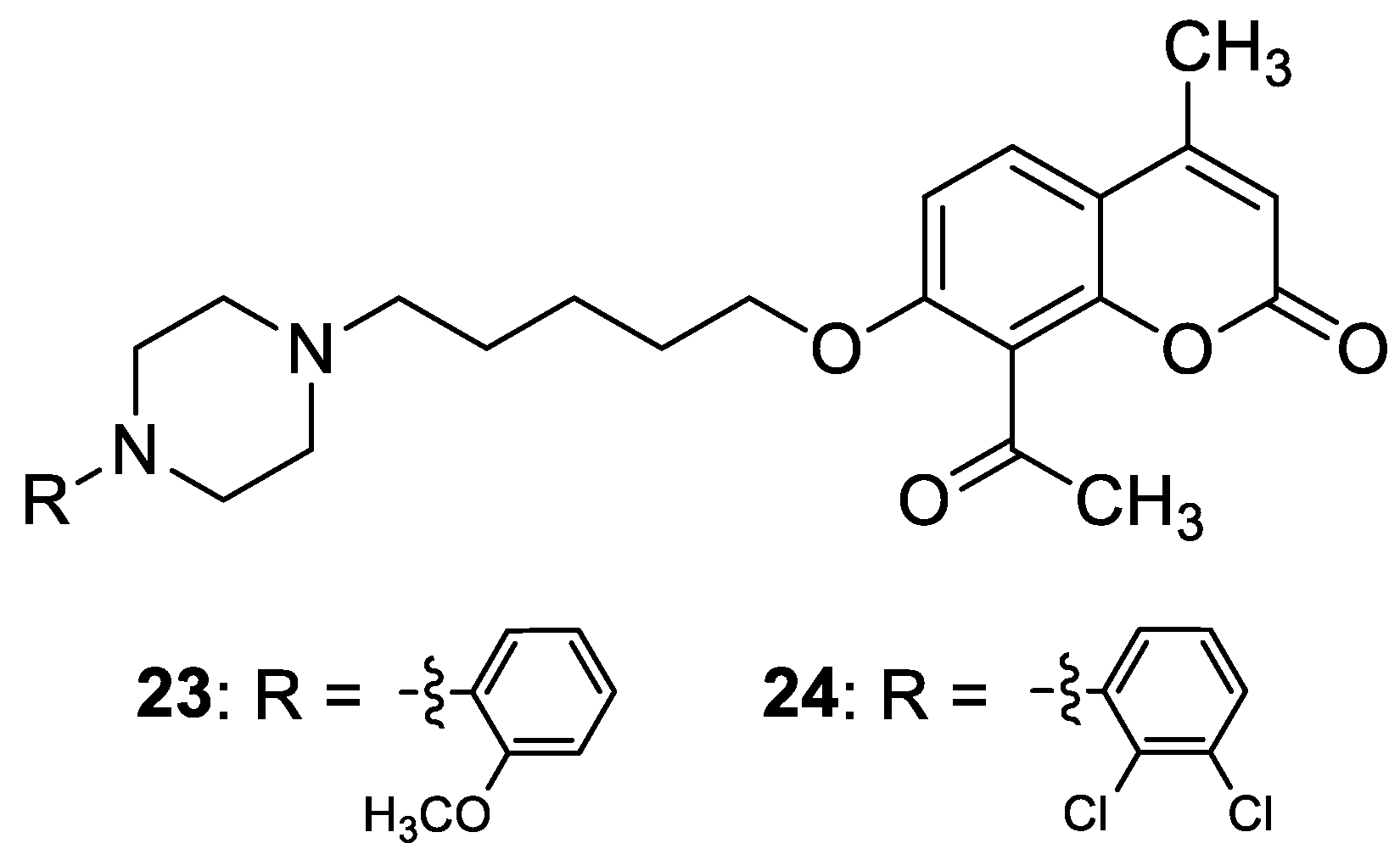
 | Treatment of neuropsychiatric diseases | 5-HT1A | 23 | [70] |
 | Treatment of neuropsychiatric diseases | 5-HT2A | 24 | [70] |
 | Antiepileptic | GABAA | 25 | [73] |
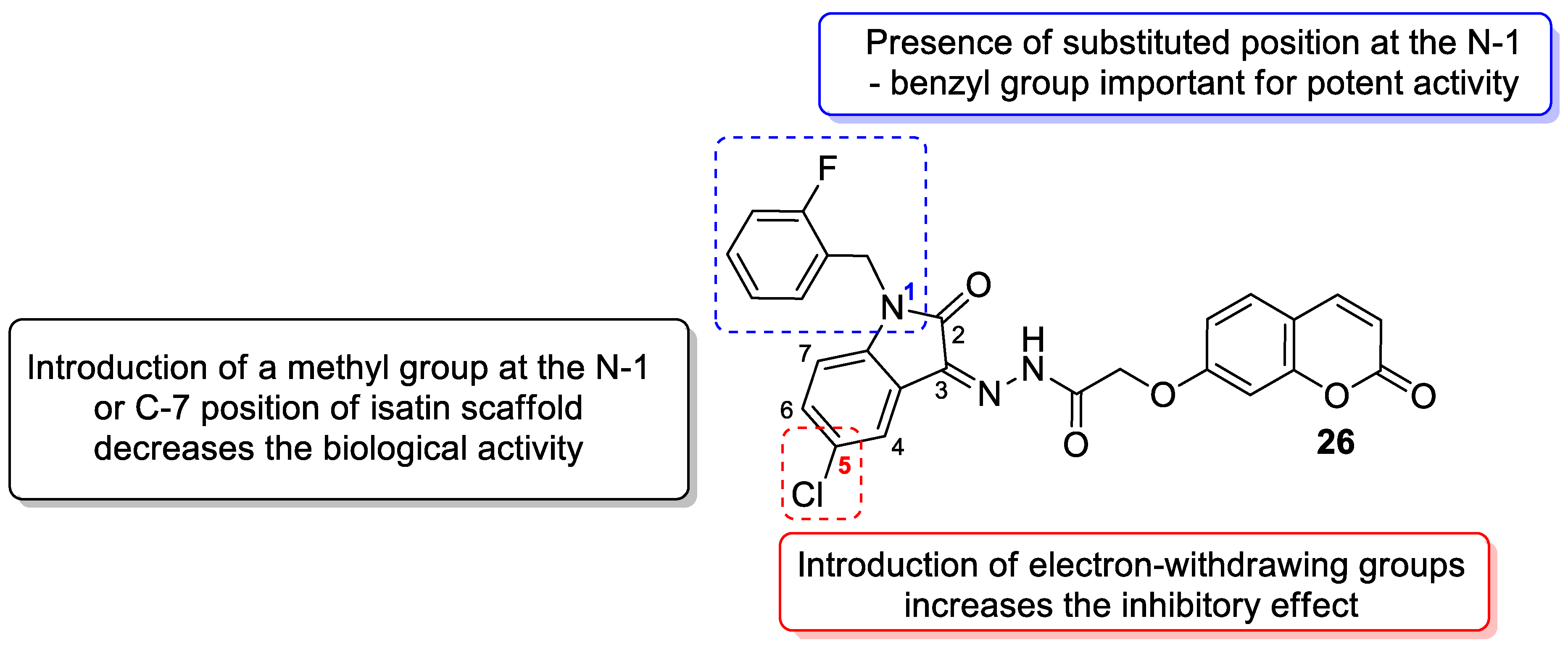
 | Antidiabetic | α-glucosidase | 26 | [79] |
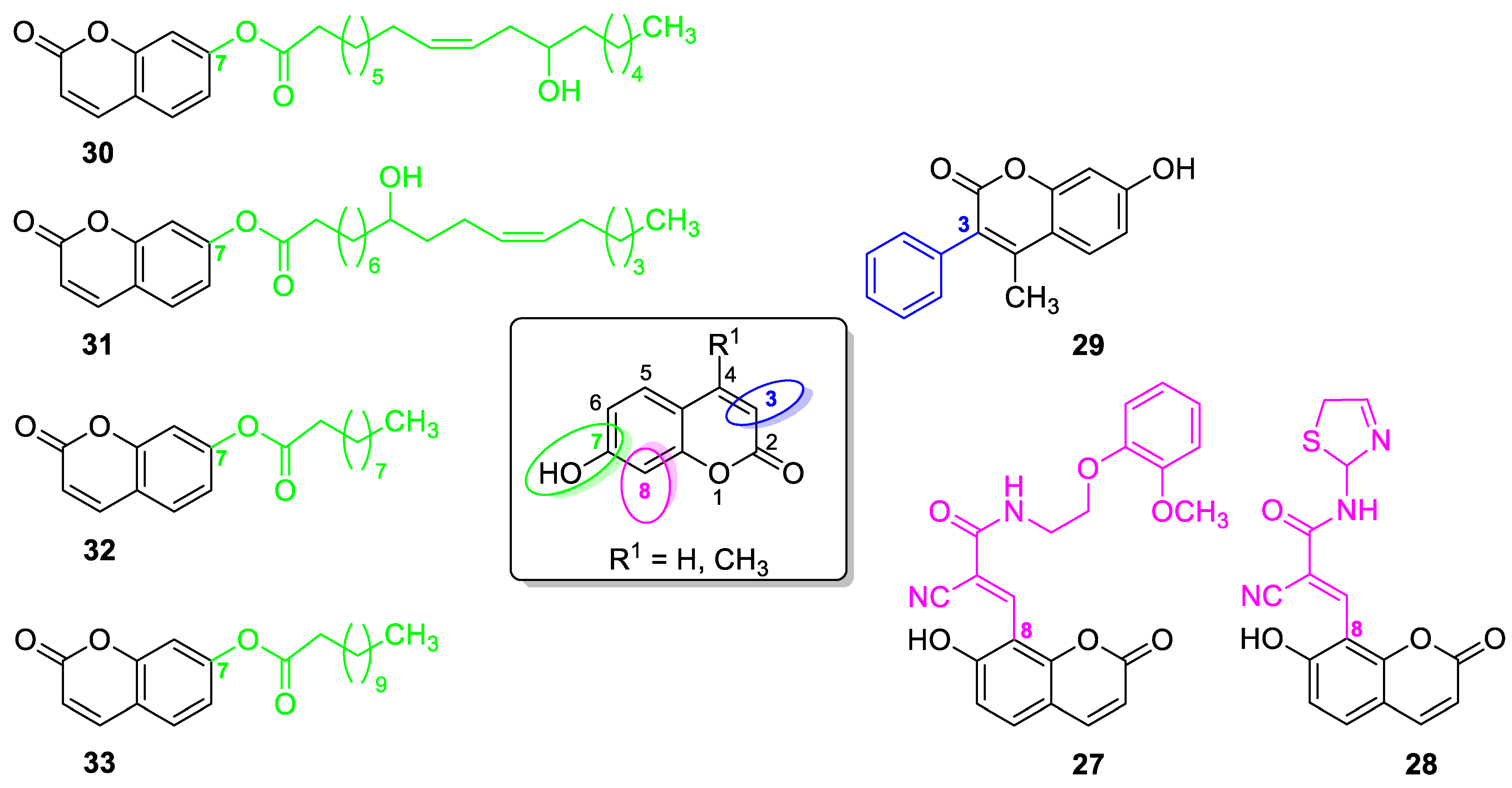
 | Antibacterial and antifungal | E. coli, S. aureus, P. aeruginosa, A. niger, and C. albicans | 27 | [88] |
 | Antibacterial and antifungal | B. subtilis, S. pyogenes, S. aureus, E. coli, C. albicans, C. parapsilosis, and C. neoformans | 31 | [91] |
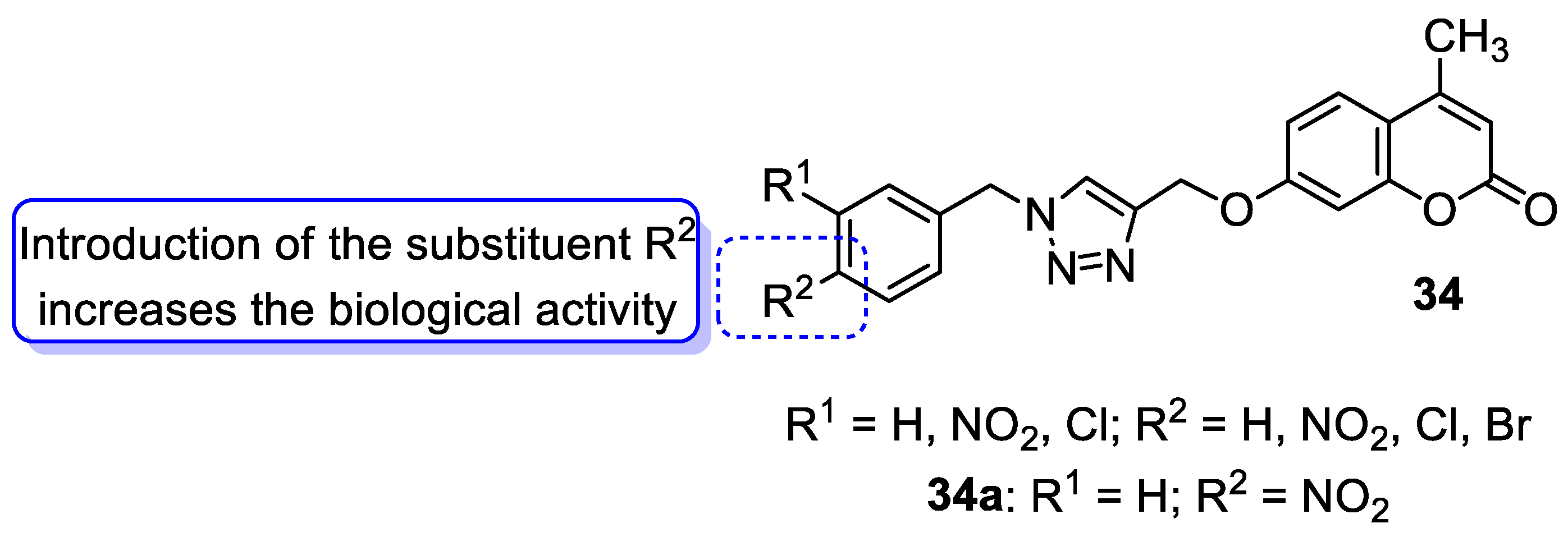
 | Antibacterial | M. luteus, B. cereus, E. coli, and P. fluorescens | 34a | [95] |
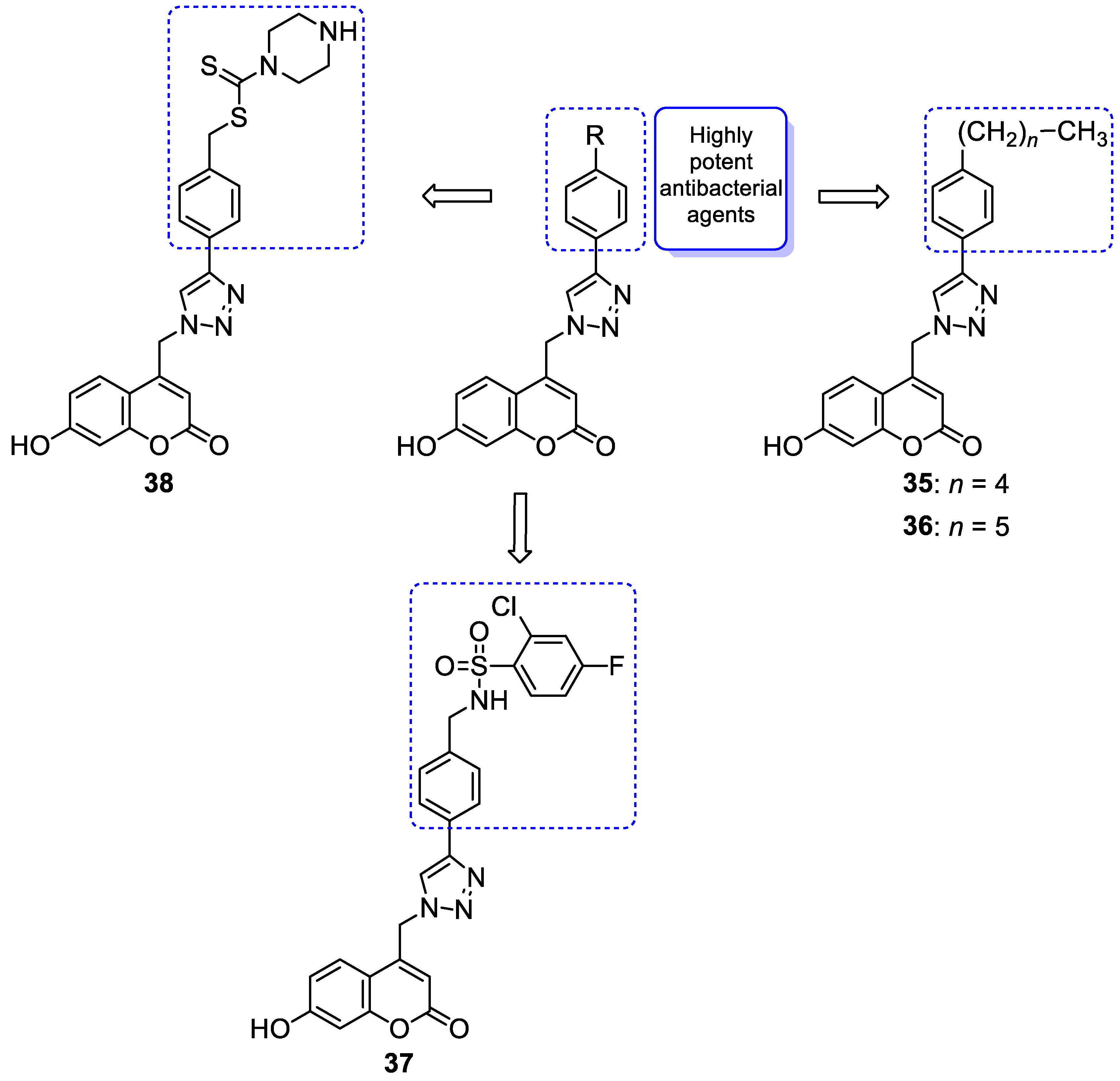
 | Antibacterial | VRA E. faecium and E. faecalis | 35 | [96] |
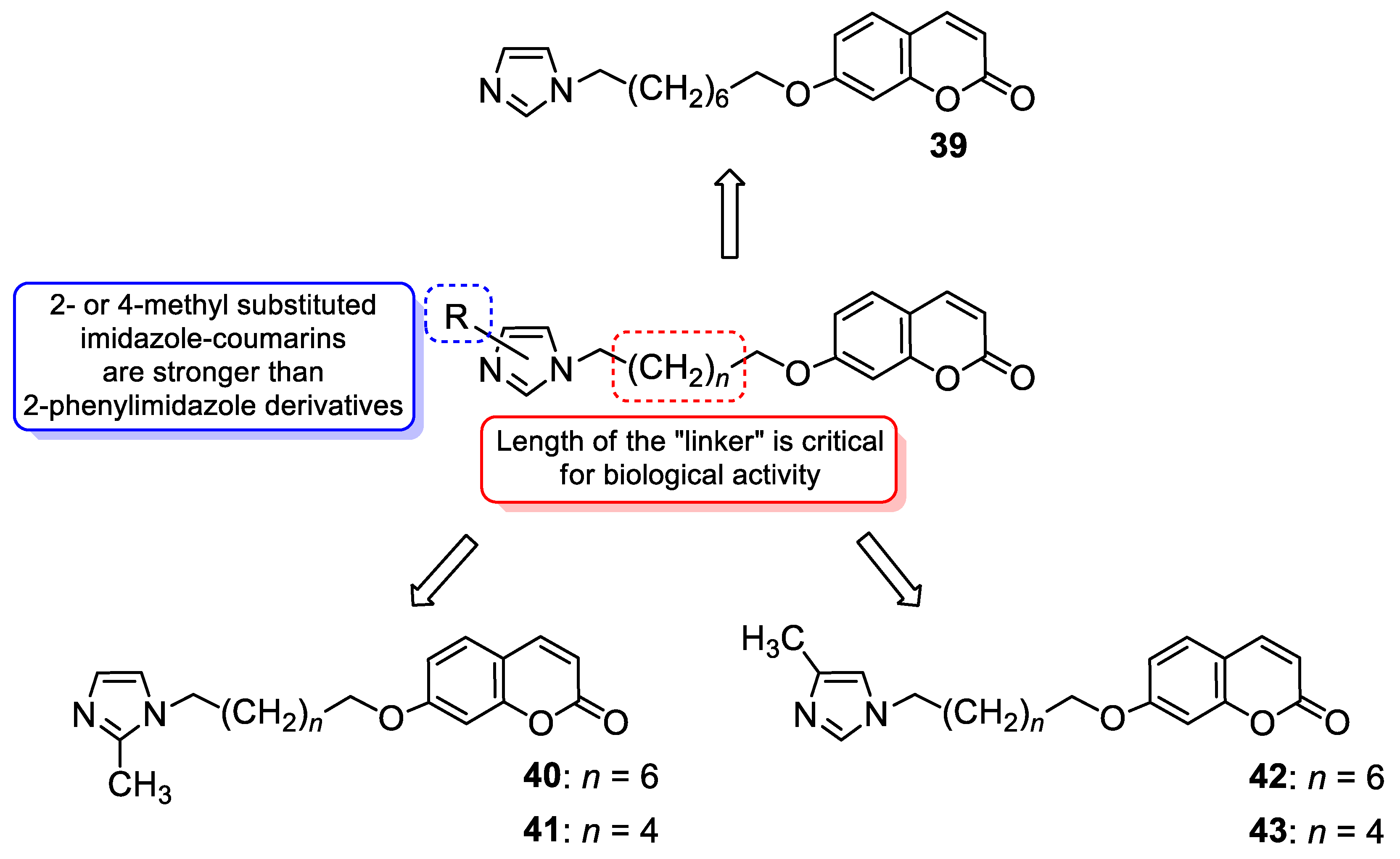
 | Antibacterial and antiviral | E. coli and infectious hematopoietic necrosis virus (IHNV) | 41 | [97] [111] |
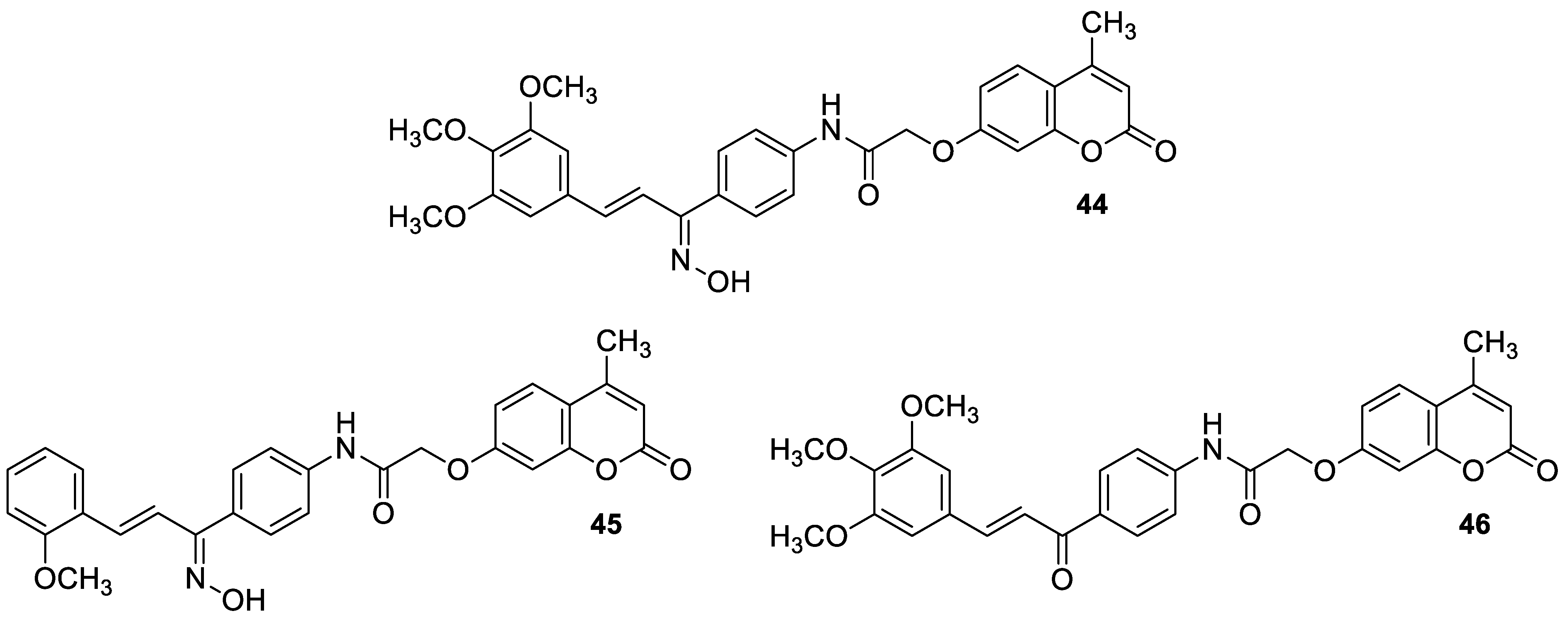
 | Antibacterial | S. aureus | 45 | [98] |
 | Antibacterial | S. aureus, E. coli, and K. pneumoniae | 46 | [98] |
 | Antibacterial and antifungal | S. aureus, B. subtilis, B. cereus, S. epidermis, P. aeruginosa, and C. albicans | 50 | [102] |
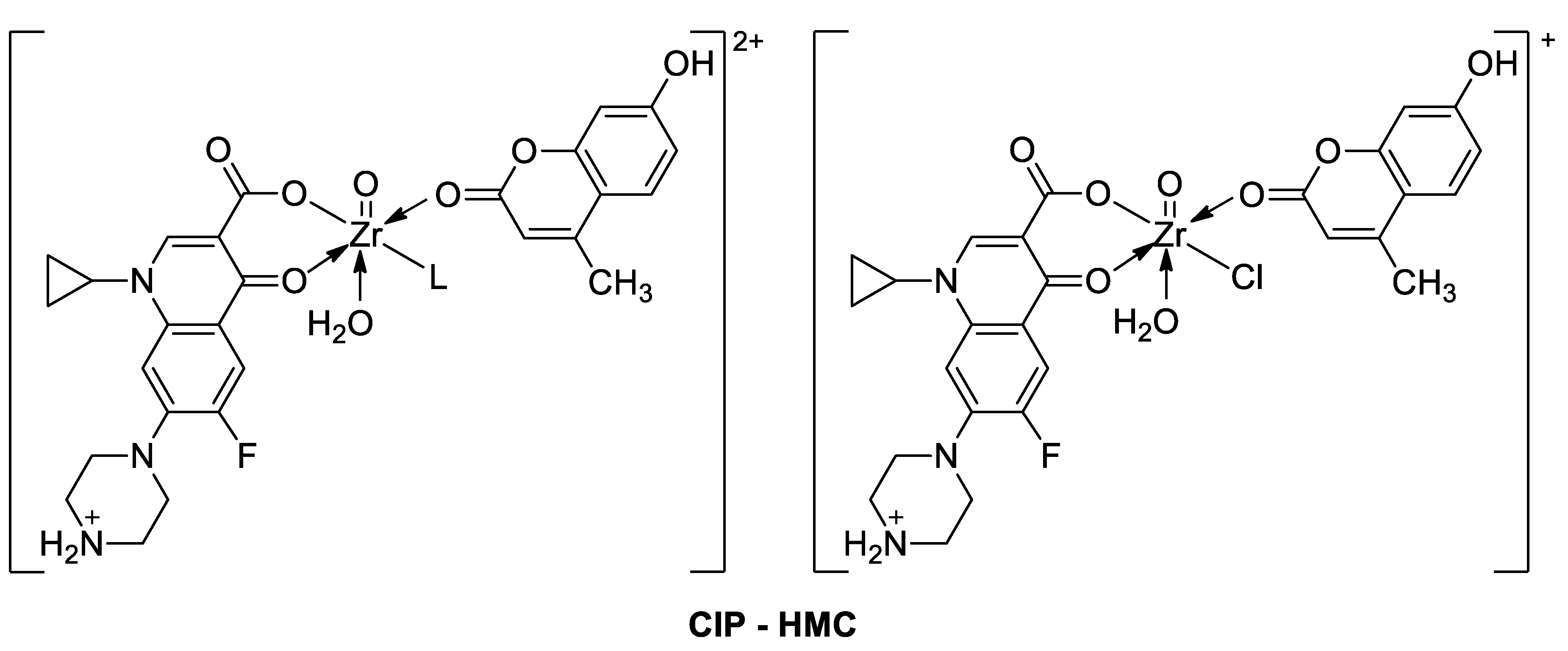
 | Antibacterial | B. subtilis, B. cereus, P. aeruginosa, and E. coli | CIP—HMC | [103] |
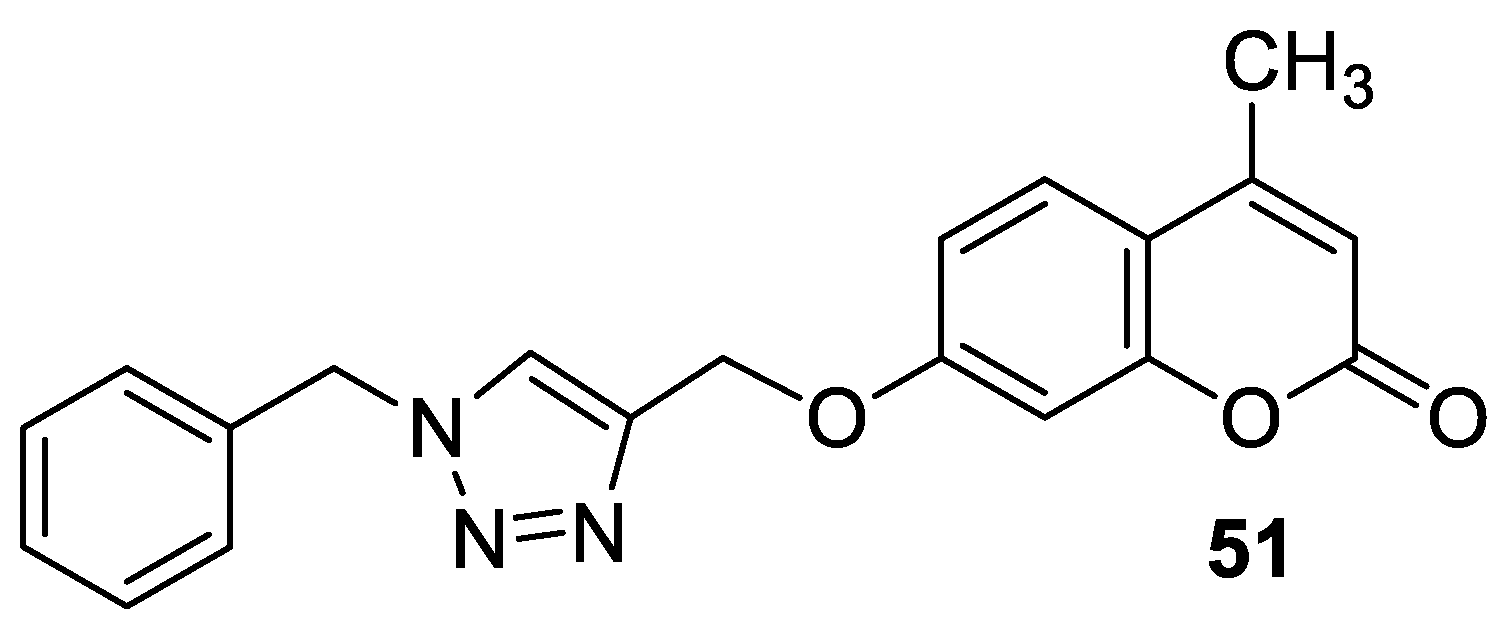
 | Antitubercular | M. tuberculosis H37Ra and Dpr E1 | 51 | [104] |
 | Antimalarial | P. falciparum | 53 | [43] |
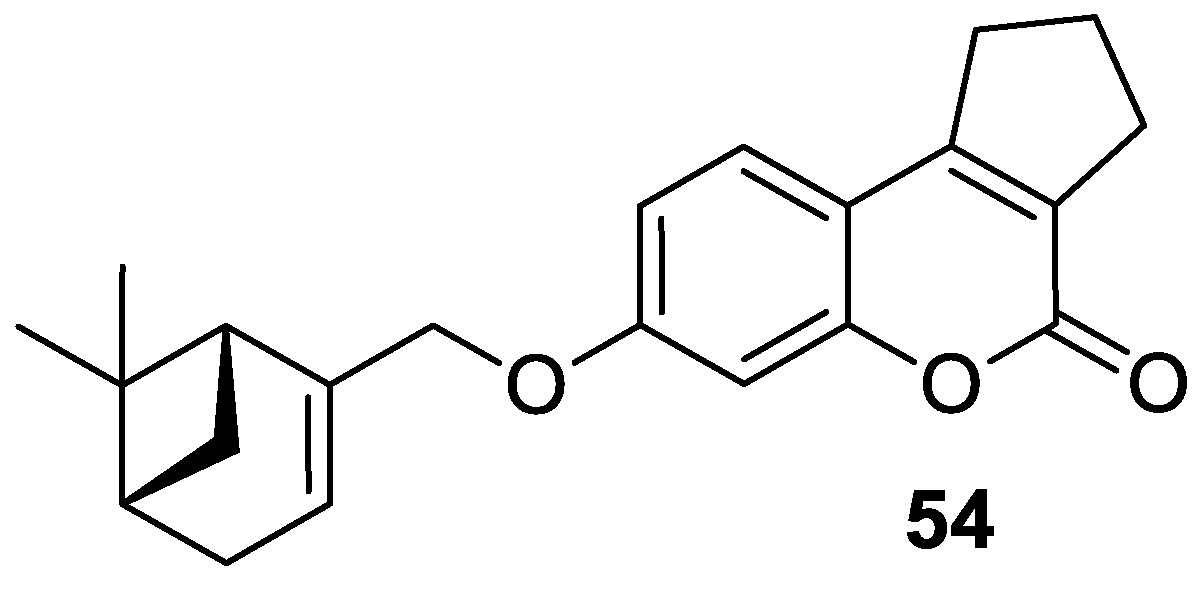
 | Antiviral | Influenza A virus (viral hemagglutinin, proton channel M2) | 54 | [110] |
 | Antiproliferative | Colon cancer cell line (HCT116), lung cancer cell line (A549), and leukemia (HL60) | 55 | [131] |
 | Anticancer | HDAC1 | 59 | [132] |
 | Anticancer | HDAC1 | 61 | [133] |
 | Anticancer | HDAC1 and cervical cancer cell line (HeLa) | 65 | [134] |
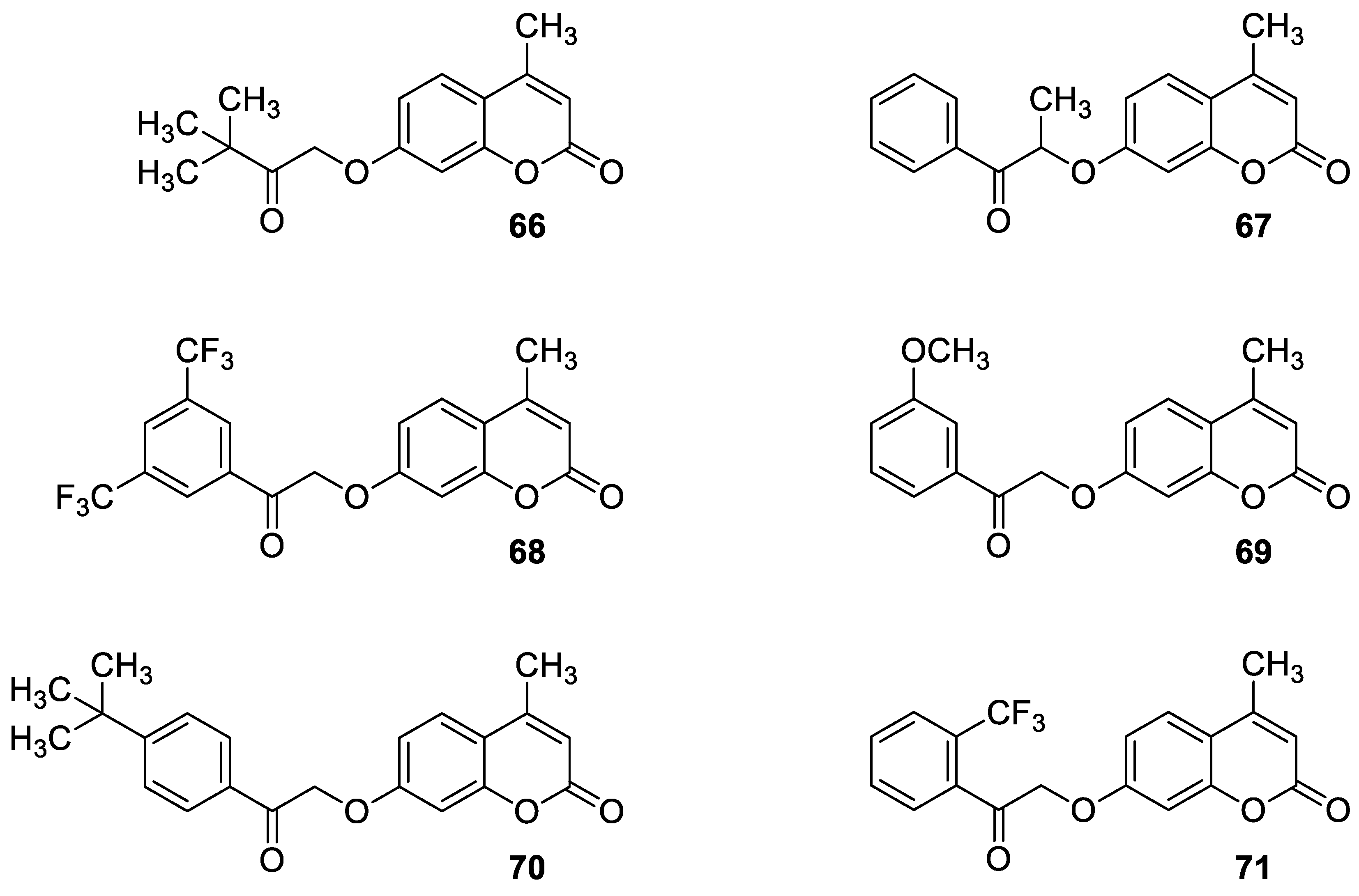
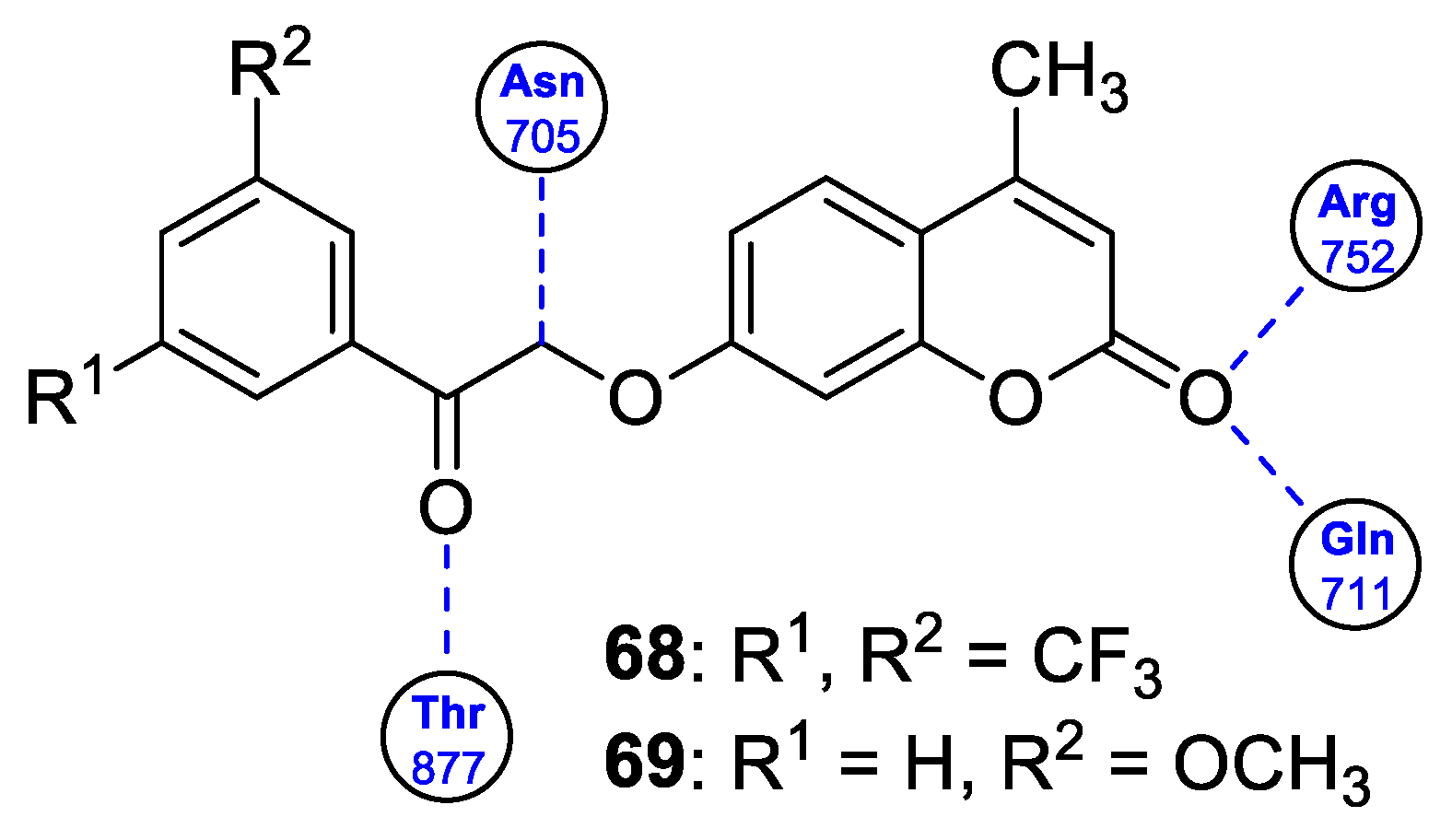
 | Anticancer | Prostate cancer cell line (22Rv1) and breast cancer cell line (MCF-7) | 68 | [138] |
 | Anticancer | Prostate cancer cell line (22Rv1) and breast cancer cell line (MCF-7) | 69 | [138] |
 | Anticancer | PI3Kα/β/δ signal pathway, lung carcinoma (A549), breast carcinoma (MCF-7), leukemia (K562), and cervical carcinoma (HeLa) | 72 | [144] |
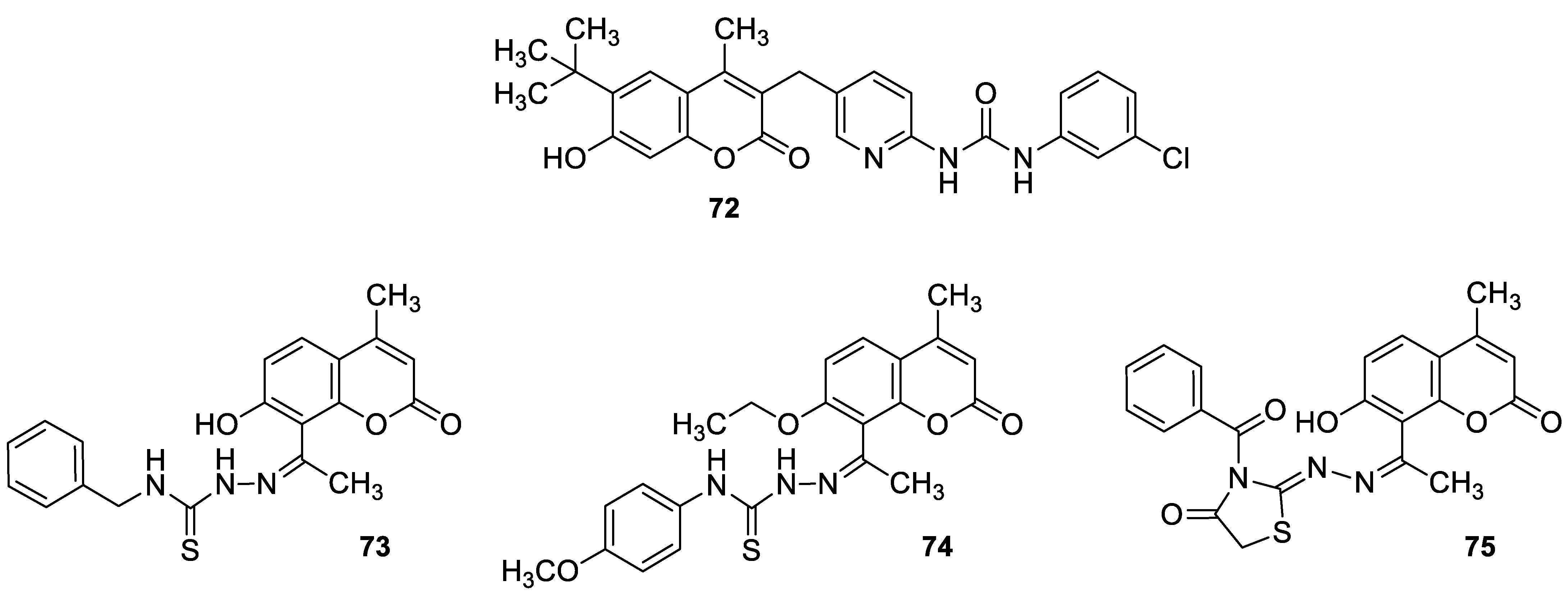
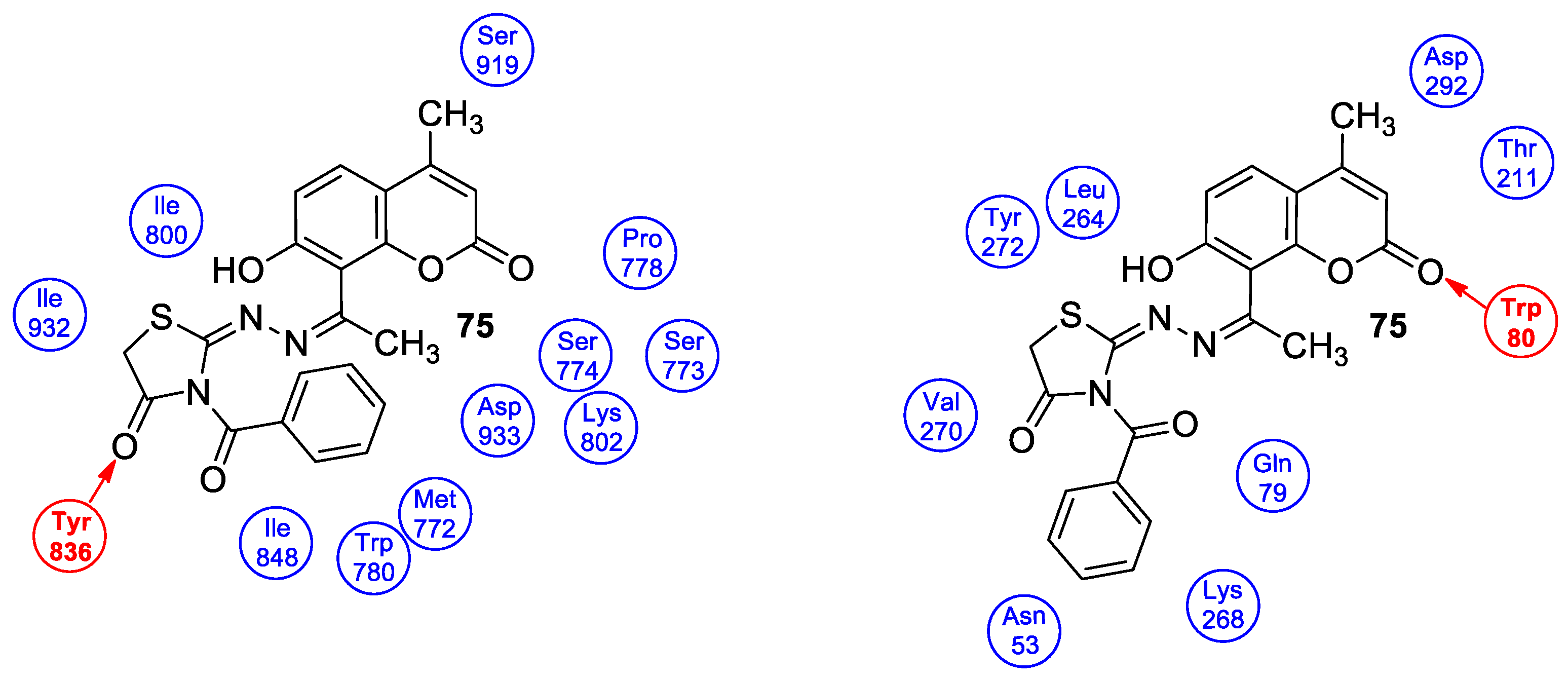
 | Cytotoxic | PI3Kα/Akt-1 signal pathway and breast carcinoma (MCF-7) | 75 | [145] |
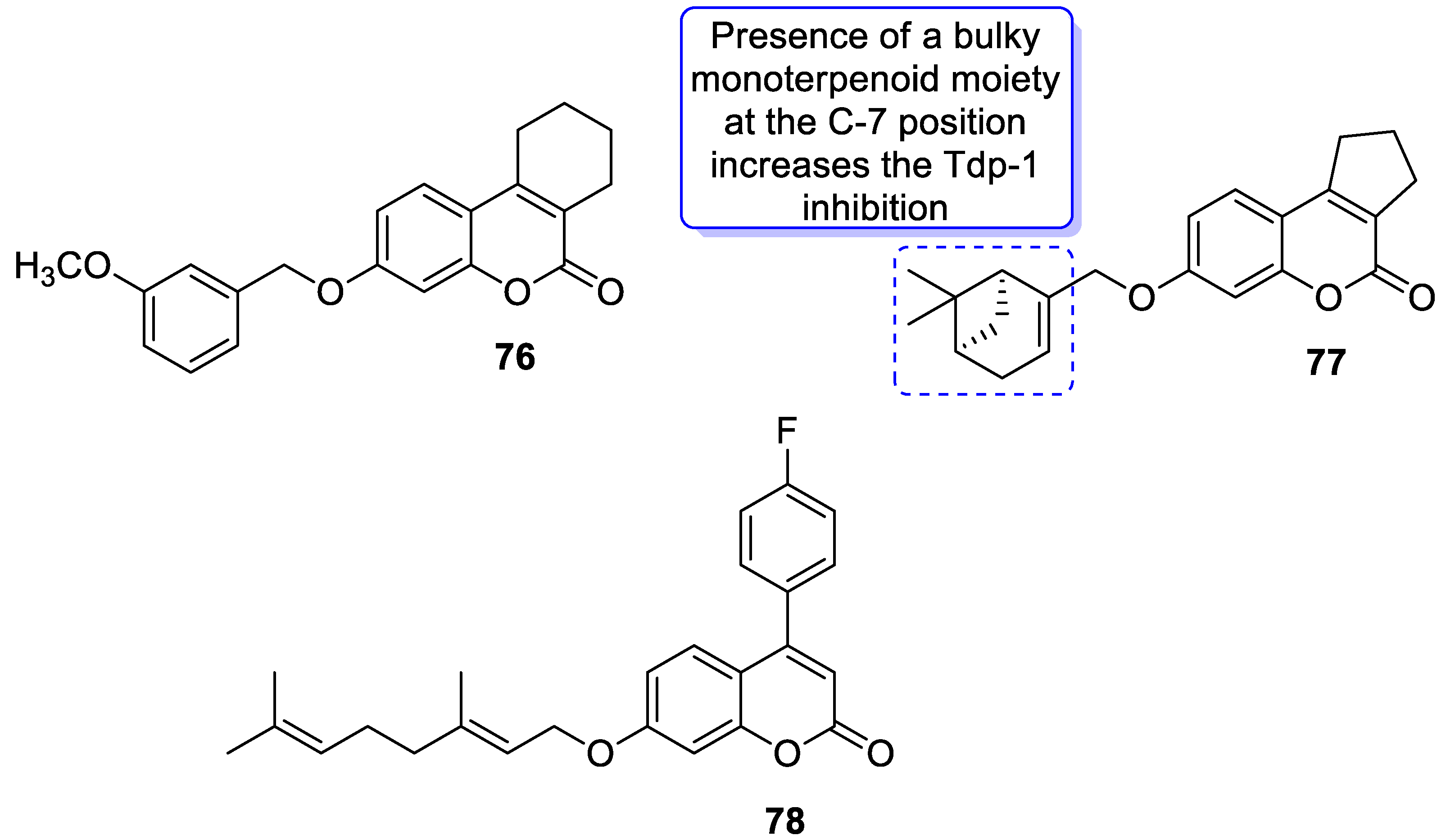
 | Anticancer | Tyrosyl-DNA phosphodiesterase (Tdp1) | 77 | [152] |
 | Anticancer | Krebs-2 carcinoma | 78 | [153] |
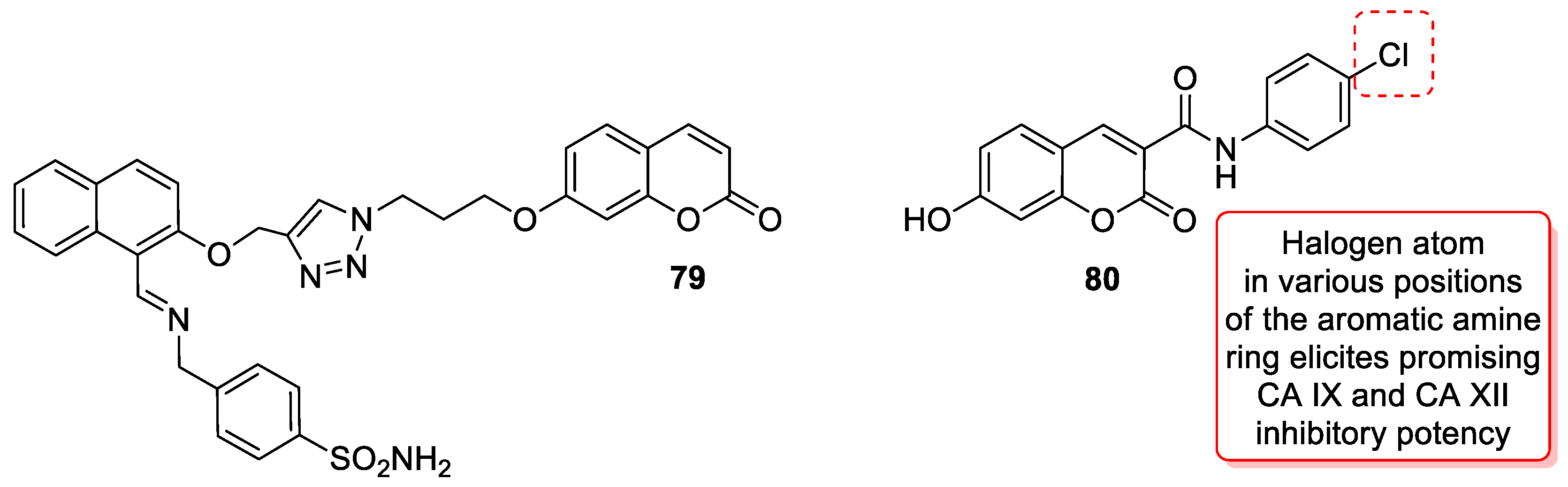
 | Anticancer | CA IX and CA XII | 80 | [162] |
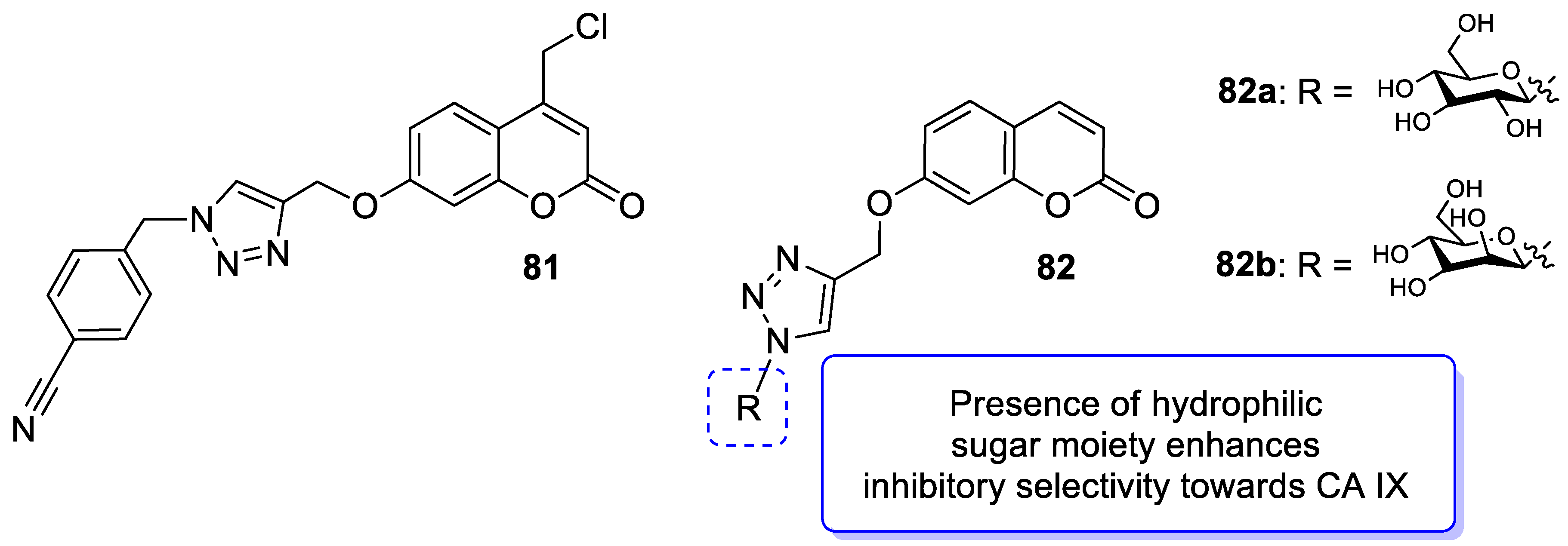
 | Anticancer | CA IX | 82a | [167] |
 | Anticancer | CA IX | 82b | [167] |
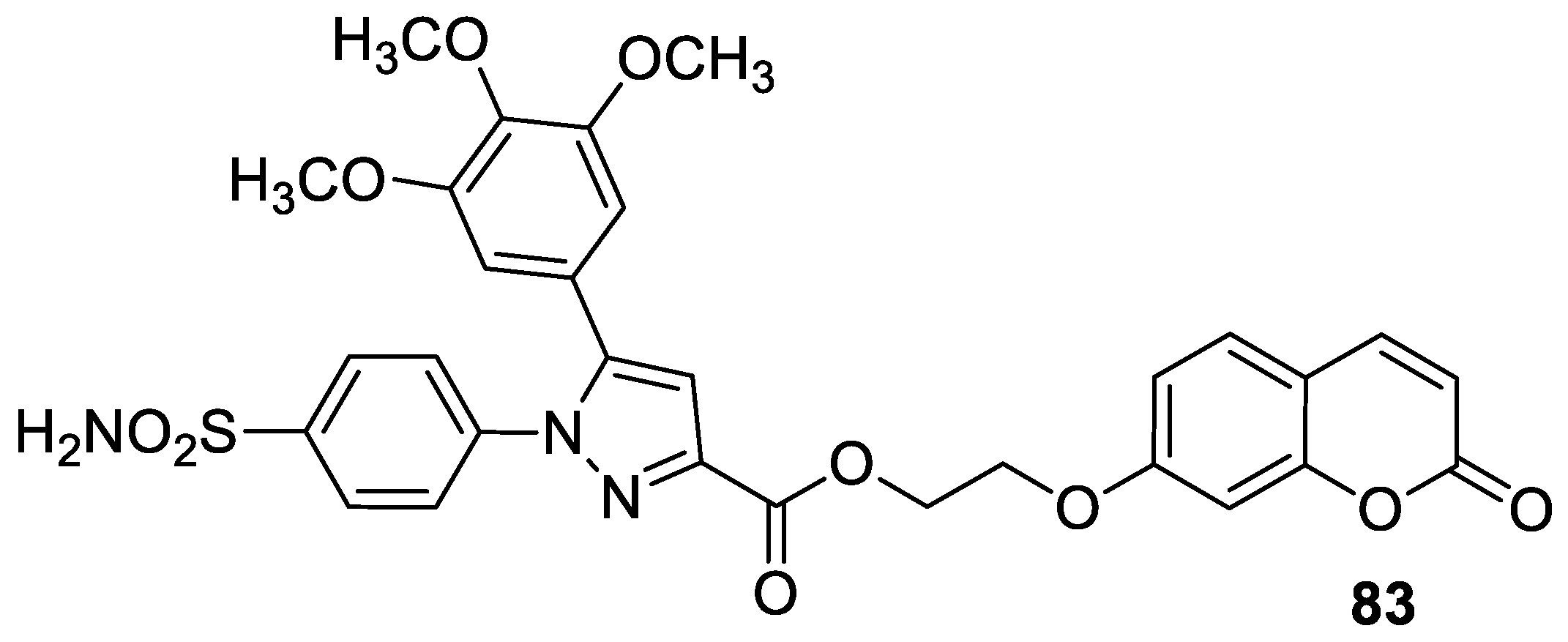
 | Anticancer | COX-2, 5-LOX, and lung carcinoma (A549) | 83 | [174] |
 | Anticancer | Androgen receptor (AR) and prostate adenocarcinoma | 85 | [177] |
 | Anticancer | ERK signal pathway, colorectal cancer (HCT-116), HepG-2 (hepatocellular carcinoma), and non-small cell lung cancer (A549) | 92 | [182] |
 | Fluorescent sensor | MIF tautomerase active site | 102 | [189] |
 | Two-photon ratiometric probe | γ-glutamyl transferase (GGT) | 103 | [190] |
 | Fluorescent non-canonical amino acid (fNCAA) | Acceptor of FRET in HTS or monitoring of drug metabolites | 108 | [194] |
 | Fluorescent probe | H2O2 | 109 | [201] |
 | Fluorescent probe | Hg2+ | 111 | [203] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kornicka, A.; Balewski, Ł.; Lahutta, M.; Kokoszka, J. Umbelliferone and Its Synthetic Derivatives as Suitable Molecules for the Development of Agents with Biological Activities: A Review of Their Pharmacological and Therapeutic Potential. Pharmaceuticals 2023, 16, 1732. https://doi.org/10.3390/ph16121732
Kornicka A, Balewski Ł, Lahutta M, Kokoszka J. Umbelliferone and Its Synthetic Derivatives as Suitable Molecules for the Development of Agents with Biological Activities: A Review of Their Pharmacological and Therapeutic Potential. Pharmaceuticals. 2023; 16(12):1732. https://doi.org/10.3390/ph16121732
Chicago/Turabian StyleKornicka, Anita, Łukasz Balewski, Monika Lahutta, and Jakub Kokoszka. 2023. "Umbelliferone and Its Synthetic Derivatives as Suitable Molecules for the Development of Agents with Biological Activities: A Review of Their Pharmacological and Therapeutic Potential" Pharmaceuticals 16, no. 12: 1732. https://doi.org/10.3390/ph16121732
APA StyleKornicka, A., Balewski, Ł., Lahutta, M., & Kokoszka, J. (2023). Umbelliferone and Its Synthetic Derivatives as Suitable Molecules for the Development of Agents with Biological Activities: A Review of Their Pharmacological and Therapeutic Potential. Pharmaceuticals, 16(12), 1732. https://doi.org/10.3390/ph16121732