

Kaurenoic Acid Reduces Ongoing Chronic Constriction Injury-Induced Neuropathic Pain: Nitric Oxide Silencing of Dorsal Root Ganglia Neurons

 , , ,
, , ,  ,
,  and
and 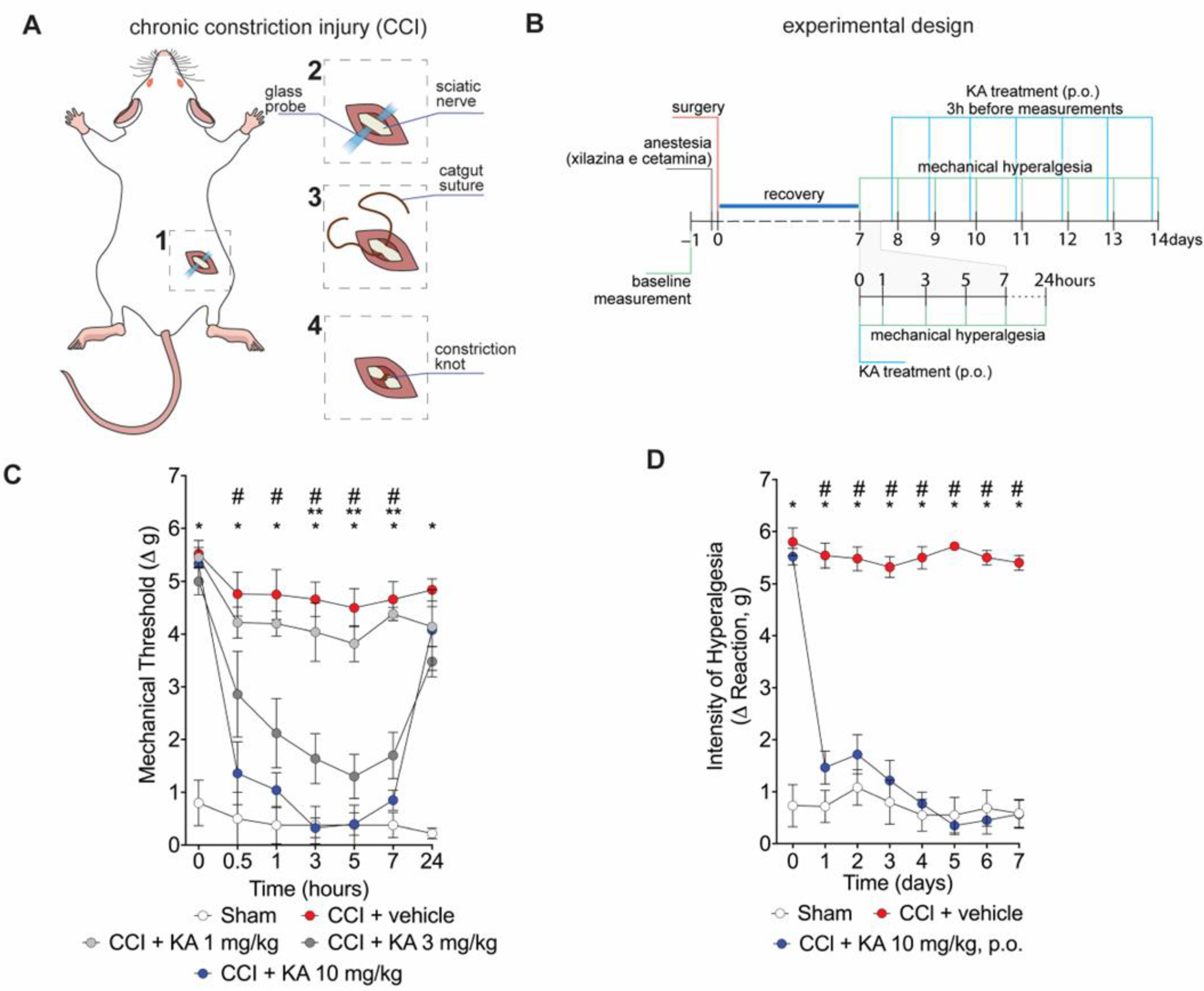{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. KA Reduces CCI-Induced Mechanical Hyperalgesia in Mice
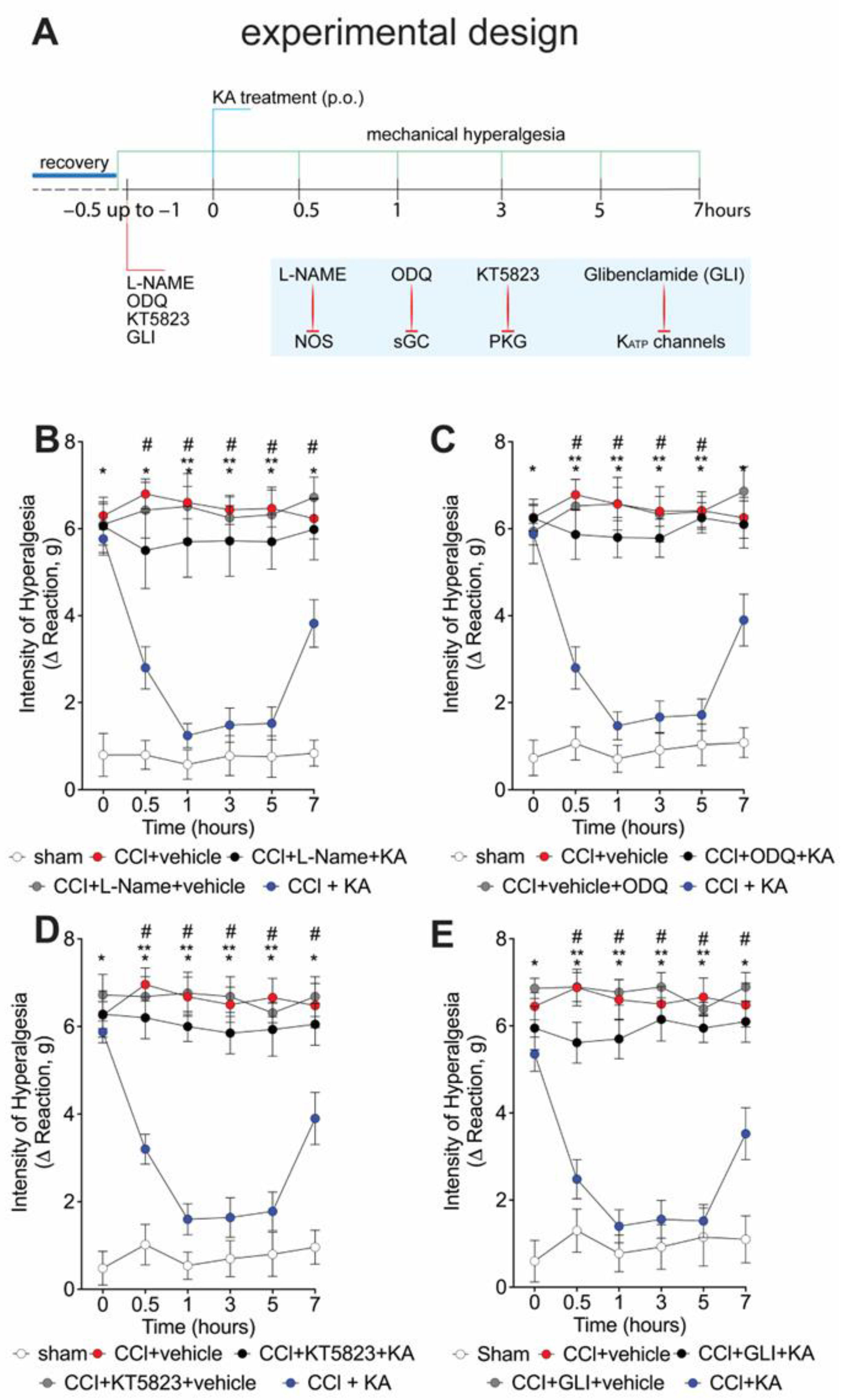
2.2. The Analgesic Effect of KA in CCI-Induced Mechanical Hyperalgesia Depends on Activating the NO-cGMP-PKG-ATP-Sensitive Potassium Channel Signaling Pathway
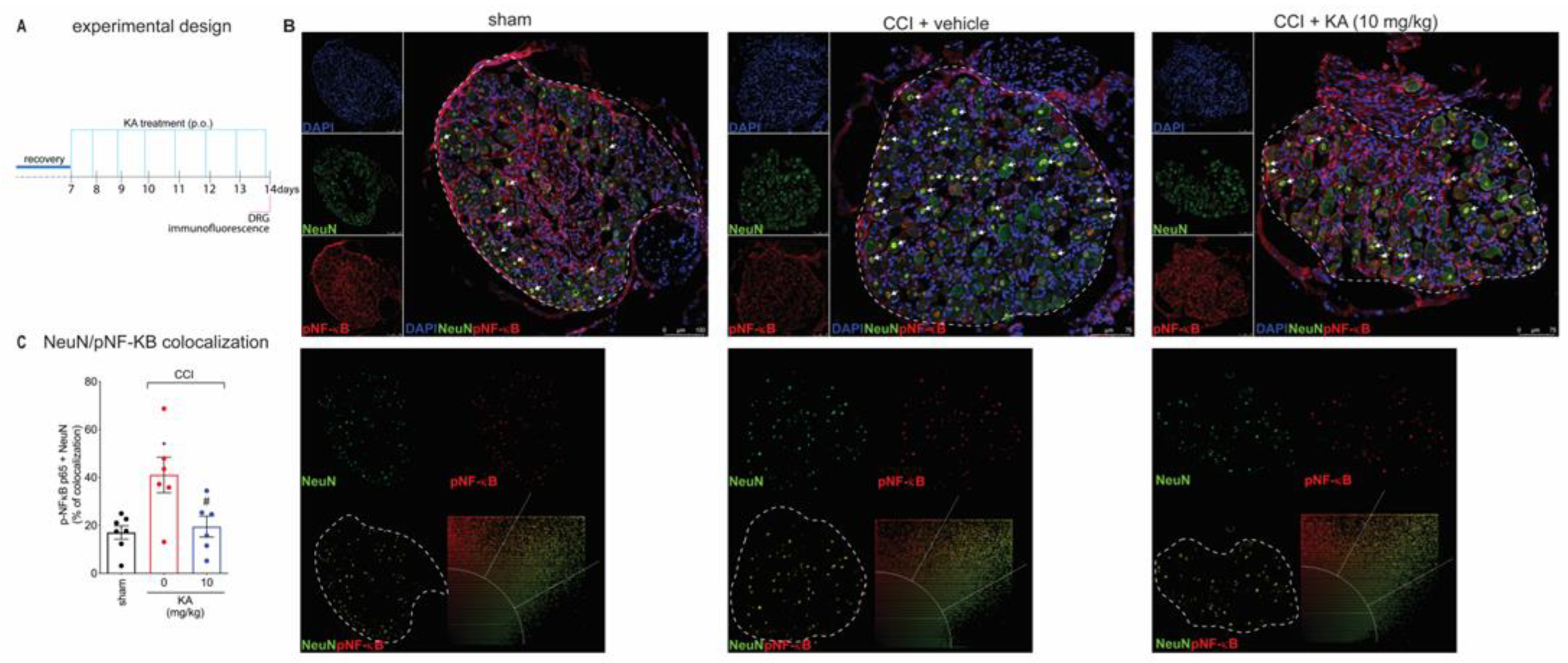
2.3. KA Reduces CCI-Induced Dorsal Root Ganglia (DRG) Activation
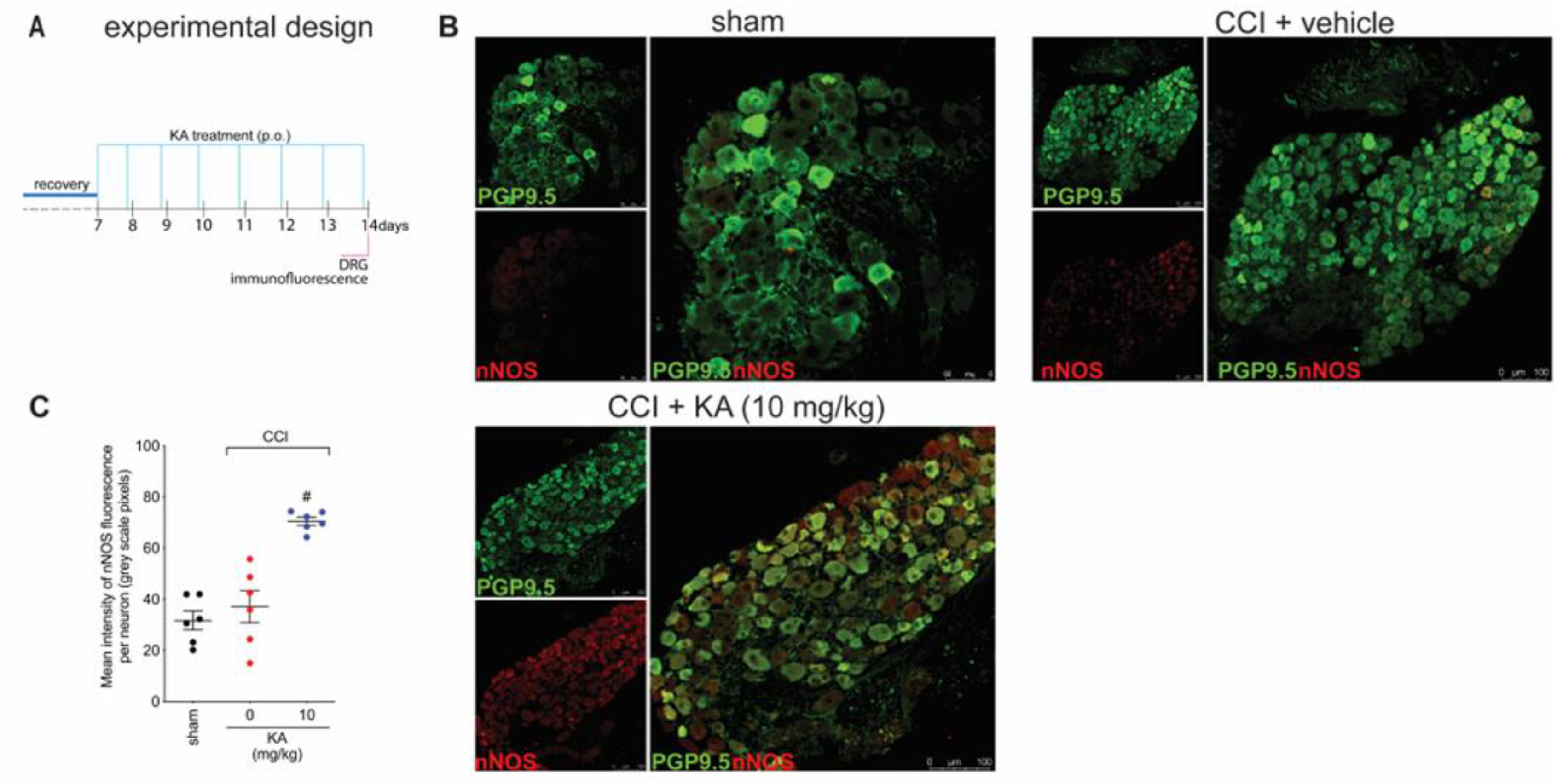
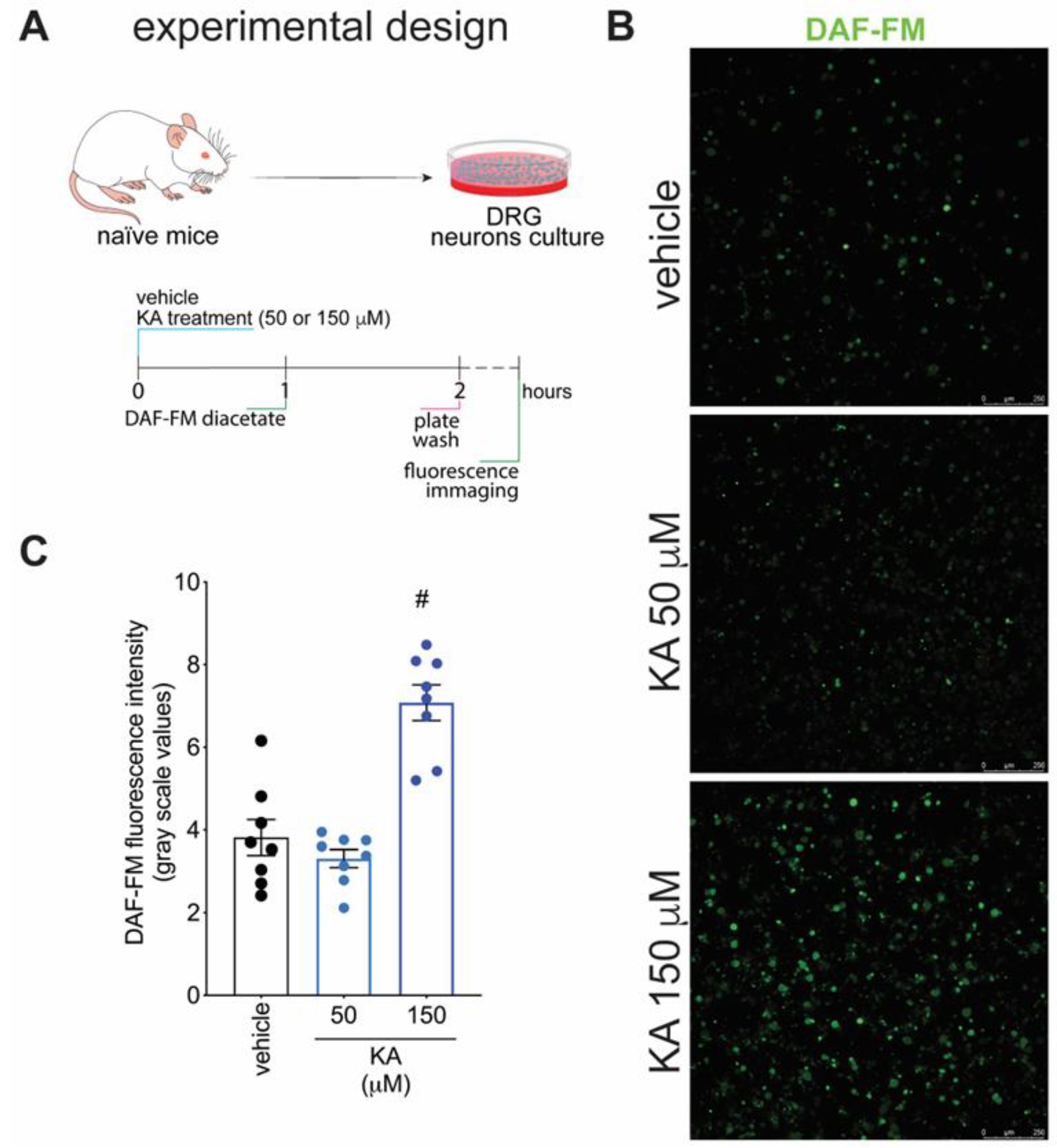
2.4. KA Increases the Expression of Neuronal Nitric Oxide Synthase (nNOS) In Vivo and the Production of NO in DRG Neuron Culture (In Vitro)
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Extraction and Isolation of Kaurenoic Acid
4.3. Drugs and Stimuli
4.4. Model of Chronic Constriction Injury (CCI)
4.5. Experimental Protocols
4.6. Mechanical Hyperalgesia
4.7. Immunofluorescence Staining
4.8. Intracellular Nitric Oxide Detection
4.9. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gao, Y.J.; Ji, R.R. Chemokines, Neuronal-Glial Interactions, and Central Processing of Neuropathic Pain. Pharmacol. Ther. 2010, 126, 56–68. [Google Scholar] [CrossRef] [Green Version]
- Woolf, C.J.; Mannion, R.J. Neuropathic Pain: Aetiology, Symptoms, Mechanisms, and Management. Lancet 1999, 353, 1959–1964. [Google Scholar] [CrossRef]
- Campbell, J.N.; Meyer, R.A. Mechanisms of Neuropathic Pain. Neuron 2006, 52, 77–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinho-Ribeiro, F.A.; Verri, W.A.; Chiu, I.M. Nociceptor Sensory Neuron–Immune Interactions in Pain and Inflammation. Trends Immunol. 2017, 38, 5–19. [Google Scholar] [CrossRef] [Green Version]
- Austin, P.J.; Moalem-Taylor, G. The Neuro-Immune Balance in Neuropathic Pain: Involvement of Inflammatory Immune Cells, Immune-like Glial Cells and Cytokines. J. Neuroimmunol. 2010, 229, 26–50. [Google Scholar] [CrossRef] [PubMed]
- Pottorf, T.S.; Rotterman, T.M.; McCallum, W.M.; Haley-Johnson, Z.A.; Alvarez, F.J. The Role of Microglia in Neuroinflammation of the Spinal Cord after Peripheral Nerve Injury. Cells 2022, 11, 2083. [Google Scholar] [CrossRef]
- Scholz, J.; Woolf, C.J. The Neuropathic Pain Triad: Neurons, Immune Cells and Glia. Nat. Neurosci. 2007, 10, 1361–1368. [Google Scholar] [CrossRef] [PubMed]
- Colloca, L.; Ludman, T.; Bouhassira, D.; Baron, R.; Dickenson, A.H.; Yarnitsky, D.; Freeman, R.; Truini, A.; Attal, N.; Finnerup, N.B.; et al. Neuropathic Pain. Nat. Rev. Dis. Prim. 2017, 3, 17002. [Google Scholar] [CrossRef] [Green Version]
- Cherif, F.; Zouari, H.G.; Cherif, W.; Hadded, M.; Cheour, M.; Damak, R. Depression Prevalence in Neuropathic Pain and Its Impact on the Quality of Life. Pain Res. Manag. 2020, 2020, 7408508. [Google Scholar] [CrossRef]
- Zarpelon, A.C.; Rodrigues, F.C.; Lopes, A.H.; Souza, G.R.; Carvalho, T.T.; Pinto, L.G.; Xu, D.; Ferreira, S.H.; Alves-Filho, J.C.; McInnes, I.B.; et al. Spinal Cord Oligodendrocyte-Derived Alarmin IL-33 Mediates Neuropathic Pain. FASEB J. 2016, 30, 54–65. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves dos Santos, G.; Delay, L.; Yaksh, T.L.; Corr, M. Neuraxial Cytokines in Pain States. Front. Immunol. 2020, 10, 3061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dworkin, R.H.; Backonja, M.; Rowbotham, M.C.; Allen, R.R.; Argoff, C.R.; Bennett, G.J.; Bushnell, M.C.; Farrar, J.T.; Galer, B.S.; Haythornthwaite, J.A.; et al. Advances in Neuropathic Pain: Diagnosis, Mechanisms, and Treatment Recommendations. Arch. Neurol. 2003, 60, 1524–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Velzen, M.; Dahan, A.; Niesters, M. Neuropathic Pain: Challenges and Opportunities. Front. Pain Res. 2020, 1, 1. [Google Scholar] [CrossRef] [PubMed]
- Dworkin, R.H.; O’Connor, A.B.; Audette, J.; Baron, R.; Gourlay, G.K.; Haanpää, M.L.; Kent, J.L.; Krane, E.J.; LeBel, A.A.; Levy, R.M.; et al. Recommendations for the Pharmacological Management of Neuropathic Pain: An Overview and Literature Update. Mayo Clin. Proc. 2010, 85, S3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tirapelli, C.R.; Ambrosio, S.R.; da Costa, F.B.; de Oliveira, A.M. Diterpenes: A Therapeutic Promise for Cardiovascular Diseases. Recent Pat. Cardiovasc. Drug Discov. 2008, 3, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Aquila, S.; Weng, Z.Y.; Zeng, Y.Q.; Sun, H.D.; Ríos, J.L. Inhibition of NF-ΚB Activation and INOS Induction by Ent-Kaurane Diterpenoids in LPS-Stimulated RAW264.7 Murine Macrophages. J. Nat. Prod. 2009, 72, 1269–1272. [Google Scholar] [CrossRef] [PubMed]
- Lizarte Neto, F.S.; Tirapelli, D.P.C.; Ambrosio, S.R.; Tirapelli, C.R.; Oliveira, F.M.; Novais, P.C.; Peria, F.M.; Oliveira, H.F.; Carlotti Junior, C.G.; Tirapelli, L.F. Kaurene Diterpene Induces Apoptosis in U87 Human Malignant Glioblastoma Cells by Suppression of Anti-Apoptotic Signals and Activation of Cysteine Proteases. Brazilian J. Med. Biol. Res. Rev. Bras. Pesqui. Med. Biol. 2013, 46, 71–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, G.E.; Adams, L.S.; Rosales, J.C.; Jacobs, H.; Heber, D.; Seeram, N.P. Kaurene Diterpenes from Laetia Thamnia Inhibit the Growth of Human Cancer Cells in Vitro. Cancer Lett. 2006, 244, 190–194. [Google Scholar] [CrossRef]
- Fernandes, V.C.; Pereira, S.I.V.; Coppede, J.; Martins, J.S.; Rizo, W.F.; Beleboni, R.O.; Marins, M.; Pereira, P.S.; Pereira, A.M.S.; Fachin, A.L. The Epimer of Kaurenoic Acid from Croton Antisyphiliticus Is Cytotoxic toward B-16 and HeLa Tumor Cells through Apoptosis Induction. Genet. Mol. Res. 2013, 12, 1005–1011. [Google Scholar] [CrossRef]
- Borghi, S.M.; Domiciano, T.P.; Rasquel-Oliveira, F.S.; Ferraz, C.R.; Bussmann, A.J.C.; Vignoli, J.A.; Camilios-Neto, D.; Ambrósio, S.R.; Arakawa, N.S.; Casagrande, R.; et al. Sphagneticola Trilobata (L.) Pruski-Derived Kaurenoic Acid Prevents Ovalbumin-Induced Asthma in Mice: Effect on Th2 Cytokines, STAT6/GATA-3 Signaling, NFκB/Nrf2 Redox Sensitive Pathways, and Regulatory T Cell Phenotype Markers. J. Ethnopharmacol. 2022, 283, 114708. [Google Scholar] [CrossRef]
- Tirapelli, C.R.; Ambrosio, S.R.; Da Costa, F.B.; Coutinho, S.T.; De Oliveira, D.C.R.; De Oliveira, A.M. Analysis of the Mechanisms Underlying the Vasorelaxant Action of Kaurenoic Acid in the Isolated Rat Aorta. Eur. J. Pharmacol. 2004, 492, 233–241. [Google Scholar] [CrossRef]
- Borghi, S.M.; Mizokami, S.S.; Carvalho, T.T.; Rasquel-Oliveira, F.S.; Ferraz, C.R.; Fattori, V.; Hayashida, T.H.; Peron, J.P.S.; Camilios-Neto, D.; Ambrosio, S.R.; et al. The Diterpene from Sphagneticola Trilobata (L.) Pruski, Kaurenoic Acid, Reduces Lipopolysaccharide-Induced Peritonitis and Pain in Mice. J. Ethnopharmacol. 2021, 273, 113980. [Google Scholar] [CrossRef]
- Mizokami, S.S.; Arakawa, N.S.; Ambrosio, S.R.; Zarpelon, A.C.; Casagrande, R.; Cunha, T.M.; Ferreira, S.H.; Cunha, F.Q.; Verri, W.A. Kaurenoic Acid from Sphagneticola Trilobata Inhibits Inflammatory Pain: Effect on Cytokine Production and Activation of the NO-Cyclic GMP-Protein Kinase G-ATP-Sensitive Potassium Channel Signaling Pathway. J. Nat. Prod. 2012, 75, 896–904. [Google Scholar] [CrossRef]
- Boller, S.; Soldi, C.; Marques, M.C.A.; Santos, E.P.; Cabrini, D.A.; Pizzolatti, M.G.; Zampronio, A.R.; Otuki, M.F. Anti-Inflammatory Effect of Crude Extract and Isolated Compounds from Baccharis Illinita DC in Acute Skin Inflammation. J. Ethnopharmacol. 2010, 130, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Block, L.C.; Santos, A.R.S.; De Souza, M.M.; Scheidt, C.; Yunes, R.A.; Santos, M.A.; Monache, F.D.; Filho, V.C. Chemical and Pharmacological Examination of Antinociceptive Constituents of Wedelia Paludosa. J. Ethnopharmacol. 1998, 61, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Sachs, D.; Cunha, F.Q.; Ferreira, S.H. Peripheral Analgesic Blockade of Hypernociception: Activation of Arginine/NO/CGMP/Protein Kinase G/ATP-Sensitive K+ Channel Pathway. Proc. Natl. Acad. Sci. USA 2004, 101, 3680–3685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunha, T.M.; Roman-Campos, D.; Lotufo, C.M.; Duarte, H.L.; Souza, G.R.; Verri, W.A., Jr.; Souza, G.R.; Funez, M.I.; Dias, Q.M.; Schivo, I.R.; et al. Morphine Peripheral Analgesia Depends on Activation of the PI3Kgamma/AKT/NNOS/NO/KATP Signaling Pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 4442–4447. [Google Scholar] [CrossRef] [Green Version]
- Lima, F.O.; Souza, G.R.; Verri, W.A., Jr.; Parada, C.A.; Ferreira, S.H.; Cunha, F.Q.; Cunha, T.M. Direct Blockade of Inflammatory Hypernociception by Peripheral A1 Adenosine Receptors: Involvement of the NO/CGMP/PKG/KATP Signaling Pathway. Pain 2010, 151, 506–515. [Google Scholar] [CrossRef]
- Finnerup, N.B.; Kuner, R.; Jensen, T.S. Neuropathic Pain: Frommechanisms to Treatment. Physiol. Rev. 2021, 101, 259–301. [Google Scholar] [CrossRef]
- McLachlan, E.M.; Jänig, W.; Devor, M.; Michaelis, M. Peripheral Nerve Injury Triggers Noradrenergic Sprouting within Dorsal Root Ganglia. Nature 1993, 363, 543–546. [Google Scholar] [CrossRef]
- Kawano, T.; Zoga, V.; Kimura, M.; Liang, M.Y.; Wu, H.E.; Gemes, G.; Bruce, J.B.; Kwok, W.M.; Hogan, Q.H.; Sarantopoulos, C.D. Nitric Oxide Activates ATP-Sensitive Potassium Channels in Mammalian Sensory Neurons: Action by Direct S-Nitrosylation. Mol. Pain 2009, 5, 12. [Google Scholar] [CrossRef] [Green Version]
- Brito, G.A.C.; Sachs, D.; Cunha, F.Q.; Vale, M.L.; Lotufo, C.M.C.; Ferreira, S.H.; Ribeiro, R.A. Peripheral Antinociceptive Effect of Pertussis Toxin: Activation of the Arginine/NO/CGMP/PKG/ATP-Sensitive K Channel Pathway. Eur. J. Neurosci. 2006, 24, 1175–1181. [Google Scholar] [CrossRef]
- Keilhoff, G.; Fansa, H.; Wolf, G. Neuronal Nitric Oxide Synthase Is the Dominant Nitric Oxide Supplier for the Survival of Dorsal Root Ganglia after Peripheral Nerve Axotomy. J. Chem. Neuroanat. 2002, 24, 181–187. [Google Scholar] [CrossRef]
- Choi, R.J.; Shin, E.M.; Jung, H.A.; Choi, J.S.; Kim, Y.S. Inhibitory Effects of Kaurenoic Acid from Aralia Continentalis on LPS-Induced Inflammatory Response in RAW264.7 Macrophages. Phytomedicine 2011, 18, 677–682. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-ΚB Signaling in Inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Hartung, J.E.; Eskew, O.; Wong, T.; Tchivileva, I.E.; Oladosu, F.A.; O’Buckley, S.C.; Nackley, A.G. Nuclear Factor-Kappa B Regulates Pain and COMT Expression in a Rodent Model of Inflammation. Brain. Behav. Immun. 2015, 50, 196–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Li, X.; Zhou, Y.; Wang, Y.; Liu, X. Exogenous TIPE2 Inhibit TAK1 to Improve Inflammation and Neuropathic Pain Induced by Sciatic Nerve Injury Through Inactivating NF-ΚB and JNK. Neurochem. Res. 2022, 47, 3167–3177. [Google Scholar] [CrossRef]
- Zaninelli, T.H.; Fattori, V.; Saraiva-Santos, T.; Badaro-Garcia, S.; Staurengo-Ferrari, L.; Andrade, K.C.; Artero, N.A.; Ferraz, C.R.; Bertozzi, M.M.; Rasquel-Oliveira, F.; et al. RvD1 Disrupts Nociceptor Neuron and Macrophage Activation, and Neuroimmune Communication Reducing Pain and Inflammation in Gouty Arthritis in Mice. Br. J. Pharmacol. 2022, 179, 4500–4515. [Google Scholar] [CrossRef] [PubMed]
- Soylemezoglu, F.; Onder, S.; Tezel, G.G.; Berker, M. Neuronal Nuclear Antigen (NeuN): A New Tool in the Diagnosis of Central Neurocytoma. Pathol. Res. Pract. 2003, 199, 463–468. [Google Scholar] [CrossRef]
- Paiva, L.A.; Gurgel, L.A.; Silva, R.M.; Tome, A.R.; Gramosa, N.V.; Silveira, E.R.; Santos, F.A.; Rao, V.S. Anti-Inflammatory Effect of Kaurenoic Acid, a Diterpene from Copaifera Langsdorffi on Acetic Acid-Induced Colitis in Rats. Vasc. Pharmacol. 2002, 39, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Kingery, W.S.; Vallin, J.A. The Development of Chronic Mechanical Hyperalgesia, Autotomy and Collateral Sprouting Following Sciatic Nerve Section in Rat. Pain 1989, 38, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Cury, Y.; Picolo, G.; Gutierrez, V.P.; Ferreira, S.H. Pain and Analgesia: The Dual Effect of Nitric Oxide in the Nociceptive System. Nitric Oxide 2011, 25, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Cano, B.L.; Moreira, M.R.; Goulart, M.O.; dos Santos Gonçalves, N.; Veneziani, R.C.S.; Bastos, J.K.; Ambrósio, S.R.; dos Santos, R.A. Comparative Study of the Cytotoxicity and Genotoxicity of Kaurenoic Acid and Its Semi-Synthetic Derivatives Methoxy Kaurenoic Acid and Kaurenol in CHO-K1 Cells. Food Chem. Toxicol. 2017, 102, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Vargas, F.D.S.; De Almeida, P.D.O.; Aranha, E.S.P.; Boleti, A.P.D.A.; Newton, P.; De Vasconcellos, M.C.; Veiga, V.F.; Lima, E.S. Biological Activities and Cytotoxicity of Diterpenes from Copaifera Spp. Oleoresins. Molecules 2015, 20, 6194–6210. [Google Scholar] [CrossRef] [Green Version]
- da Costa, R.M.; Bastos, J.K.; Costa, M.C.A.; Ferreira, M.M.C.; Mizuno, C.S.; Caramori, G.F.; Nagurniak, G.R.; Simão, M.R.; dos Santos, R.A.; Veneziani, R.C.S.; et al. In Vitro Cytotoxicity and Structure-Activity Relationship Approaches of Ent-Kaurenoic Acid Derivatives against Human Breast Carcinoma Cell Line. Phytochemistry 2018, 156, 214–223. [Google Scholar] [CrossRef]
- Costa-Lotufo, L.V.; Cunha, G.M.A.; Farias, P.A.M.; Viana, G.S.B.; Cunha, K.M.A.; Pessoa, C.; Moraes, M.O.; Silveira, E.R.; Gramosa, N.V.; Rao, V.S.N. The Cytotoxic and Embryotoxic Effects of Kaurenoic Acid, a Diterpene Isolated from Copaifera Langsdorffii Oleo-Resin. Toxicon 2002, 40, 1231–1234. [Google Scholar] [CrossRef]
- Bürger, C.; Fischer, D.R.; Cordenunzzi, A.; Paula De Borba Batschauer, A.; Filho, C.; Roberto, A.; Soares, S. Acute and Subacute Toxicity of the Hydroalcoholic Extract from Wedelia Paludosa (Acmela Brasiliensis) (Asteraceae) in Mice. J Pharm Pharm. Sci 2005, 8, 370–373. [Google Scholar]
- Marcondes-Alves, L.; Fattori, V.; Borghi, S.M.; Lourenco-Gonzalez, Y.; Bussmann, A.J.C.; Hirooka, E.Y.; Casagrande, R.; Verri, W.A., Jr.; Arakawa, N.S. Kaurenoic Acid Extracted from Sphagneticola Trilobata Reduces Acetaminophen-Induced Hepatotoxicity through Inhibition of Oxidative Stress and pro-Inflammatory Cytokine Production in Mice. Nat. Prod. Res. 2019, 33, 921–924. [Google Scholar] [CrossRef]
- Okuyama, E.; Nishimura, S.; Yamazaki, M. Analgesic Principles from Aralia Cordata Thunb. Chem. Pharm. Bull. 1991, 39, 405–407. [Google Scholar] [CrossRef] [Green Version]
- De Andrade, B.B.; Moreira, M.R.; Ambrosio, S.R.; Furtado, N.A.J.C.; Cunha, W.R.; Heleno, V.C.G.; Silva, A.N.; Simão, M.R.; Da Rocha, E.M.P.; Martins, C.H.G.; et al. Evaluation of Ent-Kaurenoic Acid Derivatives for Their Anticariogenic Activity. Nat. Prod. Commun. 2011, 6, 777–780. [Google Scholar] [CrossRef] [Green Version]
- Bennett, G.J.; Xie, Y.K. A Peripheral Mononeuropathy in Rat That Produces Disorders of Pain Sensation like Those Seen in Man. Pain 1988, 33, 87–107. [Google Scholar] [CrossRef] [PubMed]
- Perner, C.; Sokol, C.L. Protocol for Dissection and Culture of Murine Dorsal Root Ganglia Neurons to Study Neuropeptide Release. STAR Protoc. 2021, 2, 100333. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.J.; Choi, G.W.; Yang, S.J.; Lee, Y.B.; Cho, H.Y. Pharmacokinetic Profile of Kaurenoic Acid after Oral Administration of Araliae Continentalis Radix Extract Powder to Humans. Pharmaceutics 2018, 10, 253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdfelder, E.; FAul, F.; Buchner, A.; Lang, A.G. Statistical Power Analyses Using G*Power 3.1: Tests for Correlation and Regression Analyses. Behav. Res. Methods 2009, 41, 1149–1160. [Google Scholar] [CrossRef] [Green Version]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaninelli, T.H.; Mizokami, S.S.; Bertozzi, M.M.; Saraiva-Santos, T.; Pinho-Ribeiro, F.A.; de Oliveira, G.I.; Streck, R.; Araújo, E.J.A.; Arakawa, N.S.; Borghi, S.M.; et al. Kaurenoic Acid Reduces Ongoing Chronic Constriction Injury-Induced Neuropathic Pain: Nitric Oxide Silencing of Dorsal Root Ganglia Neurons. Pharmaceuticals 2023, 16, 343. https://doi.org/10.3390/ph16030343
Zaninelli TH, Mizokami SS, Bertozzi MM, Saraiva-Santos T, Pinho-Ribeiro FA, de Oliveira GI, Streck R, Araújo EJA, Arakawa NS, Borghi SM, et al. Kaurenoic Acid Reduces Ongoing Chronic Constriction Injury-Induced Neuropathic Pain: Nitric Oxide Silencing of Dorsal Root Ganglia Neurons. Pharmaceuticals. 2023; 16(3):343. https://doi.org/10.3390/ph16030343
Chicago/Turabian StyleZaninelli, Tiago H., Sandra S. Mizokami, Mariana M. Bertozzi, Telma Saraiva-Santos, Felipe A. Pinho-Ribeiro, Gabriele Inácio de Oliveira, Renata Streck, Eduardo J. A. Araújo, Nilton S. Arakawa, Sergio M. Borghi, and et al. 2023. "Kaurenoic Acid Reduces Ongoing Chronic Constriction Injury-Induced Neuropathic Pain: Nitric Oxide Silencing of Dorsal Root Ganglia Neurons" Pharmaceuticals 16, no. 3: 343. https://doi.org/10.3390/ph16030343
APA StyleZaninelli, T. H., Mizokami, S. S., Bertozzi, M. M., Saraiva-Santos, T., Pinho-Ribeiro, F. A., de Oliveira, G. I., Streck, R., Araújo, E. J. A., Arakawa, N. S., Borghi, S. M., Casagrande, R., & Verri, W. A. (2023). Kaurenoic Acid Reduces Ongoing Chronic Constriction Injury-Induced Neuropathic Pain: Nitric Oxide Silencing of Dorsal Root Ganglia Neurons. Pharmaceuticals, 16(3), 343. https://doi.org/10.3390/ph16030343