
Targeting Protein-Protein Interactions to Inhibit Cyclin-Dependent Kinases
Abstract
:1. Introduction
2. Biology and Structure of CDKs
3. Examples of PPI Inhibition in CDKs
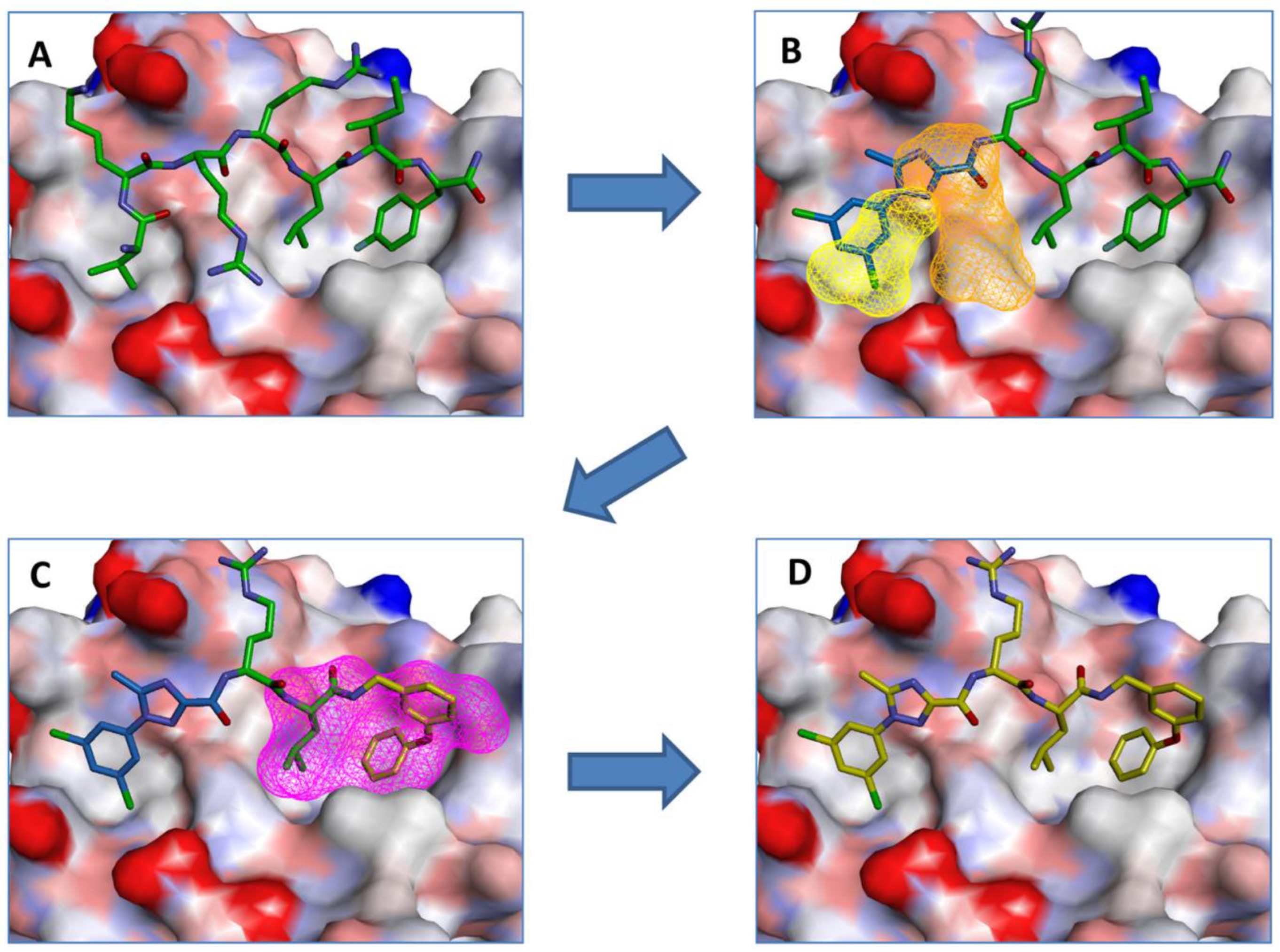
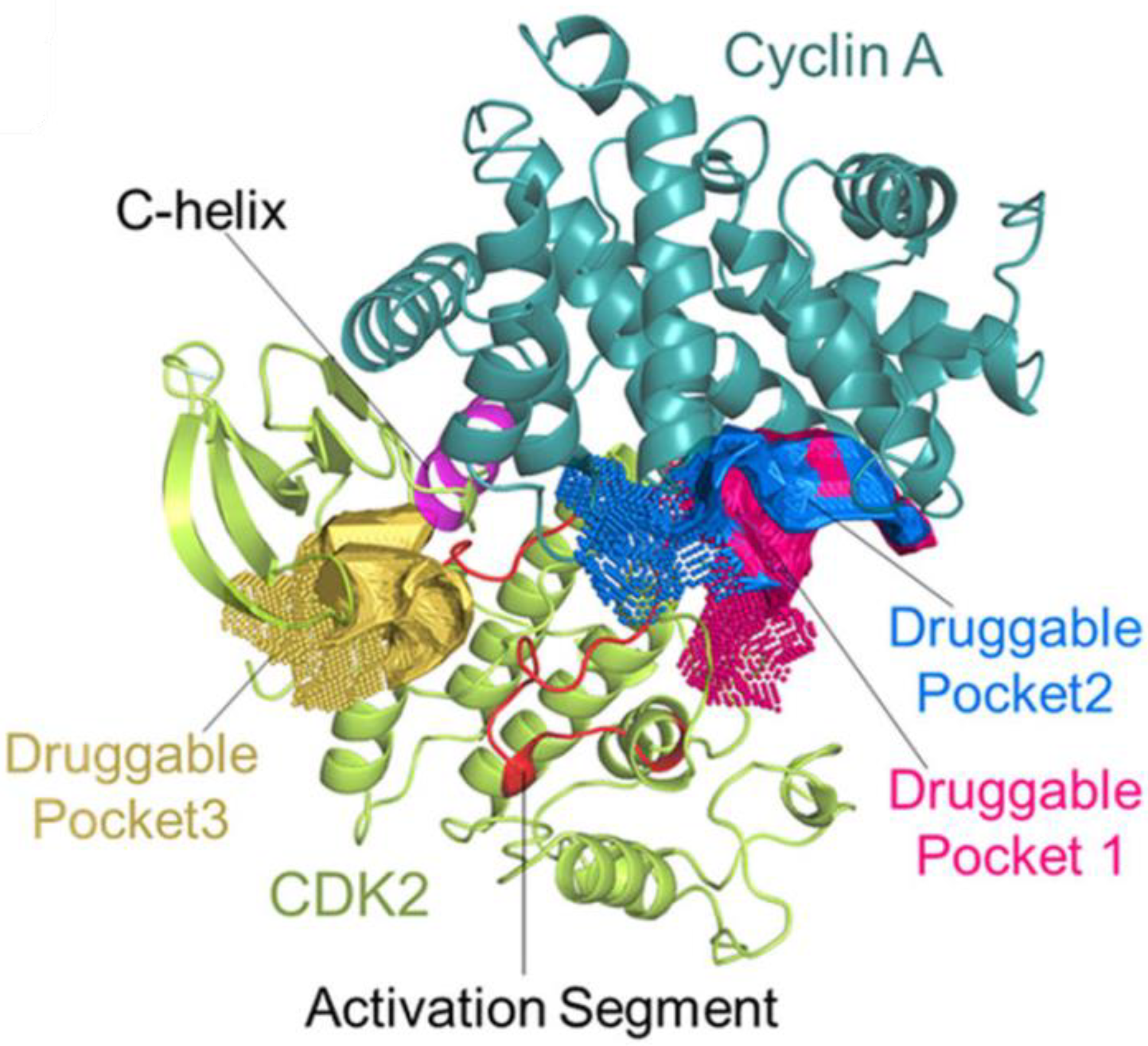
3.1. CDK2/Cyclin A
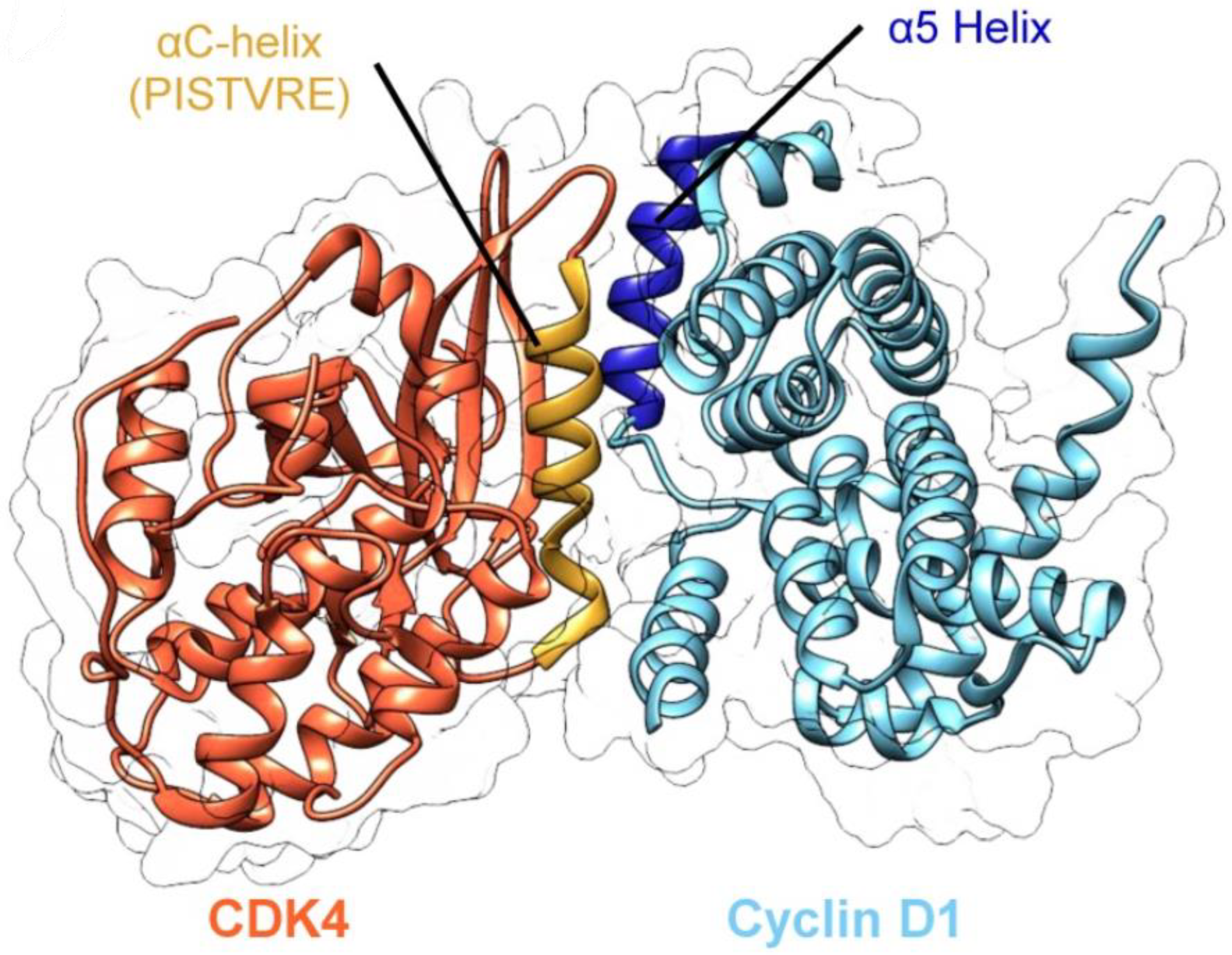
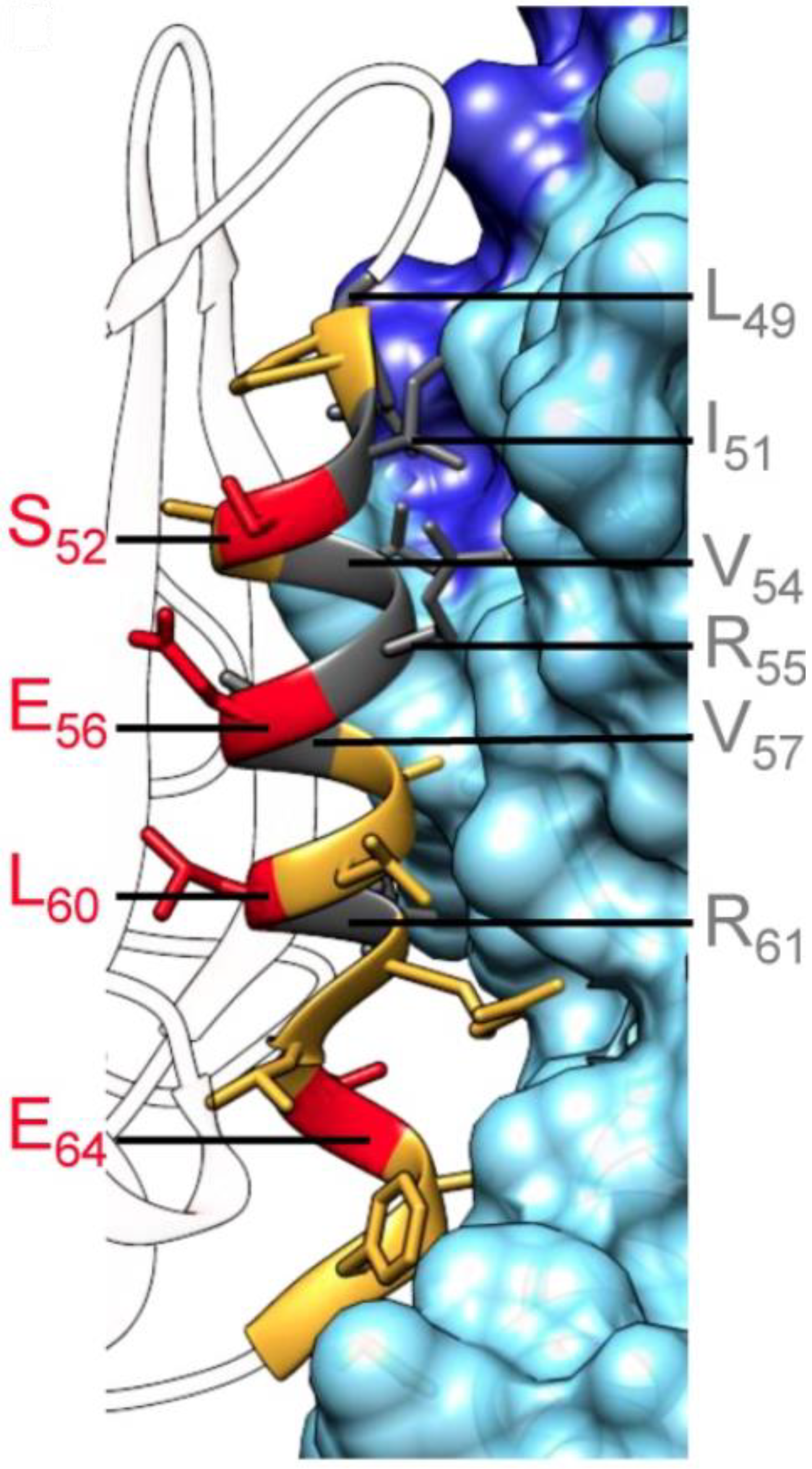
3.2. CDK4/Cyclin D
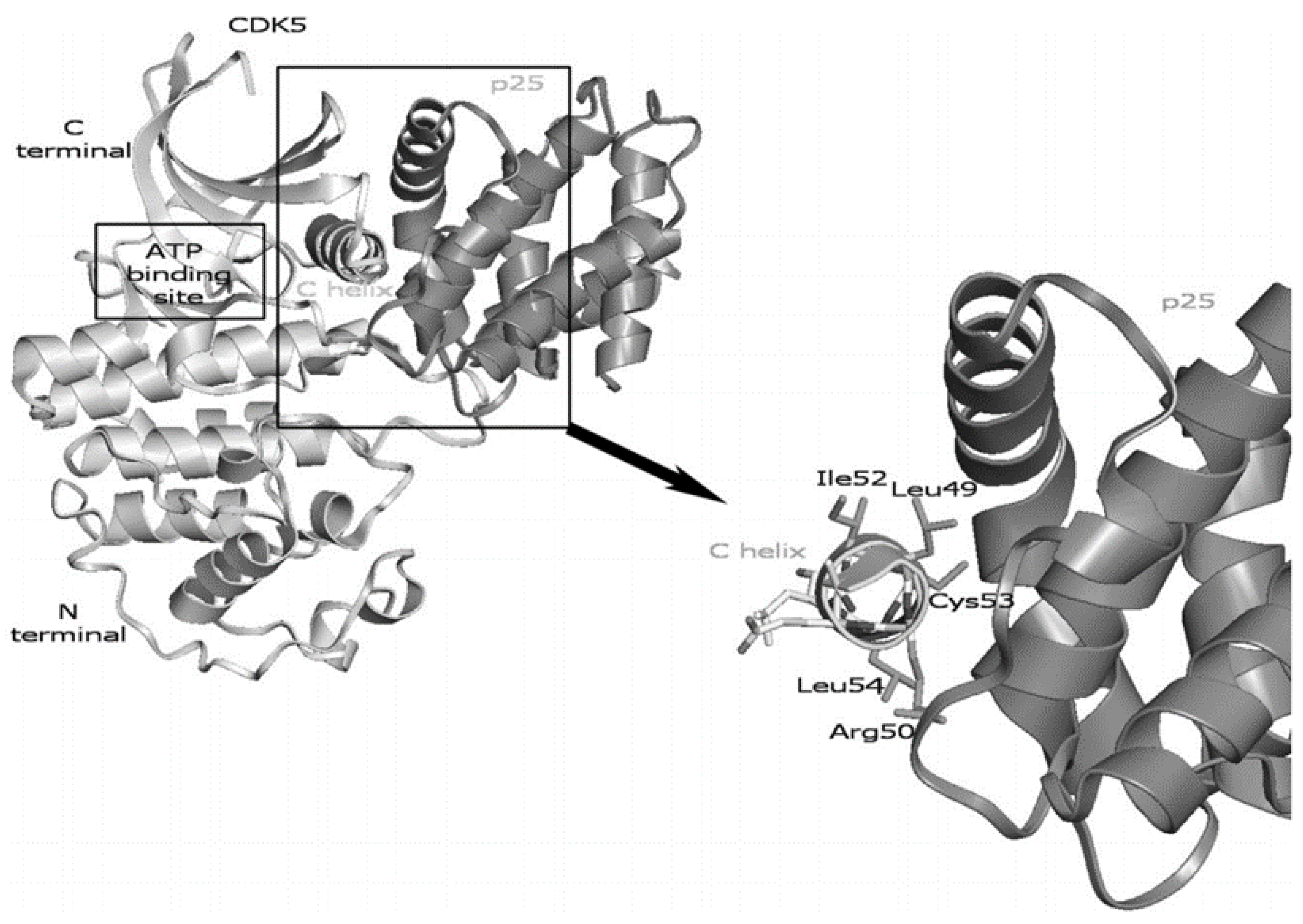
3.3. CDK5/p25
3.4. CDK9/Cyclin T
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Choi, Y.J.; Anders, L. Signaling through cyclin D-dependent kinases. Oncogene 2014, 33, 1890–1903. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.K.; Diehl, J.A. Nuclear cyclin D1: An oncogenic driver in human cancer. J. Cell. Physiol. 2009, 220, 292–296. [Google Scholar] [CrossRef] [Green Version]
- Swanton, C. Cell-cycle targeted therapies. Lancet Oncol. 2004, 5, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Crown, J.P.; Lang, I.; Boer, K.; Bondarenko, I.M.; Kulyk, S.O.; Ettl, J.; Patel, R.; Pinter, T.; Schmidt, M.; et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): A randomised phase 2 study. Lancet Oncol. 2015, 16, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.S.; Sonke, G.S.; Hart, L.; Campone, M.; Petrakova, K.; Winer, E.P.; Janni, W.; et al. Overall Survival with Ribociclib plus Letrozole in Advanced Breast Cancer. N. Engl. J. Med. 2022, 386, 942–950. [Google Scholar] [CrossRef]
- Sledge, G.W., Jr.; Toi, M.; Neven, P.; Sohn, J.; Inoue, K.; Pivot, X.; Burdaeva, O.; Okera, M.; Masuda, N.; Kaufman, P.A.; et al. MONARCH 2: Abemaciclib in Combination with Fulvestrant in Women With HR+/HER2− Advanced Breast Cancer Who Had Progressed while Receiving Endocrine Therapy. J. Clin. Oncol. 2017, 35, 2875–2884. [Google Scholar] [CrossRef]
- Hart, L.L.; Ferrarotto, R.; Andric, Z.G.; Beck, J.T.; Subramanian, J.; Radosavljevic, D.Z.; Zaric, B.; Hanna, W.T.; Aljumaily, R.; Owonikoko, T.K.; et al. Myelopreservation with Trilaciclib in Patients Receiving Topotecan for Small Cell Lung Cancer: Results from a Randomized, Double-Blind, Placebo-Controlled Phase II Study. Adv. Ther. 2021, 38, 350–365. [Google Scholar] [CrossRef]
- Narasimha, A.M.; Kaulich, M.; Shapiro, G.S.; Choi, Y.J.; Sicinski, P.; Dowdy, S.F. Cyclin D activates the Rb tumor suppressor by mono-phosphorylation. Elife 2014, 3, e02872. [Google Scholar] [CrossRef] [PubMed]
- Cadoo, K.A.; Gucalp, A.; Traina, T.A. Palbociclib: An evidence-based review of its potential in the treatment of breast cancer. Breast Cancer 2014, 6, 123–133. [Google Scholar]
- Kalra, S.; Joshi, G.; Munshi, A.; Kumar, R. Structural insights of cyclin dependent kinases: Implications in design of selective inhibitors. Eur. J. Med. Chem. 2017, 142, 424–458. [Google Scholar] [CrossRef]
- Klein, M.A. Cyclin-dependent kinase inhibition: An opportunity to target protein-protein interactions. Adv. Protein Chem. Struct. Biol. 2020, 121, 115–141. [Google Scholar] [PubMed]
- Malumbres, M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Do, P.A.; Lee, C.H. The Role of CDK5 in Tumours and Tumour Microenvironments. Cancers 2020, 13, 101. [Google Scholar] [CrossRef]
- Mandal, R.; Becker, S.; Strebhardt, K. Targeting CDK9 for Anti-Cancer Therapeutics. Cancers 2021, 13, 2181. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.J.; Endicott, J.A. Structural insights into the functional diversity of the CDK-cyclin family. Open Biol. 2018, 8, 180112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Premnath, P.N.; Craig, S.N.; Liu, S.; McInnes, C. Benzamide capped peptidomimetics as non-ATP competitive inhibitors of CDK2 using the REPLACE strategy. Bioorganic Med. Chem. Lett. 2016, 26, 3754–3760. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Premnath, P.N.; Bolger, J.K.; Perkins, T.L.; Kirkland, L.O.; Kontopidis, G.; McInnes, C. Optimization of non-ATP competitive CDK/cyclin groove inhibitors through REPLACE-mediated fragment assembly. J. Med. Chem. 2013, 56, 1573–1582. [Google Scholar] [CrossRef] [Green Version]
- Premnath, P.N.; Liu, S.; Perkins, T.; Abbott, J.; Anderson, E.; McInnes, C. Fragment based discovery of arginine isosteres through REPLACE: Towards non-ATP competitive CDK inhibitors. Bioorganic Med. Chem. 2014, 22, 616–622. [Google Scholar] [CrossRef] [Green Version]
- Premnath, P.N.; Craig, S.; McInnes, C. Development of Inhibitors of Protein-protein Interactions through REPLACE: Application to the Design and Development Non-ATP Competitive CDK Inhibitors. J. Vis. Exp. 2015, 105, e52441. [Google Scholar]
- Kontopidis, G.; Andrews, M.J.; McInnes, C.; Cowan, A.; Powers, H.; Innes, L.; Plater, A.; Griffiths, G.; Paterson, D.; Zheleva, D.I.; et al. Insights into cyclin groove recognition: Complex crystal structures and inhibitor design through ligand exchange. Structure 2003, 11, 1537–1546. [Google Scholar] [CrossRef]
- Kontopidis, G.; Andrews, M.J.; McInnes, C.; Plater, A.; Innes, L.; Renachowski, S.; Cowan, A.; Fischer, P.M. Truncation and optimisation of peptide inhibitors of cyclin-dependent kinase 2-cyclin a through structure-guided design. ChemMedChem 2009, 4, 1120–1128. [Google Scholar] [CrossRef] [PubMed]
- Andrews, M.J.; Kontopidis, G.; McInnes, C.; Plater, A.; Innes, L.; Cowan, A.; Jewsbury, P.; Fischer, P. REPLACE: A strategy for iterative design of cyclin-binding groove inhibitors. ChemBioChem 2006, 7, 1909–1915. [Google Scholar] [CrossRef] [PubMed]
- Premnath, P.N.; Craig, S.N.; Liu, S.; Anderson, E.L.; Grigoroudis, A.I.; Kontopidis, G.; Perkins, T.L.; Wyatt, M.D.; Pittman, D.L.; McInnes, C. Iterative conversion of cyclin binding groove peptides into druglike CDK inhibitors with antitumor activity. J. Med. Chem. 2015, 58, 433–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Gan, Y.; Li, H.; Yin, J.; He, X.; Lin, L.; Xu, S.; Fang, Z.; Kim, B.-W.; Gao, L.; et al. Inhibition of the CDK2 and Cyclin A complex leads to autophagic degradation of CDK2 in cancer cells. Nat. Commun. 2022, 13, 2835. [Google Scholar] [CrossRef]
- Nagasundaram, N.; Zhu, H.; Liu, J.; Karthick, V.; Doss, G.P.C.; Chakraborty, C.; Chen, L. Analysing the Effect of Mutation on Protein Function and Discovering Potential Inhibitors of CDK4: Molecular Modelling and Dynamics Studies. PLoS ONE 2015, 10, e0133969. [Google Scholar]
- Bouclier, C.; Simon, M.; Laconde, G.; Pellerano, M.; Diot, S.; Lantuejoul, S.; Busser, B.; Vanwonterghem, L.; Vollaire, J.; Josserand, V.; et al. Stapled peptide targeting the CDK4/Cyclin D interface combined with Abemaciclib inhibits KRAS mutant lung cancer growth. Theranostics 2020, 10, 2008–2028. [Google Scholar] [CrossRef]
- Zhang, B.; Corbel, C.; Gueritte, F.; Couturier, C.; Bach, S.; Tan, V.B. An in silico approach for the discovery of CDK5/p25 interaction inhibitors. Biotechnol. J. 2011, 6, 871–881. [Google Scholar] [CrossRef]
- Zhang, B.; Su, Z.C.; Tay, T.E.; Tan, V.B. Mechanism of CDK5 activation revealed by steered molecular dynamics simulations and energy calculations. J. Mol. Model. 2010, 16, 1159–1168. [Google Scholar] [CrossRef]
- Corbel, C.; Wang, Q.; Bousserouel, H.; Hamdi, A.; Zhang, B.; Lozach, O.; Ferandin, Y.; Tan, V.B.C.; Guéritte, F.; Colas, P.; et al. First BRET-based screening assay performed in budding yeast leads to the discovery of CDK5/p25 interaction inhibitors. Biotechnol. J. 2011, 6, 860–870. [Google Scholar] [CrossRef]
- Romano, G.; Giordano, A. Role of the cyclin-dependent kinase 9-related pathway in mammalian gene expression and human diseases. Cell Cycle 2008, 7, 3664–3668. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.; Yang, G.J.; Wang, W.; Ma, D.L.; Leung, C.H. Discovery of a tetrahydroisoquinoline-based CDK9-cyclin T1 protein-protein interaction inhibitor as an anti-proliferative and anti-migration agent against triple-negative breast cancer cells. Genes Dis. 2022, 9, 1674–1688. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.S.; Qu, Y.Q.; Wu, J.; Yang, G.J.; Liu, H.; Wang, W.; Huang, Q.; Chen, F.; Li, G.; Wong, C.-Y.; et al. Inhibition of the CDK9-cyclin T1 protein-protein interaction as a new approach against triple-negative breast cancer. Acta Pharm. Sin. B 2022, 12, 1390–1405. [Google Scholar] [CrossRef] [PubMed]
- Randjelovic, J.; Eric, S.; Savic, V. Computational study and peptide inhibitors design for the CDK9—Cyclin T1 complex. J. Mol. Model. 2013, 19, 1711–1725. [Google Scholar] [CrossRef] [PubMed]
- Randjelovic, J.; Eric, S.; Savic, V. In silico design of small molecule inhibitors of CDK9/cyclin T1 interaction. J. Mol. Graph. Model. 2014, 50, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Brenke, R.; Kozakov, D.; Chuang, G.Y.; Beglov, D.; Hall, D.; Landon, M.R.; Mattos, C.; Vajda, S. Fragment-based identification of druggable ‘hot spots’ of proteins using Fourier domain correlation techniques. Bioinformatics 2009, 25, 621–627. [Google Scholar] [CrossRef] [Green Version]
- Koes, D.; Khoury, K.; Huang, Y.; Wang, W.; Bista, M.; Popowicz, G.M.; Wolf, S.; Holak, T.A.; Dömling, A.; Camacho, C.J. Enabling large-scale design, synthesis and validation of small molecule protein-protein antagonists. PLoS ONE 2012, 7, e32839. [Google Scholar] [CrossRef] [Green Version]
- Koes, D.R.; Camacho, C.J. ZINCPharmer: Pharmacophore search of the ZINC database. Nucleic Acids Res. 2012, 40, W409–W414. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Complex | Key Biologic Role | FDA-Approved Drugs |
|---|---|---|
| CDK2/Cyclin A | Cell Cycle | None |
| CDK4/Cyclin D and CDK6/Cyclin D | Cell Cycle | palbociclib, abemaciclib, ribociclib, trilaciclib |
| CDK5/p25, CDK5/p35 CDK9/Cyclin T | Neuronal function Transcription | None None |
| Scaffold | R1 | R2 | R3 | R4 |
|---|---|---|---|---|
 | H | RLNpfF | ||
 | OH |  | H | RLIF |
 | OCH3 |  | H | RLIF |
| Name | Peptide Sequence |
|---|---|
| aC-helix (native from CDK4) | PISTVREVALLRRLEAFE |
| P2 | Ac-GLPISTVRS5VALS5RRLEAFE-NH2 |
| P2short | Ac-CLPISTVRS5VALS5RRL-NH2 |
| P2shortA | ALPISTVRS5VALS5RRL-NH2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klein, M. Targeting Protein-Protein Interactions to Inhibit Cyclin-Dependent Kinases. Pharmaceuticals 2023, 16, 519. https://doi.org/10.3390/ph16040519
Klein M. Targeting Protein-Protein Interactions to Inhibit Cyclin-Dependent Kinases. Pharmaceuticals. 2023; 16(4):519. https://doi.org/10.3390/ph16040519
Chicago/Turabian StyleKlein, Mark. 2023. "Targeting Protein-Protein Interactions to Inhibit Cyclin-Dependent Kinases" Pharmaceuticals 16, no. 4: 519. https://doi.org/10.3390/ph16040519
APA StyleKlein, M. (2023). Targeting Protein-Protein Interactions to Inhibit Cyclin-Dependent Kinases. Pharmaceuticals, 16(4), 519. https://doi.org/10.3390/ph16040519