
Ketamine and the Disinhibition Hypothesis: Neurotrophic Factor-Mediated Treatment of Depression



Abstract
:1. Introduction
2. Review
2.1. Ketamine’s Unique Pharmacological Properties
2.2. Ketamine and Its Metabolites Show Differences in Pharmacology and Antidepressant Effects
2.2.1. Arketamine
2.2.2. Esketamine
2.2.3. Norketamine
2.2.4. Hydroxynorketamine
2.3. Neuroplasticity and Neuroprotection Mediate Ketamine’s Complex Antidepressant Mechanism of Action
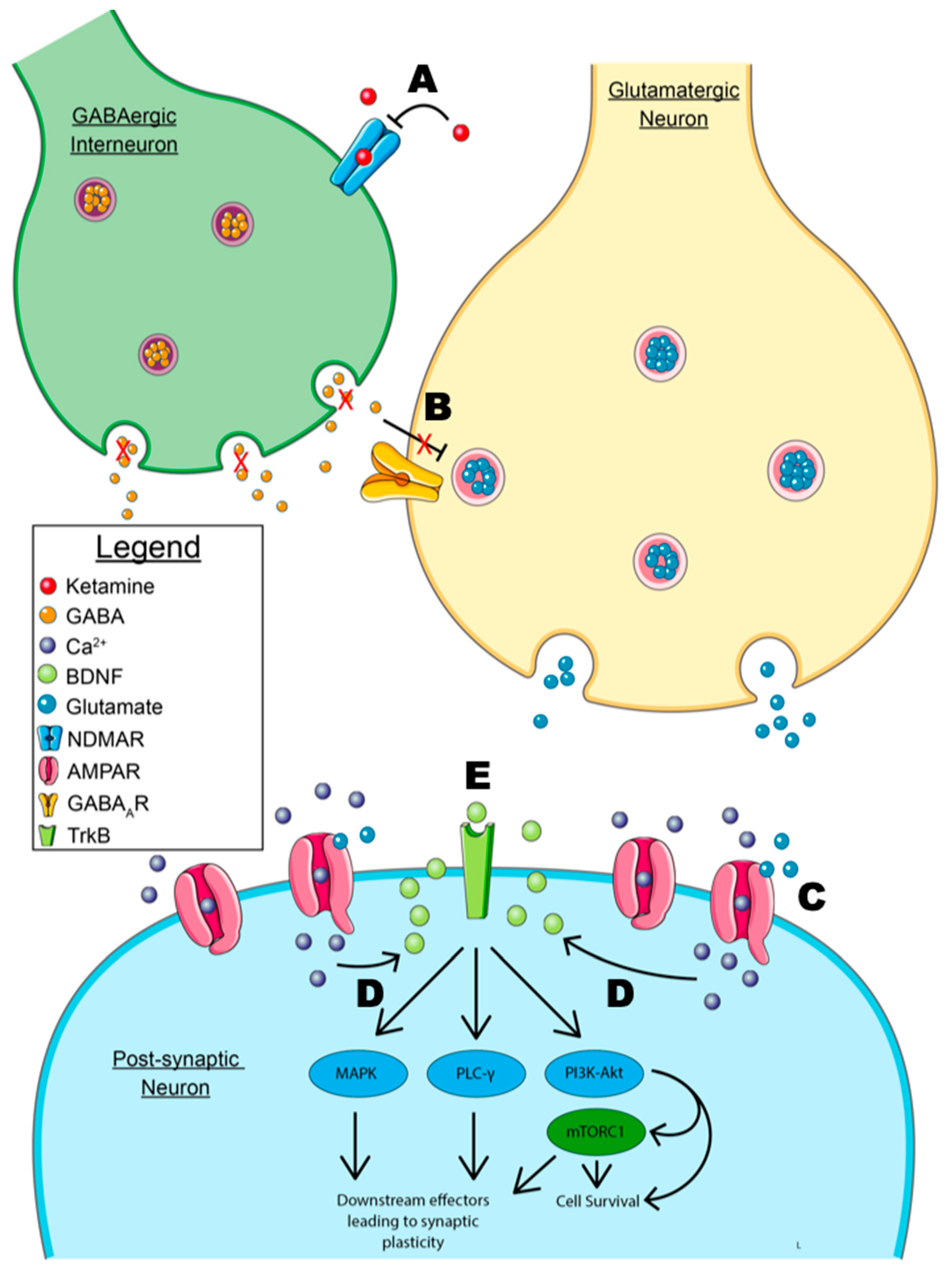
2.4. Ketamine and the Disinhibition Hypothesis
2.5. Other Potential Growth Factors Involved in Ketamine’s Action
2.5.1. VEGF
2.5.2. IGF-1
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization Depression. Available online: https://www.who.int/news-room/fact-sheets/detail/depression (accessed on 6 March 2023).
- National Institute of Mental Health Major Depression. Available online: https://www.nimh.nih.gov/health/statistics/major-depression (accessed on 5 March 2023).
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders; American Psychiatric Association Publishing: Washington DC, USA, 2022; ISBN 0-89042-575-2. [Google Scholar]
- Angst, J.; Angst, F.; Stassen, H.H. Suicide Risk in Patients with Major Depressive Disorder. J. Clin. Psychiatry 1999, 60 (Suppl. S2), 57–62; discussion 75–76, 113–116. [Google Scholar] [PubMed]
- Karrouri, R.; Hammani, Z.; Benjelloun, R.; Otheman, Y. Major Depressive Disorder: Validated Treatments and Future Challenges. World J. Clin. Cases 2021, 9, 9350–9367. [Google Scholar] [CrossRef] [PubMed]
- Clevenger, S.S.; Malhotra, D.; Dang, J.; Vanle, B.; IsHak, W.W. The Role of Selective Serotonin Reuptake Inhibitors in Preventing Relapse of Major Depressive Disorder. Ther. Adv. Psychopharmacol. 2018, 8, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Kirwin, J.L.; Gören, J.L. Duloxetine: A Dual Serotonin-Norepinephrine Reuptake Inhibitor for Treatment of Major Depressive Disorder. Pharmacotherapy 2005, 25, 396–410. [Google Scholar] [CrossRef] [PubMed]
- Sverre, K.T.; Nissen, E.R.; Farver-Vestergaard, I.; Johannsen, M.; Zachariae, R. Comparing the Efficacy of Mindfulness-Based Therapy and Cognitive-Behavioral Therapy for Depression in Head-to-Head Randomized Controlled Trials: A Systematic Review and Meta-Analysis of Equivalence. Clin. Psychol. Rev. 2023, 100, 102234. [Google Scholar] [CrossRef]
- Bassa, A.; Sagués, T.; Porta-Casteràs, D.; Serra, P.; Martínez-Amorós, E.; Palao, D.; Cano, M.; Cardoner, N. The Neurobiological Basis of Cognitive Side Effects of Electroconvulsive Therapy: A Systematic Review. Brain Sci. 2021, 11, 1273. [Google Scholar] [CrossRef]
- Cipriani, A.; Furukawa, T.A.; Salanti, G.; Chaimani, A.; Atkinson, L.Z.; Ogawa, Y.; Leucht, S.; Ruhe, H.G.; Turner, E.H.; Higgins, J.P.T.; et al. Comparative Efficacy and Acceptability of 21 Antidepressant Drugs for the Acute Treatment of Adults with Major Depressive Disorder: A Systematic Review and Network Meta-Analysis. Lancet 2018, 391, 1357–1366. [Google Scholar] [CrossRef]
- Voineskos, D.; Daskalakis, Z.J.; Blumberger, D.M. Management of Treatment-Resistant Depression: Challenges and Strategies. Neuropsychiatr. Dis. Treat. 2020, 16, 221–234. [Google Scholar] [CrossRef]
- Machado-Vieira, R.; Baumann, J.; Wheeler-Castillo, C.; Latov, D.; Henter, I.; Salvadore, G.; Zarate, C. The Timing of Antidepressant Effects: A Comparison of Diverse Pharmacological and Somatic Treatments. Pharmaceuticals 2010, 3, 19–41. [Google Scholar] [CrossRef]
- Kennedy, S.H.; Lam, R.W.; McIntyre, R.S.; Tourjman, S.V.; Bhat, V.; Blier, P.; Hasnain, M.; Jollant, F.; Levitt, A.J.; MacQueen, G.M.; et al. Canadian Network for Mood and Anxiety Treatments (CANMAT) 2016 Clinical Guidelines for the Management of Adults with Major Depressive Disorder. Can. J. Psychiatry 2016, 61, 540–560. [Google Scholar] [CrossRef]
- Pandarakalam, J.P. Challenges of Treatment-Resistant Depression. Psychiatr. Danub. 2018, 30, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Kverno, K.S.; Mangano, E. Treatment-Resistant Depression: Approaches to Treatment. J. Psychosoc. Nurs. Ment. Health Serv. 2021, 59, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Zhdanava, M.; Pilon, D.; Ghelerter, I.; Chow, W.; Joshi, K.; Lefebvre, P.; Sheehan, J.J. The Prevalence and National Burden of Treatment-Resistant Depression and Major Depressive Disorder in the United States. J. Clin. Psychiatry 2021, 82, 29169. [Google Scholar] [CrossRef]
- Kendler, K.S. The Origin of Our Modern Concept of Depression-The History of Melancholia from 1780–1880: A Review. JAMA Psychiatry 2020, 77, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Moncrieff, J.; Cooper, R.E.; Stockmann, T.; Amendola, S.; Hengartner, M.P.; Horowitz, M.A. The Serotonin Theory of Depression: A Systematic Umbrella Review of the Evidence. Mol. Psychiatry 2022. [Google Scholar] [CrossRef] [PubMed]
- Ayala-Lopez, N.; Watts, S.W. Physiology and Pharmacology of Neurotransmitter Transporters. In Comprehensive Physiology; Wiley: Hoboken, NJ, USA, 2021; pp. 2279–2295. [Google Scholar]
- Sansone, R.A.; Sansone, L.A. Serotonin Norepinephrine Reuptake Inhibitors: A Pharmacological Comparison. Innov. Clin. Neurosci. 2014, 11, 37–42. [Google Scholar] [PubMed]
- Xue, W.; Wang, P.; Li, B.; Li, Y.; Xu, X.; Yang, F.; Yao, X.; Chen, Y.Z.; Xu, F.; Zhu, F. Identification of the Inhibitory Mechanism of FDA Approved Selective Serotonin Reuptake Inhibitors: An Insight from Molecular Dynamics Simulation Study. Phys. Chem. Chem. Phys. 2016, 18, 3260–3271. [Google Scholar] [CrossRef]
- Nakamura, S. Perspective Chapter: Depression as a Disorder of Monoamine Axon Degeneration May Hold an Answer to Two Antidepressant Questions-Delayed Clinical Efficacy and Treatment-Resistant Depression. In COVID-19 Pandemic, Mental Health and Neuroscience-New Scenarios for Understanding and Treatment; IntechOpen: London, UK, 2023. [Google Scholar]
- Mosiołek, A.; Mosiołek, J.; Jakima, S.; Pięta, A.; Szulc, A. Effects of Antidepressant Treatment on Neurotrophic Factors (BDNF and IGF-1) in Patients with Major Depressive Disorder (MDD). J. Clin. Med. 2021, 10, 3377. [Google Scholar] [CrossRef]
- Price, R.B.; Duman, R. Neuroplasticity in Cognitive and Psychological Mechanisms of Depression: An Integrative Model. Mol. Psychiatry 2020, 25, 530–543. [Google Scholar] [CrossRef]
- Duman, R.S. Pathophysiology of Depression and Innovative Treatments: Remodeling Glutamatergic Synaptic Connections. Dialogues Clin. Neurosci. 2014, 16, 11–27. [Google Scholar] [CrossRef]
- Duman, R.S.; Aghajanian, G.K.; Sanacora, G.; Krystal, J.H. Synaptic Plasticity and Depression: New Insights from Stress and Rapid-Acting Antidepressants. Nat. Med. 2016, 22, 238–249. [Google Scholar] [CrossRef]
- Li, N.; Liu, R.-J.; Dwyer, J.M.; Banasr, M.; Lee, B.; Son, H.; Li, X.-Y.; Aghajanian, G.; Duman, R.S. Glutamate N-Methyl-D-Aspartate Receptor Antagonists Rapidly Reverse Behavioral and Synaptic Deficits Caused by Chronic Stress Exposure. Biol. Psychiatry 2011, 69, 754–761. [Google Scholar] [CrossRef] [PubMed]
- Dogra, S.; Conn, P.J. Targeting Metabotropic Glutamate Receptors for the Treatment of Depression and Other Stress-Related Disorders. Neuropharmacology 2021, 196, 108687. [Google Scholar] [CrossRef] [PubMed]
- Deyama, S.; Kaneda, K. Role of Neurotrophic and Growth Factors in the Rapid and Sustained Antidepressant Actions of Ketamine. Neuropharmacology 2023, 224, 109335. [Google Scholar] [CrossRef] [PubMed]
- Ly, C.; Greb, A.C.; Vargas, M.V.; Duim, W.C.; Grodzki, A.C.G.; Lein, P.J.; Olson, D.E. Transient Stimulation with Psychoplastogens Is Sufficient to Initiate Neuronal Growth. ACS Pharmacol. Transl. Sci. 2021, 4, 452–460. [Google Scholar] [CrossRef]
- Domino, E.F.; Warner, D.S. Taming the Ketamine Tiger. Anesthesiology 2010, 113, 678–684. [Google Scholar] [CrossRef]
- Cohen, B.D. Comparison of Phencyclidine Hydrochloride (Sernyl) with Other Drugs. Arch. Gen. Psychiatry 1962, 6, 395. [Google Scholar] [CrossRef]
- Sleigh, J.; Harvey, M.; Voss, L.; Denny, B. Ketamine–More Mechanisms of Action than Just NMDA Blockade. Trends Anaesth. Crit. Care 2014, 4, 76–81. [Google Scholar] [CrossRef]
- Mion, G. History of Anaesthesia -The Ketamine Story–Past, Present and Future. Eur. J. Anaesthesiol 2017, 34, 571–575. [Google Scholar] [CrossRef]
- Hashimoto, K. Rapid-acting Antidepressant Ketamine, Its Metabolites and Other Candidates: A Historical Overview and Future Perspective. Psychiatry Clin. Neurosci. 2019, 73, 613–627. [Google Scholar] [CrossRef]
- Tyler, M.W.; Yourish, H.B.; Ionescu, D.F.; Haggarty, S.J. Classics in Chemical Neuroscience: Ketamine. ACS Chem. Neurosci. 2017, 8, 1122–1134. [Google Scholar] [CrossRef] [PubMed]
- Berman, R.M.; Cappiello, A.; Anand, A.; Oren, D.A.; Heninger, G.R.; Charney, D.S.; Krystal, J.H. Antidepressant Effects of Ketamine in Depressed Patients. Biol. Psychiatry 2000, 47, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Zarate, C.A.; Singh, J.B.; Carlson, P.J.; Brutsche, N.E.; Ameli, R.; Luckenbaugh, D.A.; Charney, D.S.; Manji, H.K. A Randomized Trial of an N-Methyl-D-Aspartate Antagonist in Treatment-Resistant Major Depression. Arch. Gen. Psychiatry 2006, 63, 856. [Google Scholar] [CrossRef] [PubMed]
- Su, T.-P.; Chen, M.-H.; Li, C.-T.; Lin, W.-C.; Hong, C.-J.; Gueorguieva, R.; Tu, P.-C.; Bai, Y.-M.; Cheng, C.-M.; Krystal, J.H. Dose-Related Effects of Adjunctive Ketamine in Taiwanese Patients with Treatment-Resistant Depression. Neuropsychopharmacology 2017, 42, 2482–2492. [Google Scholar] [CrossRef] [PubMed]
- Price, R.B.; Nock, M.K.; Charney, D.S.; Mathew, S.J. Effects of Intravenous Ketamine on Explicit and Implicit Measures of Suicidality in Treatment-Resistant Depression. Biol. Psychiatry 2009, 66, 522–526. [Google Scholar] [CrossRef]
- Larkin, G.L.; Beautrais, A.L. A Preliminary Naturalistic Study of Low-Dose Ketamine for Depression and Suicide Ideation in the Emergency Department. Int. J. Neuropsychopharmacol. 2011, 14, 1127–1131. [Google Scholar] [CrossRef]
- FDA. FDA Approves New Nasal Spray Medication for Treatment-Resistant Depression; Available Only at a Certified Doctor’s Office or Clinic. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-new-nasal-spray-medication-treatment-resistant-depression-available-only-certified (accessed on 9 May 2023).
- Zheng, W.; Zhou, Y.-L.; Liu, W.-J.; Wang, C.-Y.; Zhan, Y.-N.; Li, H.-Q.; Chen, L.-J.; Li, M.D.; Ning, Y.-P. Rapid and Longer-Term Antidepressant Effects of Repeated-Dose Intravenous Ketamine for Patients with Unipolar and Bipolar Depression. J. Psychiatr. Res. 2018, 106, 61–68. [Google Scholar] [CrossRef]
- Zarate, C.A.; Brutsche, N.E.; Ibrahim, L.; Franco-Chaves, J.; Diazgranados, N.; Cravchik, A.; Selter, J.; Marquardt, C.A.; Liberty, V.; Luckenbaugh, D.A. Replication of Ketamine’s Antidepressant Efficacy in Bipolar Depression: A Randomized Controlled Add-on Trial. Biol. Psychiatry 2012, 71, 939–946. [Google Scholar] [CrossRef]
- d’Andrea, G.; Pettorruso, M.; Di Lorenzo, G.; Mancusi, G.; McIntyre, R.S.; Martinotti, G. Rethinking Ketamine and Esketamine Action: Are They Antidepressants with Mood-Stabilizing Properties? Eur. Neuropsychopharmacol. 2023, 70, 49–55. [Google Scholar] [CrossRef]
- Diazgranados, N.; Ibrahim, L.; Brutsche, N.E.; Newberg, A.; Kronstein, P.; Khalife, S.; Kammerer, W.A.; Quezado, Z.; Luckenbaugh, D.A.; Salvadore, G.; et al. A Randomized Add-on Trial of an N-Methyl-D-Aspartate Antagonist in Treatment-Resistant Bipolar Depression. Arch. Gen. Psychiatry 2010, 67, 793–802. [Google Scholar] [CrossRef]
- Lee, Y.; Syeda, K.; Maruschak, N.A.; Cha, D.S.; Mansur, R.B.; Wium-Andersen, I.K.; Woldeyohannes, H.O.; Rosenblat, J.D.; McIntyre, R.S. A New Perspective on the Anti-Suicide Effects With Ketamine Treatment: A Procognitive Effect. J. Clin. Psychopharmacol. 2016, 36, 50–56. [Google Scholar] [CrossRef]
- Duman, R.S.; Deyama, S.; Fogaça, M.V. Role of BDNF in the Pathophysiology and Treatment of Depression: Activity-dependent Effects Distinguish Rapid-acting Antidepressants. Eur. J. Neurosci. 2021, 53, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Aleksandrova, L.R.; Wang, Y.T.; Phillips, A.G. Ketamine and Its Metabolite, (2R,6R)-HNK, Restore Hippocampal LTP and Long-Term Spatial Memory in the Wistar-Kyoto Rat Model of Depression. Mol. Brain 2020, 13, 92. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information PubChem Compound Summary for CID 3821, Ketamine. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Ketamine (accessed on 5 March 2023).
- Djoumbou Feunang, Y.; Eisner, R.; Knox, C.; Chepelev, L.; Hastings, J.; Owen, G.; Fahy, E.; Steinbeck, C.; Subramanian, S.; Bolton, E.; et al. ClassyFire: Automated Chemical Classification with a Comprehensive, Computable Taxonomy. J. Cheminform. 2016, 8, 61. [Google Scholar] [CrossRef] [PubMed]
- Hastings, J.; Owen, G.; Dekker, A.; Ennis, M.; Kale, N.; Muthukrishnan, V.; Turner, S.; Swainston, N.; Mendes, P.; Steinbeck, C. ChEBI in 2016: Improved Services and an Expanding Collection of Metabolites. Nucleic Acids Res. 2016, 44, D1214–D1219. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A Major Update to the DrugBank Database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Botanas, C.J.; de la Peña, J.B.; Kim, H.J.; Lee, Y.S.; Cheong, J.H. Methoxetamine: A Foe or Friend? Neurochem. Int. 2019, 122, 1–7. [Google Scholar] [CrossRef]
- Wishart, D.S.; Guo, A.; Oler, E.; Wang, F.; Anjum, A.; Peters, H.; Dizon, R.; Sayeeda, Z.; Tian, S.; Lee, B.L.; et al. HMDB 5.0: The Human Metabolome Database for 2022. Nucleic Acids Res. 2022, 50, D622–D631. [Google Scholar] [CrossRef]
- Anis, N.A.; Berry, S.C.; Burton, N.R.; Lodge, D. The Dissociative Anaesthetics, Ketamine and Phencyclidine, Selectively Reduce Excitation of Central Mammalian Neurones by N-Methyl-Aspartate. Br. J. Pharmacol. 1983, 79, 565–575. [Google Scholar] [CrossRef]
- Thomson, A.M.; West, D.C.; Lodge, D. An N-Methylaspartate Receptor-Mediated Synapse in Rat Cerebral Cortex: A Site of Action of Ketamine? Nature 1985, 313, 479–481. [Google Scholar] [CrossRef]
- Zanos, P.; Moaddel, R.; Morris, P.J.; Georgiou, P.; Fischell, J.; Elmer, G.I.; Alkondon, M.; Yuan, P.; Pribut, H.J.; Singh, N.S.; et al. NMDAR Inhibition-Independent Antidepressant Actions of Ketamine Metabolites. Nature 2016, 533, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Sinner, B.; Graf, B.M. Ketamine. In Modern Anesthetics; Schüttler, J., Schwilden, H., Eds.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 313–333. ISBN 978-3-540-74806-9. [Google Scholar]
- Appadu, B.L.; Lambert, D.G. Interaction of i.v. Anaesthetic Agents with 5-HT3 Receptors. Br. J. Anaesth. 1996, 76, 271–273. [Google Scholar] [CrossRef] [PubMed]
- Ho, K.K.; Flood, P. Single Amino Acid Residue in the Extracellular Portion of Transmembrane Segment 2 in the Nicotinic A7 Acetylcholine Receptor Modulates Sensitivity to Ketamine. Anesthesiology 2004, 100, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Durieux, M.E. Inhibition by Ketamine of Muscarinic Acetylcholine Receptor Function. Anesth. Analg. 1995, 81, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudzade, S.; Goudarzi, S.; Mohammad Jafari, R.; Shafaroodi, H.; Dehpour, A.R.; Sanatkar, M. The N-methyl-D-aspartate Receptor Antagonist Ketamin Exerts Analgesic Effects via Modulation of the Nitric Oxide Pathway. Fundam. Clin. Pharmacol. 2022, 36, 956–965. [Google Scholar] [CrossRef] [PubMed]
- Soares, P.C.L.R.; Corrêa, J.M.X.; Niella, R.V.; de Oliveira, J.N.S.; Costa, B.A.; Silva Junior, A.C.; Sena, A.S.; Pinto, T.M.; Munhoz, A.D.; Martins, L.A.F.; et al. Continuous Infusion of Ketamine and Lidocaine Either with or without Maropitant as an Adjuvant Agent for Analgesia in Female Dogs Undergoing Mastectomy. Vet. Med. Int. 2021, 2021, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Seeman, P.; Guan, H.-C.; Hirbec, H. Dopamine D2 High Receptors Stimulated by Phencyclidines, Lysergic Acid Diethylamide, Salvinorin A, and Modafinil. Synapse 2009, 63, 698–704. [Google Scholar] [CrossRef]
- Hustveit, O.; Maurset, A.; Øye, I. Interaction of the Chiral Forms of Ketamine with Opioid, Phencyclidine, σ and Muscarinic Receptors. Pharmacol. Toxicol. 1995, 77, 355–359. [Google Scholar] [CrossRef]
- Kohrs, R.; Durieux, M.E. Ketamine-Teaching an Old Drug New Tricks. Anesth. Analg. 1998, 87, 1186–1193. [Google Scholar] [CrossRef]
- Salt, P.J.; Barnes, P.K.; Beswick, F.J. Inhibition of Neuronal and Extraneuronal Uptake of Noradrenaline by Ketamine in the Isolated Perfused Rat Heart. Br. J. Anaesth. 1979, 51, 835–838. [Google Scholar] [CrossRef]
- Vadivelu, N.; Schermer, E.; Kodumudi, V.; Belani, K.; Urman, R.; Kaye, A. Role of Ketamine for Analgesia in Adults and Children. J. Anaesthesiol. Clin. Pharmacol. 2016, 32, 298. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Vlisides, P.E. Ketamine: 50 Years of Modulating the Mind. Front Hum Neurosci. 2016, 10, 612. [Google Scholar] [CrossRef] [PubMed]
- Kharasch, E.D.; Labroo, R. Metabolism of Ketamine Stereoisomers by Human Liver Microsomes. Anesthesiology 1992, 77, 1201–1207. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Chang, L.; Hashimoto, K. Molecular Mechanisms Underlying the Antidepressant Actions of Arketamine: Beyond the NMDA Receptor. Mol. Psychiatry 2022, 27, 559–573. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Yang, J.; Luo, A.; Hashimoto, K. Molecular and Cellular Mechanisms Underlying the Antidepressant Effects of Ketamine Enantiomers and Its Metabolites. Transl. Psychiatry 2019, 9, 280. [Google Scholar] [CrossRef]
- Clements, J.A.; Nimmo, W.S.; Grant, I.S. Bioavailability, Pharmacokinetics, and Analgesic Activity of Ketamine in Humans. J. Pharm. Sci. 1982, 71, 539–542. [Google Scholar] [CrossRef]
- McIntyre, R.S.; Rosenblat, J.D.; Nemeroff, C.B.; Sanacora, G.; Murrough, J.W.; Berk, M.; Brietzke, E.; Dodd, S.; Gorwood, P.; Ho, R.; et al. Synthesizing the Evidence for Ketamine and Esketamine in Treatment-Resistant Depression: An International Expert Opinion on the. Available Evidence and Implementation. Am. J. Psychiatry 2021, 178, 383–399. [Google Scholar] [CrossRef]
- Ihmsen, H. Stereoselective Pharmacokinetics of Ketamine: R(–)-Ketamine Inhibits the Elimination of S(+)-Ketamine. Clin. Pharmacol. Ther. 2001, 70, 431–438. [Google Scholar] [CrossRef]
- Kaka, J.S.; Hayton, W.L. Pharmacokinetics of Ketamine and Two Metabolites in the Dog. J. Pharm. Biopharm. 1980, 8, 193–202. [Google Scholar] [CrossRef]
- Hess, E.M.; Riggs, L.M.; Michaelides, M.; Gould, T.D. Mechanisms of Ketamine and Its Metabolites as Antidepressants. Biochem. Pharmacol. 2022, 197, 114892. [Google Scholar] [CrossRef]
- Fanta, S.; Kinnunen, M.; Backman, J.T.; Kalso, E. Population Pharmacokinetics of S-Ketamine and Norketamine in Healthy Volunteers after Intravenous and Oral Dosing. Eur. J. Clin. Pharmacol. 2015, 71, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Highland, J.N.; Zanos, P.; Riggs, L.M.; Georgiou, P.; Clark, S.M.; Morris, P.J.; Moaddel, R.; Thomas, C.J.; Zarate, C.A.; Pereira, E.F.R.; et al. Hydroxynorketamines: Pharmacology and Potential Therapeutic Applications. Pharmacol. Rev. 2021, 73, 763–791. [Google Scholar] [CrossRef] [PubMed]
- Weiss, M.; Siegmund, W. Pharmacokinetic Modeling of Ketamine Enantiomers and Their Metabolites After Administration of Prolonged-Release Ketamine with Emphasis on 2,6-Hydroxynorketamines. Clin. Pharmacol. Drug Dev. 2022, 11, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Highland, J.N.; Morris, P.J.; Konrath, K.M.; Riggs, L.M.; Hagen, N.R.; Zanos, P.; Powels, C.F.; Moaddel, R.; Thomas, C.J.; Wang, A.Q.; et al. Hydroxynorketamine Pharmacokinetics and Antidepressant Behavioral Effects of (2,6)- and (5R)-Methyl-(2R,6R)-Hydroxynorketamines. ACS Chem. Neurosci. 2022, 13, 510–523. [Google Scholar] [CrossRef] [PubMed]
- Lumsden, E.W.; Troppoli, T.A.; Myers, S.J.; Zanos, P.; Aracava, Y.; Kehr, J.; Lovett, J.; Kim, S.; Wang, F.-H.; Schmidt, S.; et al. Antidepressant-Relevant Concentrations of the Ketamine Metabolite (2R,6R)-Hydroxynorketamine Do Not Block NMDA Receptor Function. Proc. Natl. Acad. Sci. USA 2019, 116, 5160–5169. [Google Scholar] [CrossRef] [PubMed]
- Zanos, P.; Highland, J.N.; Stewart, B.W.; Georgiou, P.; Jenne, C.E.; Lovett, J.; Morris, P.J.; Thomas, C.J.; Moaddel, R.; Zarate, C.A.; et al. (2R,6R)-Hydroxynorketamine Exerts MGlu 2 Receptor-Dependent Antidepressant Actions. Proc. Natl. Acad. Sci. USA 2019, 116, 6441–6450. [Google Scholar] [CrossRef]
- Malhotra, M.D.A. Ketamine-Induced Exacerbation of Psychotic Symptoms and Cognitive Impairment in Neuroleptic-Free Schizophrenics. Neuropsychopharmacology 1997, 17, 141–150. [Google Scholar] [CrossRef]
- Short, B.; Fong, J.; Galvez, V.; Shelker, W.; Loo, C.K. Side-Effects Associated with Ketamine Use in Depression: A Systematic Review. Lancet Psychiatry 2018, 5, 65–78. [Google Scholar] [CrossRef]
- Morrison, R.L.; Fedgchin, M.; Singh, J.; Van Gerven, J.; Zuiker, R.; Lim, K.S.; van der Ark, P.; Wajs, E.; Xi, L.; Zannikos, P.; et al. Effect of Intranasal Esketamine on Cognitive Functioning in Healthy Participants: A Randomized, Double-Blind, Placebo-Controlled Study. Psychopharmacology 2018, 235, 1107–1119. [Google Scholar] [CrossRef]
- Wajs, E.; Aluisio, L.; Holder, R.; Daly, E.J.; Lane, R.; Lim, P.; George, J.E.; Morrison, R.L.; Sanacora, G.; Young, A.H.; et al. Esketamine Nasal Spray Plus Oral Antidepressant in Patients With Treatment-Resistant Depression. J. Clin. Psychiatry 2020, 81, 10773. [Google Scholar] [CrossRef]
- Leal, G.C.; Bandeira, I.D.; Correia-Melo, F.S.; Telles, M.; Mello, R.P.; Vieira, F.; Lima, C.S.; Jesus-Nunes, A.P.; Guerreiro-Costa, L.N.F.; Marback, R.F.; et al. Intravenous Arketamine for Treatment-Resistant Depression: Open-Label Pilot Study. Eur. Arch. Psychiatry Clin. Neurosci. 2021, 271, 577–582. [Google Scholar] [CrossRef]
- Zhang, J.; Yao, W.; Hashimoto, K. Arketamine, a New Rapid-Acting Antidepressant: A Historical Review and Future Directions. Neuropharmacology 2022, 218, 109219. [Google Scholar] [CrossRef]
- Popova, V.; Daly, E.J.; Trivedi, M.; Cooper, K.; Lane, R.; Lim, P.; Mazzucco, C.; Hough, D.; Thase, M.E.; Shelton, R.C.; et al. Efficacy and Safety of Flexibly Dosed Esketamine Nasal Spray Combined with a Newly Initiated Oral Antidepressant in Treatment-Resistant Depression: A Randomized Double-Blind Active-Controlled Study. Am. J. Psychiatry 2019, 176, 428–438. [Google Scholar] [CrossRef]
- Monteggia, L.M.; Gideons, E.; Kavalali, E.T. The Role of Eukaryotic Elongation Factor 2 Kinase in Rapid Antidepressant Action of Ketamine. Biol. Psychiatry 2013, 73, 1199–1203. [Google Scholar] [CrossRef]
- Yang, C.; Kobayashi, S.; Nakao, K.; Dong, C.; Han, M.; Qu, Y.; Ren, Q.; Zhang, J.; Ma, M.; Toki, H.; et al. AMPA Receptor Activation–Independent Antidepressant Actions of Ketamine Metabolite (S)-Norketamine. Biol. Psychiatry 2018, 84, 591–600. [Google Scholar] [CrossRef]
- Ebert, B.; Mikkelsen, S.; Thorkildsen, C.; Borgbjerg, F.M. Norketamine, the Main Metabolite of Ketamine, Is a Non-Competitive NMDA Receptor Antagonist in the Rat Cortex and Spinal Cord. Eur. J. Pharmacol. 1997, 333, 99–104. [Google Scholar] [CrossRef]
- Lin, J.-W.; Lin, Y.-C.; Liu, J.-M.; Liu, S.-H.; Fang, K.-M.; Hsu, R.-J.; Huang, C.-F.; Chang, K.-Y.; Lee, K.-I.; Chang, K.-C.; et al. Norketamine, the Main Metabolite of Ketamine, Induces Mitochondria-Dependent and ER Stress-Triggered Apoptotic Death in Urothelial Cells via a Ca2+-Regulated ERK1/2-Activating Pathway. Int. J. Mol. Sci. 2022, 23, 4666. [Google Scholar] [CrossRef]
- Raja, S.; Mack, M. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04711005 (accessed on 8 March 2023).
- Ma, L.; Hashimoto, K. The Role of Hippocampal KCNQ2 Channel in Antidepressant Actions of Ketamine. Neuron 2022, 110, 2201–2203. [Google Scholar] [CrossRef]
- Lopez, J.P.; Lücken, M.D.; Brivio, E.; Karamihalev, S.; Kos, A.; De Donno, C.; Benjamin, A.; Yang, H.; Dick, A.L.W.; Stoffel, R.; et al. Ketamine Exerts Its Sustained Antidepressant Effects via Cell-Type-Specific Regulation of Kcnq2. Neuron 2022, 110, 2283–2298.e9. [Google Scholar] [CrossRef]
- Fukumoto, K.; Fogaça, M.V.; Liu, R.-J.; Duman, C.; Kato, T.; Li, X.-Y.; Duman, R.S. Activity-Dependent Brain-Derived Neurotrophic Factor Signaling Is Required for the Antidepressant Actions of (2R,6R)-Hydroxynorketamine. Proc. Natl. Acad. Sci. USA 2019, 116, 297–302. [Google Scholar] [CrossRef]
- Grunebaum, M.F.; Galfalvy, H.C.; Choo, T.-H.; Parris, M.S.; Burke, A.K.; Suckow, R.F.; Cooper, T.B.; Mann, J.J. Ketamine Metabolite Pilot Study in a Suicidal Depression Trial. J. Psychiatr. Res. 2019, 117, 129–134. [Google Scholar] [CrossRef]
- Gliwińska, A.; Czubilińska-Łada, J.; Więckiewicz, G.; Świętochowska, E.; Badeński, A.; Dworak, M.; Szczepańska, M. The Role of Brain-Derived Neurotrophic Factor (BDNF) in Diagnosis and Treatment of Epilepsy, Depression, Schizophrenia, Anorexia Nervosa and Alzheimer’s Disease as Highly Drug-Resistant Diseases: A Narrative Review. Brain Sci. 2023, 13, 163. [Google Scholar] [CrossRef]
- Barritault, D.; Plouët, J.; Courty, J.; Courtois, Y. Purification, Characterization, and Biological Properties of the Eye-Derived Growth Factor from Retina: Analogies with Brain-Derived Growth Factor. J. Neurosci. Res. 1982, 8, 477–490. [Google Scholar] [CrossRef]
- Wang, C.S.; Kavalali, E.T.; Monteggia, L.M. BDNF Signaling in Context: From Synaptic Regulation to Psychiatric Disorders. Cell 2022, 185, 62–76. [Google Scholar] [CrossRef]
- Nibuya, M.; Morinobu, S.; Duman, R.S. Regulation of BDNF and TrkB MRNA in Rat Brain by Chronic Electroconvulsive Seizure and Antidepressant Drug Treatments. J. Neurosci. 1995, 15, 7539–7547. [Google Scholar] [CrossRef]
- Sen, S.; Duman, R.; Sanacora, G. Serum Brain-Derived Neurotrophic Factor, Depression, and Antidepressant Medications: Meta-Analyses and Implications. Biol. Psychiatry 2008, 64, 527–532. [Google Scholar] [CrossRef]
- Zhou, C.; Zhong, J.; Zou, B.; Fang, L.; Chen, J.; Deng, X.; Zhang, L.; Zhao, X.; Qu, Z.; Lei, Y.; et al. Meta-Analyses of Comparative Efficacy of Antidepressant Medications on Peripheral BDNF Concentration in Patients with Depression. PLoS ONE 2017, 12, e0172270. [Google Scholar] [CrossRef]
- De Simone, S.; Bosco, M.A.; La Russa, R.; Vittorio, S.; Di Fazio, N.; Neri, M.; Cipolloni, L.; Baldari, B. Suicide and Neurotrophin Factors: A Systematic Review of the Correlation between BDNF and GDNF and Self-Killing. Healthcare 2022, 11, 78. [Google Scholar] [CrossRef]
- Colucci-D’Amato, L.; Speranza, L.; Volpicelli, F. Neurotrophic Factor BDNF, Physiological Functions and Therapeutic Potential in Depression, Neurodegeneration and Brain Cancer. Int. J. Mol. Sci. 2020, 21, 7777. [Google Scholar] [CrossRef]
- Pei, Y.; Smith, A.K.; Wang, Y.; Pan, Y.; Yang, J.; Chen, Q.; Pan, W.; Bao, F.; Zhao, L.; Tie, C.; et al. The Brain-Derived Neurotrophic-Factor (BDNF) Val66met Polymorphism Is Associated with Geriatric Depression: A Meta-Analysis. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2012, 159B, 560–566. [Google Scholar] [CrossRef]
- Zhao, M.; Chen, L.; Yang, J.; Han, D.; Fang, D.; Qiu, X.; Yang, X.; Qiao, Z.; Ma, J.; Wang, L.; et al. BDNF Val66Met Polymorphism, Life Stress and Depression: A Meta-Analysis of Gene-Environment Interaction. J. Affect. Disord. 2018, 227, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Zhou, C.; Xue, S.; Bai, J.; Yu, S.; Li, X.; Wang, H.; Tan, Q. Mechanism of Repetitive Transcranial Magnetic Stimulation for Depression. Shanghai Arch. Psychiatry 2018, 30, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Dieni, S.; Matsumoto, T.; Dekkers, M.; Rauskolb, S.; Ionescu, M.S.; Deogracias, R.; Gundelfinger, E.D.; Kojima, M.; Nestel, S.; Frotscher, M.; et al. BDNF and Its Pro-Peptide Are Stored in Presynaptic Dense Core Vesicles in Brain Neurons. J. Cell Biol. 2012, 196, 775–788. [Google Scholar] [CrossRef]
- Balkowiec, A.; Katz, D.M. Cellular Mechanisms Regulating Activity-Dependent Release of Native Brain-Derived Neurotrophic Factor from Hippocampal Neurons. J. Neurosci. 2002, 22, 10399–10407. [Google Scholar] [CrossRef]
- Soppet, D.; Escandon, E.; Maragos, J.; Middlemas, D.S.; Raid, S.W.; Blair, J.; Burton, L.E.; Stanton, B.R.; Kaplan, D.R.; Hunter, T.; et al. The Neurotrophic Factors Brain-Derived Neurotrophic Factor and Neurotrophin-3 Are Ligands for the TrkB Tyrosine Kinase Receptor. Cell 1991, 65, 895–903. [Google Scholar] [CrossRef]
- Ji, Y.; Pang, P.T.; Feng, L.; Lu, B. Cyclic AMP Controls BDNF-Induced TrkB Phosphorylation and Dendritic Spine Formation in Mature Hippocampal Neurons. Nat. Neurosci. 2005, 8, 164–172. [Google Scholar] [CrossRef]
- Andreska, T.; Lüningschrör, P.; Sendtner, M. Regulation of TrkB Cell Surface Expression-a Mechanism for Modulation of Neuronal Responsiveness to Brain-Derived Neurotrophic Factor. Cell Tissue Res. 2020, 382, 5–14. [Google Scholar] [CrossRef]
- Minichiello, L. TrkB Signalling Pathways in LTP and Learning. Nat. Rev. Neurosci. 2009, 10, 850–860. [Google Scholar] [CrossRef]
- Zakharenko, S.S.; Patterson, S.L.; Dragatsis, I.; Zeitlin, S.O.; Siegelbaum, S.A.; Kandel, E.R.; Morozov, A. Presynaptic BDNF Required for a Presynaptic but Not Postsynaptic Component of LTP at Hippocampal CA1-CA3 Synapses. Neuron 2003, 39, 975–990. [Google Scholar] [CrossRef]
- Zanos, P.; Thompson, S.M.; Duman, R.S.; Zarate, C.A.; Gould, T.D. Convergent Mechanisms Underlying Rapid Antidepressant Action. CNS Drugs 2018, 32, 197–227. [Google Scholar] [CrossRef]
- Zanos, P.; Gould, T.D. Mechanisms of Ketamine Action as an Antidepressant. Mol. Psychiatry 2018, 23, 801–811. [Google Scholar] [CrossRef]
- Moghaddam, B.; Adams, B.; Verma, A.; Daly, D. Activation of Glutamatergic Neurotransmission by Ketamine: A Novel Step in the Pathway from NMDA Receptor Blockade to Dopaminergic and Cognitive Disruptions Associated with the Prefrontal Cortex. J. Neurosci. 1997, 17, 2921–2927. [Google Scholar] [CrossRef]
- Chowdhury, G.M.I.; Zhang, J.; Thomas, M.; Banasr, M.; Ma, X.; Pittman, B.; Bristow, L.; Schaeffer, E.; Duman, R.S.; Rothman, D.L.; et al. Transiently Increased Glutamate Cycling in Rat PFC Is Associated with Rapid Onset of Antidepressant-like Effects. Mol. Psychiatry 2017, 22, 120–126. [Google Scholar] [CrossRef]
- Jang, G.; MacIver, M.B. Ketamine Produces a Long-Lasting Enhancement of CA1 Neuron Excitability. Int. J. Mol. Sci. 2021, 22, 8091. [Google Scholar] [CrossRef]
- Zhang, B.; Yang, X.; Ye, L.; Liu, R.; Ye, B.; Du, W.; Shen, F.; Li, Q.; Guo, F.; Liu, J.; et al. Ketamine Activated Glutamatergic Neurotransmission by GABAergic Disinhibition in the Medial Prefrontal Cortex. Neuropharmacology 2021, 194, 108382. [Google Scholar] [CrossRef]
- Falkenberg, T.; Lindefors, N.; Camilli, F.; Metsis, M.; Ungerstedt, U. Glutamate Release Correlates with Brain-Derived Neurotrophic Factor and TrkB MRNA Expression in the CA1 Region of Rat Hippocampus. Mol. Brain Res. 1996, 42, 317–327. [Google Scholar] [CrossRef]
- Derkach, V.A.; Oh, M.C.; Guire, E.S.; Soderling, T.R. Regulatory Mechanisms of AMPA Receptors in Synaptic Plasticity. Nat. Rev. Neurosci. 2007, 8, 101–113. [Google Scholar] [CrossRef]
- Henley, J.M.; Wilkinson, K.A. Synaptic AMPA Receptor Composition in Development, Plasticity and Disease. Nat. Rev. Neurosci. 2016, 17, 337–350. [Google Scholar] [CrossRef]
- Pham, T.H.; Defaix, C.; Nguyen, T.M.L.; Mendez-David, I.; Tritschler, L.; David, D.J.; Gardier, A.M. Cortical and Raphe GABAA, AMPA Receptors and Glial GLT-1 Glutamate Transporter Contribute to the Sustained Antidepressant Activity of Ketamine. Pharmacol. Biochem Behav. 2020, 192, 172913. [Google Scholar] [CrossRef]
- Chen, Y.; Shen, M.; Liu, X.; Xu, J.; Wang, C. The Regulation of Glutamate Transporter 1 in the Rapid Antidepressant-Like Effect of Ketamine in Mice. Front. Behav. Neurosci. 2022, 16, 789524. [Google Scholar] [CrossRef]
- El Iskandrani, K.S.; Oosterhof, C.A.; El Mansari, M.; Blier, P. Impact of Subanesthetic Doses of Ketamine on AMPA-Mediated Responses in Rats: An in Vivo Electrophysiological Study on Monoaminergic and Glutamatergic Neurons. J. Psychopharmacol. 2015, 29, 792–801. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Yang, S.; Zhang, Z.; Liu, L.; Shi, W.; Yang, S.; Li, S.; Cai, X.; Zhou, Q. Rapid and Sustained Restoration of Astrocytic Functions by Ketamine in Depression Model Mice. Biochem. Biophys. Res. Commun. 2022, 616, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Koike, H.; Iijima, M.; Chaki, S. Involvement of AMPA Receptor in Both the Rapid and Sustained Antidepressant-like Effects of Ketamine in Animal Models of Depression. Behav. Brain Res. 2011, 224, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.K.; Budac, D.P.; Bisulco, S.; Lee, A.W.; Smith, R.A.; Beenders, B.; Kelley, K.W.; Dantzer, R. NMDA Receptor Blockade by Ketamine Abrogates Lipopolysaccharide-Induced Depressive-Like Behavior in C57BL/6J Mice. Neuropsychopharmacology 2013, 38, 1609–1616. [Google Scholar] [CrossRef] [PubMed]
- Björkholm, C.; Jardemark, K.; Schilström, B.; Svensson, T.H. Ketamine-like Effects of a Combination of Olanzapine and Fluoxetine on AMPA and NMDA Receptor-Mediated Transmission in the Medial Prefrontal Cortex of the Rat. Eur. Neuropsychopharmacol. 2015, 25, 1842–1847. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Wang, N.; Yang, C.; Li, X.-M.; Zhou, Z.-Q.; Yang, J.-J. Ketamine-Induced Antidepressant Effects Are Associated with AMPA Receptors-Mediated Upregulation of MTOR and BDNF in Rat Hippocampus and Prefrontal Cortex. Eur. Psychiatry 2014, 29, 419–423. [Google Scholar] [CrossRef]
- Qu, Y.; Shan, J.; Wang, S.; Chang, L.; Pu, Y.; Wang, X.; Tan, Y.; Yamamoto, M.; Hashimoto, K. Rapid-Acting and Long-Lasting Antidepressant-like Action of (R)-Ketamine in Nrf2 Knock-out Mice: A Role of TrkB Signaling. Eur. Arch. Psychiatry Clin. Neurosci. 2021, 271, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Patapoutian, A.; Reichardt, L.F. Trk Receptors: Mediators of Neurotrophin Action. Curr. Opin. Neurobiol. 2001, 11, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Sabatini, D.M. Growing Roles for the MTOR Pathway. Curr. Opin. Cell Biol. 2005, 17, 596–603. [Google Scholar] [CrossRef]
- Wang, J.Q.; Mao, L. The ERK Pathway: Molecular Mechanisms and Treatment of Depression. Mol. Neurobiol. 2019, 56, 6197–6205. [Google Scholar] [CrossRef]
- Garro-Martínez, E.; Fullana, M.N.; Florensa-Zanuy, E.; Senserrich, J.; Paz, V.; Ruiz-Bronchal, E.; Adell, A.; Castro, E.; Díaz, Á.; Pazos, Á.; et al. MTOR Knockdown in the Infralimbic Cortex Evokes A Depressive-like State in Mouse. Int. J. Mol. Sci. 2021, 22, 8671. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Lee, B.; Liu, R.-J.; Banasr, M.; Dwyer, J.M.; Iwata, M.; Li, X.-Y.; Aghajanian, G.; Duman, R.S. MTOR-Dependent Synapse Formation Underlies the Rapid Antidepressant Effects of NMDA Antagonists. Science 2010, 329, 959–964. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, C.G.; Averill, L.A.; Gueorguieva, R.; Goktas, S.; Purohit, P.; Ranganathan, M.; D’Souza, D.C.; Formica, R.; Southwick, S.M.; Duman, R.S.; et al. Rapamycin, an Immunosuppressant and MTORC1 Inhibitor, Triples the Antidepressant Response Rate of Ketamine at 2 Weeks Following Treatment: A Double-Blind, Placebo-Controlled, Cross-over, Randomized Clinical Trial. bioRxiv 2018, 500959. [Google Scholar] [CrossRef]
- Averill, L.A.; Averill, C.L.; Gueorguieva, R.; Fouda, S.; Sherif, M.; Ahn, K.-H.; Ranganathan, M.; D’Souza, D.C.; Southwick, S.M.; Sanacora, G.; et al. MTORC1 Inhibitor Effects on Rapid Ketamine-Induced Reductions in Suicidal Ideation in Patients with Treatment-Resistant Depression. J. Affect Disord. 2022, 303, 91–97. [Google Scholar] [CrossRef]
- Nowacka, M.M.; Obuchowicz, E. Vascular Endothelial Growth Factor (VEGF) and Its Role in the Central Nervous System: A New Element in the Neurotrophic Hypothesis of Antidepressant Drug Action. Neuropeptides 2012, 46, 1–10. [Google Scholar] [CrossRef]
- Hutton, C.P.; Déry, N.; Rosa, E.; Lemon, J.A.; Rollo, C.D.; Boreham, D.R.; Fahnestock, M.; deCatanzaro, D.; Wojtowicz, J.M.; Becker, S. Synergistic Effects of Diet and Exercise on Hippocampal Function in Chronically Stressed Mice. Neuroscience 2015, 308, 180–193. [Google Scholar] [CrossRef]
- Jin, K.; Zhu, Y.; Sun, Y.; Mao, X.O.; Xie, L.; Greenberg, D.A. Vascular Endothelial Growth Factor (VEGF) Stimulates Neurogenesis In Vitro and In Vivo. Proc. Natl. Acad. Sci. USA 2002, 99, 11946–11950. [Google Scholar] [CrossRef]
- Choi, M.; Lee, S.H.; Chang, H.L.; Son, H. Hippocampal VEGF Is Necessary for Antidepressant-like Behaviors but Not Sufficient for Antidepressant-like Effects of Ketamine in Rats. Biochim. Biophys. Acta 2016, 1862, 1247–1254. [Google Scholar] [CrossRef]
- Deyama, S.; Bang, E.; Kato, T.; Li, X.-Y.; Duman, R.S. Neurotrophic and Antidepressant Actions of Brain-Derived Neurotrophic Factor Require Vascular Endothelial Growth Factor. Biol. Psychiatry 2019, 86, 143–152. [Google Scholar] [CrossRef]
- Deyama, S.; Bang, E.; Wohleb, E.S.; Li, X.-Y.; Kato, T.; Gerhard, D.M.; Dutheil, S.; Dwyer, J.M.; Taylor, S.R.; Picciotto, M.R.; et al. Role of Neuronal VEGF Signaling in the Prefrontal Cortex in the Rapid Antidepressant Effects of Ketamine. Am. J. Psychiatry 2019, 176, 388–400. [Google Scholar] [CrossRef]
- Zheng, W.; Zhou, Y.-L.; Wang, C.-Y.; Lan, X.-F.; Zhang, B.; Zhou, S.-M.; Yan, S.; Yang, M.-Z.; Nie, S.; Ning, Y.-P. Association of Plasma VEGF Levels and the Antidepressant Effects of Ketamine in Patients with Depression. Ther. Adv. Psychopharmacol. 2021, 11, 20451253211014320. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.F.; Åberg, M.A.I.; Nilsson, M.; Eriksson, P.S. Insulin-like Growth Factor-I and Neurogenesis in the Adult Mammalian Brain. Dev. Brain Res. 2002, 134, 115–122. [Google Scholar] [CrossRef] [PubMed]
- O’Kusky, J.R.; Ye, P.; D’Ercole, A.J. Insulin-like Growth Factor-I Promotes Neurogenesis and Synaptogenesis in the Hippocampal Dentate Gyrus during Postnatal Development. J. Neurosci. 2000, 20, 8435–8442. [Google Scholar] [CrossRef] [PubMed]
- Carson, M.J.; Behringer, R.R.; Brinster, R.L.; McMorris, F.A. Insulin-like Growth Factor I Increases Brain Growth and Central Nervous System Myelination in Transgenic Mice. Neuron 1993, 10, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Woods, K.A.; Camacho-Hübner, C.; Savage, M.O.; Clark, A.J.L. Intrauterine Growth Retardation and Postnatal Growth Failure Associated with Deletion of the Insulin-Like Growth Factor I Gene. N. Engl. J. Med. 1996, 335, 1363–1367. [Google Scholar] [CrossRef]
- Beck, K.D.; Powell-Braxton, L.; Widmer, H.R.; Valverde, J.; Hefti, F. Igf1 Gene Disruption Results in Reduced Brain Size, CNS Hypomyelination, and Loss of Hippocampal Granule and Striatal Parvalbumin-Containing Neurons. Neuron 1995, 14, 717–730. [Google Scholar] [CrossRef]
- Mitschelen, M.; Yan, H.; Farley, J.A.; Warrington, J.P.; Han, S.; Hereñú, C.B.; Csiszar, A.; Ungvari, Z.; Bailey-Downs, L.C.; Bass, C.E.; et al. Long-Term Deficiency of Circulating and Hippocampal Insulin-like Growth Factor I Induces Depressive Behavior in Adult Mice: A Potential Model of Geriatric Depression. Neuroscience 2011, 185, 50–60. [Google Scholar] [CrossRef]
- Tu, K.-Y.; Wu, M.-K.; Chen, Y.-W.; Lin, P.-Y.; Wang, H.-Y.; Wu, C.-K.; Tseng, P.-T. Significantly Higher Peripheral Insulin-Like Growth Factor-1 Levels in Patients With Major Depressive Disorder or Bipolar Disorder Than in Healthy Controls. Medicine 2016, 95, e2411. [Google Scholar] [CrossRef]
- Grunbaum-Novak, N.; Taler, M.; Gil-Ad, I.; Weizman, A.; Cohen, H.; Weizman, R. Relationship between Antidepressants and IGF-1 System in the Brain: Possible Role in Cognition. Eur. Neuropsychopharmacol. 2008, 18, 431–438. [Google Scholar] [CrossRef]
- Park, S.-E.; Dantzer, R.; Kelley, K.W.; McCusker, R.H. Central Administration of Insulin-like Growth Factor-I Decreases Depressive-like Behavior and Brain Cytokine Expression in Mice. J. Neuroinflammation 2011, 8, 12. [Google Scholar] [CrossRef]
- Hoshaw, B.A.; Malberg, J.E.; Lucki, I. Central Administration of IGF-I and BDNF Leads to Long-Lasting Antidepressant-like Effects. Brain Res. 2005, 1037, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Burgdorf, J.; Zhang, X.; Colechio, E.M.; Ghoreishi-Haack, N.; Gross, A.; Kroes, R.A.; Stanton, P.K.; Moskal, J.R. Insulin-Like Growth Factor I Produces an Antidepressant-Like Effect and Elicits N-Methyl-D-Aspartate Receptor Independent Long-Term Potentiation of Synaptic Transmission in Medial Prefrontal Cortex and Hippocampus. Int. J. NeuroPsychopharmacol. 2015, 19, pyv101. [Google Scholar] [CrossRef] [PubMed]
- Deyama, S.; Kondo, M.; Shimada, S.; Kaneda, K. IGF-1 Release in the Medial Prefrontal Cortex Mediates the Rapid and Sustained Antidepressant-like Actions of Ketamine. Transl. Psychiatry 2022, 12, 178. [Google Scholar] [CrossRef] [PubMed]
- Carro, E.; Nuñez, A.; Busiguina, S.; Torres-Aleman, I. Circulating Insulin-Like Growth Factor I Mediates Effects of Exercise on the Brain. J. Neurosci. 2000, 20, 2926–2933. [Google Scholar] [CrossRef]
- McMullen, E.P.; Lee, Y.; Lipsitz, O.; Lui, L.M.W.; Vinberg, M.; Ho, R.; Rodrigues, N.B.; Rosenblat, J.D.; Cao, B.; Gill, H.; et al. Strategies to Prolong Ketamine’s Efficacy in Adults with Treatment-Resistant Depression. Adv. Ther. 2021, 38, 2795–2820. [Google Scholar] [CrossRef] [PubMed]
- Mithoefer, M.C.; Feduccia, A.A.; Jerome, L.; Mithoefer, A.; Wagner, M.; Walsh, Z.; Hamilton, S.; Yazar-Klosinski, B.; Emerson, A.; Doblin, R. MDMA-Assisted Psychotherapy for Treatment of PTSD: Study Design and Rationale for Phase 3 Trials Based on Pooled Analysis of Six Phase 2 Randomized Controlled Trials. Psychopharmacology 2019, 236, 2735–2745. [Google Scholar] [CrossRef]
- Jerome, L.; Feduccia, A.A.; Wang, J.B.; Hamilton, S.; Yazar-Klosinski, B.; Emerson, A.; Mithoefer, M.C.; Doblin, R. Long-Term Follow-up Outcomes of MDMA-Assisted Psychotherapy for Treatment of PTSD: A Longitudinal Pooled Analysis of Six Phase 2 Trials. Psychopharmacology 2020, 237, 2485–2497. [Google Scholar] [CrossRef]
- Alipoor, M.; Loripoor, M.; Kazemi, M.; Farahbakhsh, F.; Sarkoohi, A. The Effect of Ketamine on Preventing Postpartum Depression. J. Med. Life 2021, 14, 87–92. [Google Scholar] [CrossRef]
- Han, Y.; Li, P.; Miao, M.; Tao, Y.; Kang, X.; Zhang, J. S-Ketamine as an Adjuvant in Patient-Controlled Intravenous Analgesia for Preventing Postpartum Depression: A Randomized Controlled Trial. BMC Anesthesiol. 2022, 22, 49. [Google Scholar] [CrossRef]
- Monks, D.T.; Palanisamy, A.; Jaffer, D.; Singh, P.M.; Carter, E.; Lenze, S. A Randomized Feasibility Pilot-Study of Intravenous and Subcutaneous Administration of Ketamine to Prevent Postpartum Depression after Planned Cesarean Delivery under Neuraxial Anesthesia. BMC Pregnancy Childbirth 2022, 22, 786. [Google Scholar] [CrossRef]
- Mohammad Shehata, I.; Masood, W.; Nemr, N.; Anderson, A.; Bhusal, K.; Edinoff, A.N.; Cornett, E.M.; Kaye, A.M.; Kaye, A.D. The Possible Application of Ketamine in the Treatment of Depression in Alzheimer’s Disease. Neurol. Int. 2022, 14, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Can, A.T.; Hermens, D.F.; Dutton, M.; Gallay, C.C.; Jensen, E.; Jones, M.; Scherman, J.; Beaudequin, D.A.; Yang, C.; Schwenn, P.E.; et al. Low Dose Oral Ketamine Treatment in Chronic Suicidality: An Open-Label Pilot Study. Transl. Psychiatry 2021, 11, 101. [Google Scholar] [CrossRef] [PubMed]
- Norbury, A.; Rutter, S.B.; Collins, A.B.; Costi, S.; Jha, M.K.; Horn, S.R.; Kautz, M.; Corniquel, M.; Collins, K.A.; Glasgow, A.M.; et al. Neuroimaging Correlates and Predictors of Response to Repeated-Dose Intravenous Ketamine in PTSD: Preliminary Evidence. Neuropsychopharmacology 2021, 46, 2266–2277. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Target | Action | Comments | Reference |
|---|---|---|---|
| Glutamate receptor ionotropic, NMDA 3A | antagonist | blocks the open ion channel directly and through negative allosteric regulation; prevents the activation of a calcium-dependent NO synthetase, which plays a role in nociception and neurotoxicity | [59] |
| 5-hydroxytryptamine receptor 3A | potentiator | increases voltage-gated potassium channel activity; binds at supratherapeutic doses, and is thought to increase the effects of the receptor through indirect mechanisms | [60] |
| α-7 nicotinic cholinergic receptor subunit | antagonist | its effects on skeletal muscle tone are not noticed unless unmasked by additional muscle relaxants; ketamine’s NMDAR antagonism additionally inhibits acetylcholine release through the receptors | [61] |
| Muscarinic acetylcholine receptor M1 | inhibitor | primarily found in the hippocampus and the cerebral cortex | [62] |
| Nitric oxide synthase | indirect inhibitor | in the brain; functions through the glutamate/NO/cGMP system; may contribute to neuroprotective, sympathetic activating, and additional analgesic effects | [63] |
| Neurokinin 1 receptor | antagonist | through noncompetitive inhibition; possibly contributes to an analgesic effect, as this receptor modulates spinal cord nociception, but the therapeutic relevance of this interaction is not fully clear | [64] |
| Dopamine D2 receptor | agonist/partial agonist | specifically binds to the high-affinity state of the receptor; binding is more than 10 times weaker than that of dopamine and phencyclidine | [65] |
| Opioid receptors | mild agonist | binding affinity from strongest to weakest: mu > kappa > delta; related to some analgesic properties and adverse side effects, particularly with the kappa receptor | [66,67] |
| Sodium-dependent noradrenaline transporter | inhibitor | blocks reuptake in the heart, leading to increased chronotropy and vasoconstriction | [68] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borsellino, P.; Krider, R.I.; Chea, D.; Grinnell, R.; Vida, T.A. Ketamine and the Disinhibition Hypothesis: Neurotrophic Factor-Mediated Treatment of Depression. Pharmaceuticals 2023, 16, 742. https://doi.org/10.3390/ph16050742
Borsellino P, Krider RI, Chea D, Grinnell R, Vida TA. Ketamine and the Disinhibition Hypothesis: Neurotrophic Factor-Mediated Treatment of Depression. Pharmaceuticals. 2023; 16(5):742. https://doi.org/10.3390/ph16050742
Chicago/Turabian StyleBorsellino, Philip, Reese I. Krider, Deanna Chea, Ryan Grinnell, and Thomas A. Vida. 2023. "Ketamine and the Disinhibition Hypothesis: Neurotrophic Factor-Mediated Treatment of Depression" Pharmaceuticals 16, no. 5: 742. https://doi.org/10.3390/ph16050742
APA StyleBorsellino, P., Krider, R. I., Chea, D., Grinnell, R., & Vida, T. A. (2023). Ketamine and the Disinhibition Hypothesis: Neurotrophic Factor-Mediated Treatment of Depression. Pharmaceuticals, 16(5), 742. https://doi.org/10.3390/ph16050742