
In Silico Characterization and Analysis of Clinically Significant Variants of Lipase-H (LIPH Gene) Protein Associated with Hypotrichosis


 ,
,  ,
,
Abstract
:1. Introduction
2. Results
2.1. Retrieval of nsSNPs of the LIPH Gene
2.2. Screening of nsSNPs of LIPH Gene
2.3. Prediction of Clinically Significant Deleterious nsSNPs of LIPH Protein
2.4. Pathological Annotation of Clinically Significant Deleterious nsSNPs on LIPH Protein
2.5. Influence of Medically Significant Variations on LIPH Protein Robustness
2.6. Evolutionary Conservation Analysis of LIPH Protein
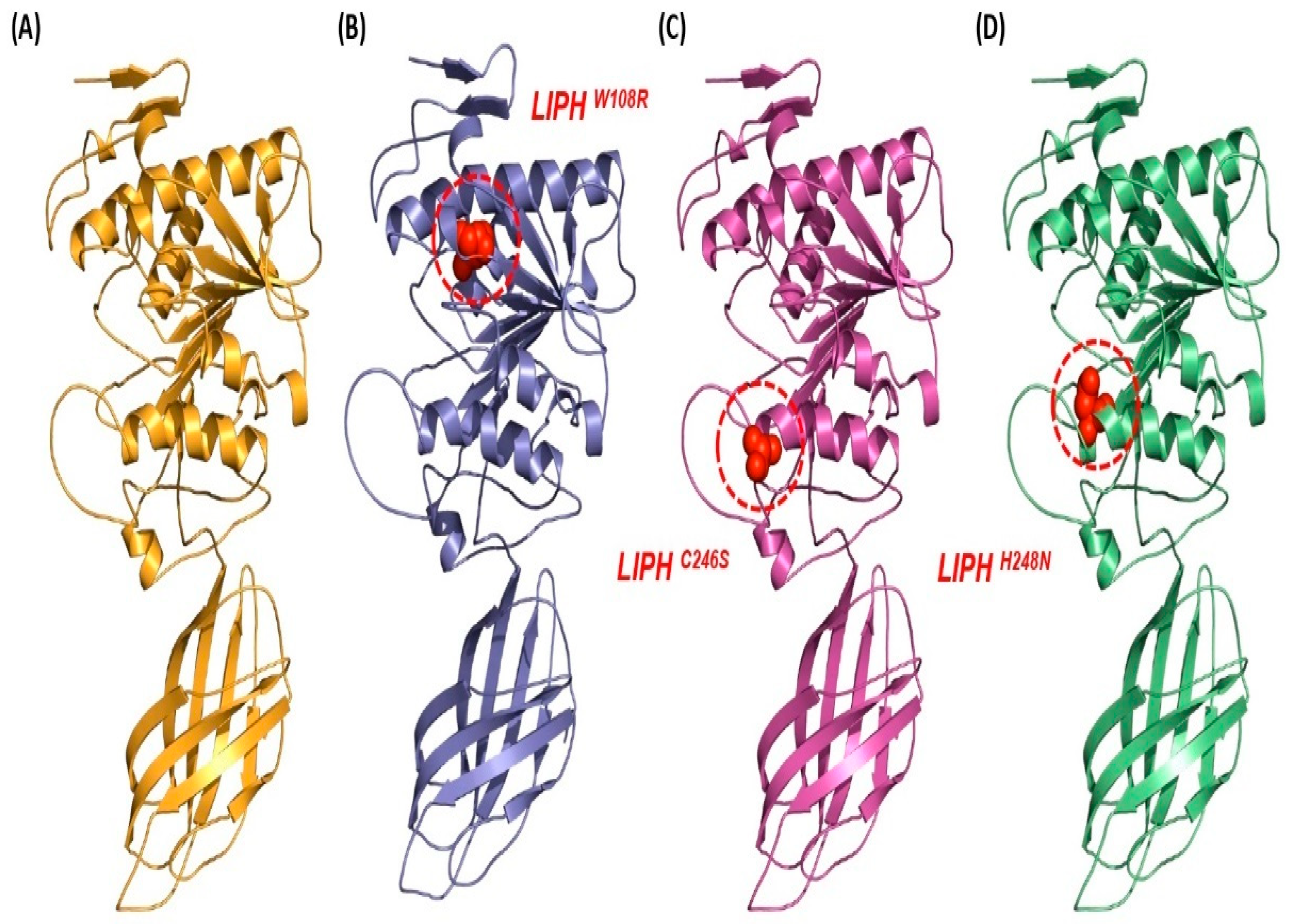
2.7. Comparative Analysis of LIPHWT and LIPHMT Structures
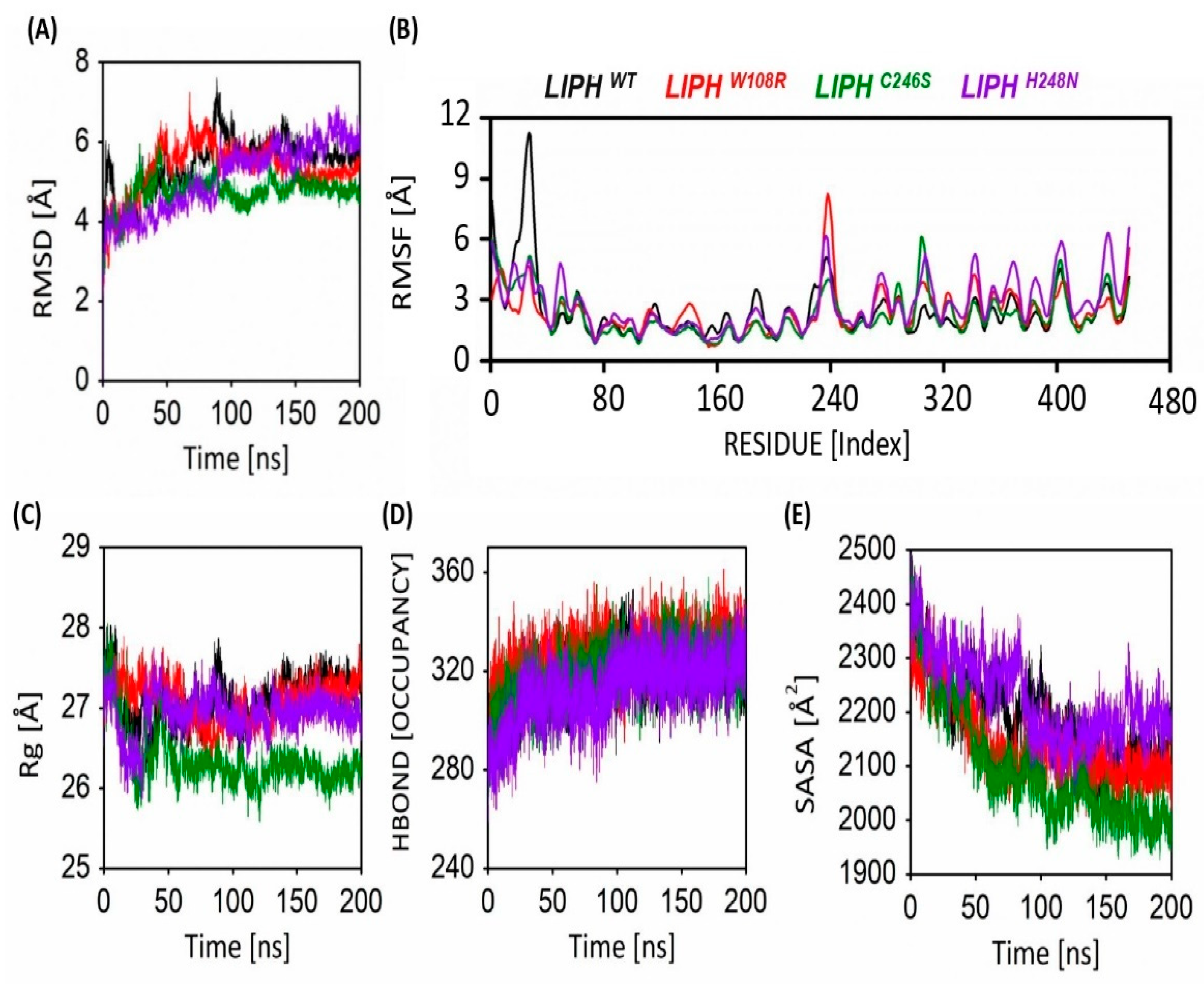
2.8. Stability Analysis of LIPHWT and LIPHMTs Protein Models
2.9. Flexibility Analysis of LIPHWT and LIPHMTs
2.10. Gyration Analysis of LIPHWT and LIPHMTs
2.11. Hydrogen Bond Occupancy
2.12. Solvent-Accessible Surface Area of LIPHWT and LIPHMTs
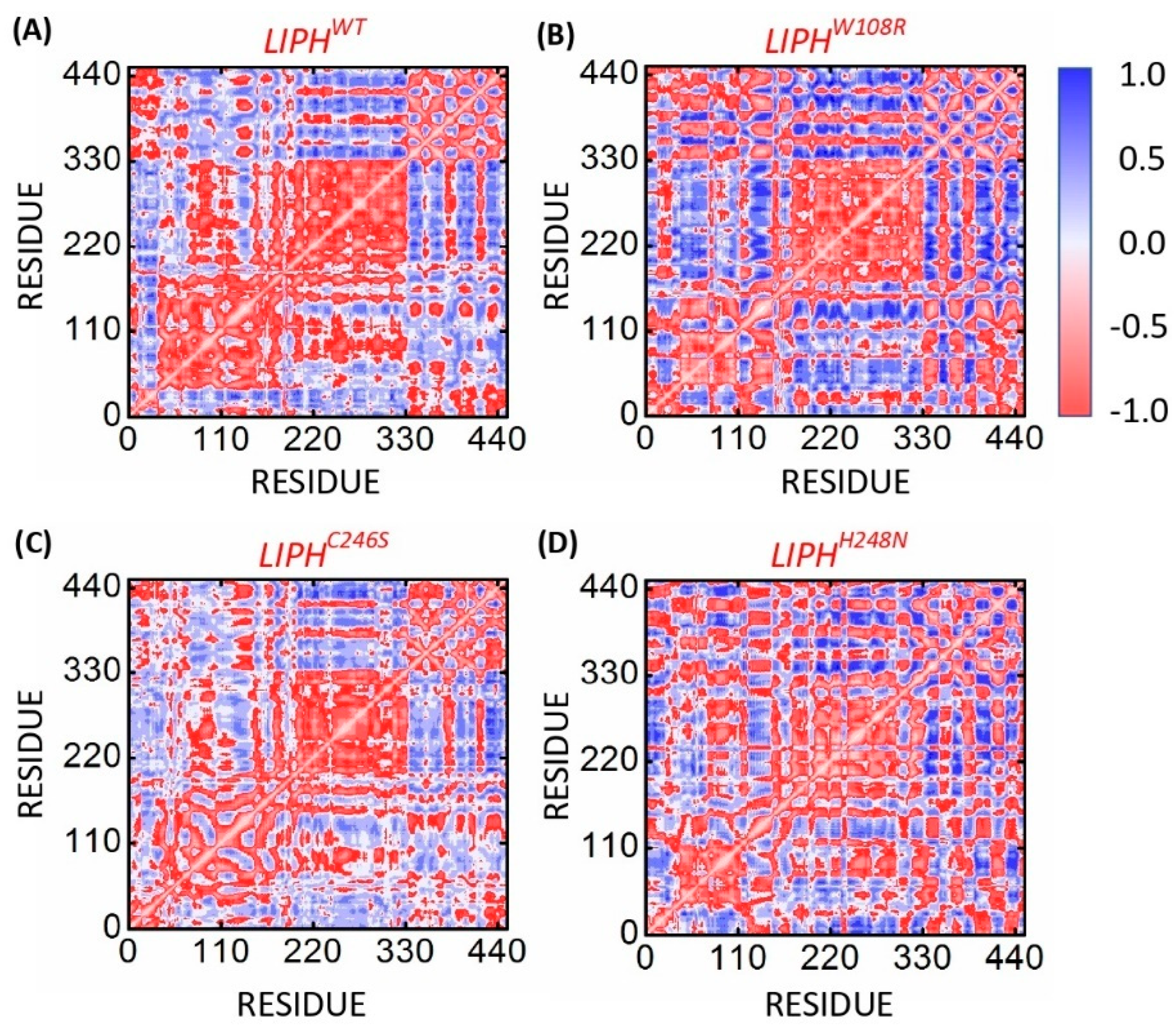
2.13. Conformational Analysis of LIPHWT and LIPHMTs
Comparative Functional Analysis of LIPHWT and Mutant Models
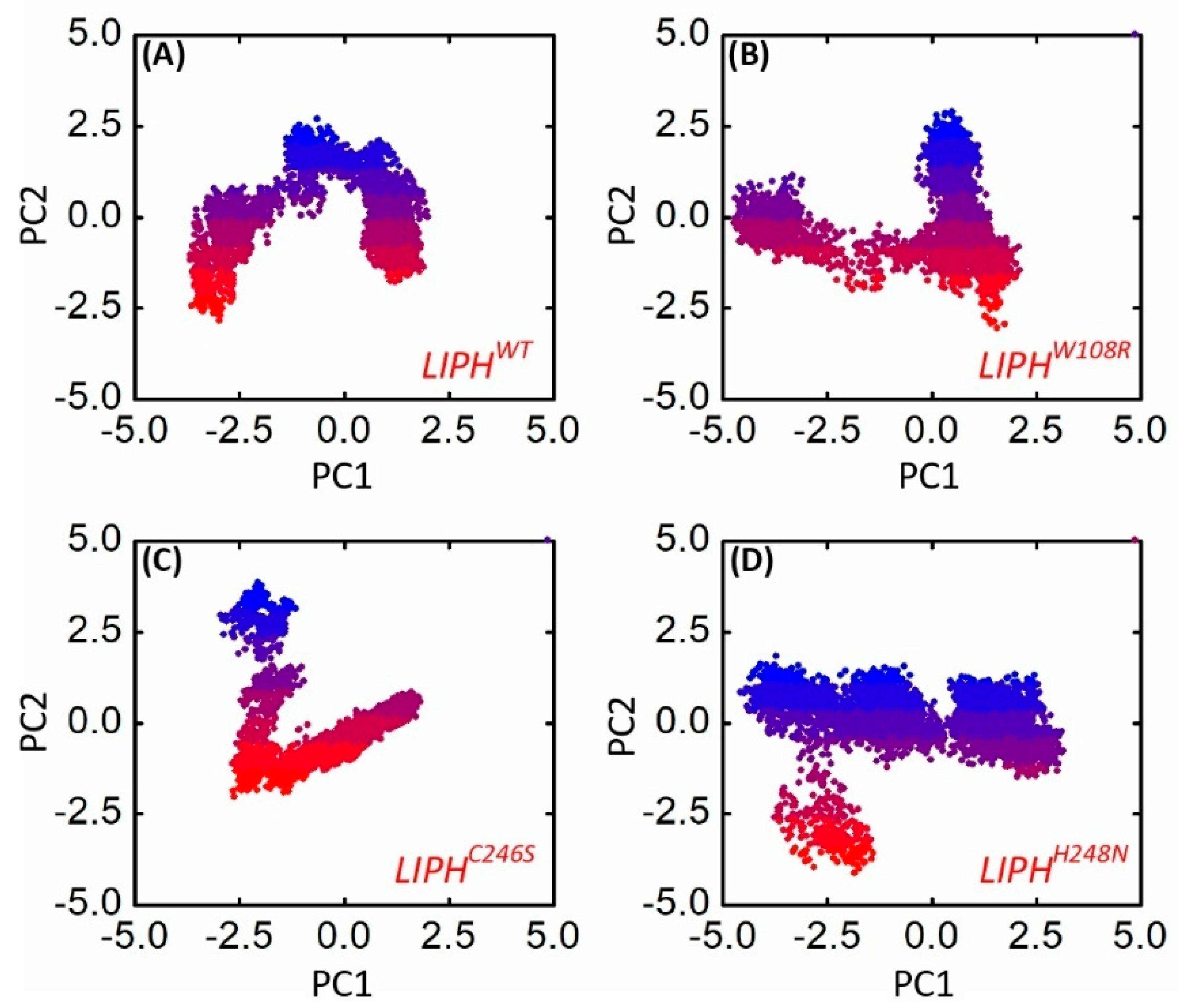
2.14. Dimensionality Reduction Using PCA
3. Discussion
4. Materials and Methods
4.1. Retrieval of LIPH Sequence and nsSNPs
4.2. Identification of Deleterious nsSNPs
4.3. Validation of Deleterious nsSNPs of LIPH Gene
4.4. Structure Stability of Mutant Protein
4.5. Structure Prediction and Validation
4.6. Computer Simulation of Molecular Dynamics
4.7. Dynamic Cross-Correlation Map (DCCM)
4.8. Essential Dynamics Using Principal Components Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bakmazian, A. The Man behind the Beard: Perception of Men’s Trustworthiness as a Function of Facial Hair. Psychology 2014, 5, 185–191. [Google Scholar] [CrossRef]
- Khan, S.; Habib, R.; Mir, H.; Naz, G.; Ayub, M.; Shafique, S.; Yamin, T.; Ali, N.; Basit, S.; Wasif, N. Mutations in the LPAR6 and LIPH Genes Underlie Autosomal Recessive Hypotrichosis/Woolly Hair in 17 Consanguineous Families from Pakistan. Clin. Exp. Dermatol. 2011, 36, 652–654. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, H.; Aoki, J.; Hiramatsu, T.; Ishida, M.; Bandoh, K.; Nagai, Y.; Taguchi, R.; Inoue, K.; Arai, H. A Novel Phosphatidic Acid-Selective Phospholipase A1That Produces Lysophosphatidic Acid. J. Biol. Chem. 2002, 277, 34254–34263. [Google Scholar] [CrossRef]
- Naz, G.; Khan, B.; Ali, G.; Azeem, Z.; Wali, A.; Ansar, M.; Ahmad, W. Novel Missense Mutations in Lipase H (LIPH) Gene Causing Autosomal Recessive Hypotrichosis (LAH2). J. Dermatol. Sci. 2009, 54, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Shinkuma, S.; Akiyama, M.; Inoue, A.; Aoki, J.; Natsuga, K.; Nomura, T.; Arita, K.; Abe, R.; Ito, K.; Nakamura, H. Prevalent LIPH Founder Mutations Lead to Loss of P2Y5 Activation Ability of PA-PLA1α in Autosomal Recessive Hypotrichosis. Hum. Mutat. 2010, 31, 602–610. [Google Scholar]
- Ng, P.C.; Henikoff, S. SIFT: Predicting Amino Acid Changes That Affect Protein Function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the Functional Impact of Protein Mutations: Application to Cancer Genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef]
- Capriotti, E.; Calabrese, R.; Fariselli, P.; Martelli, P.L.; Altman, R.B.; Casadio, R. WS-SNPs&GO: A Web Server for Predicting the Deleterious Effect of Human Protein Variants Using Functional Annotation. BMC Genom. 2013, 14, S6. [Google Scholar]
- Capriotti, E.; Fariselli, P. PhD-SNPg: A Webserver and Lightweight Tool for Scoring Single Nucleotide Variants. Nucleic Acids Res. 2017, 45, W247–W252. [Google Scholar] [CrossRef]
- Capriotti, E.; Fariselli, P.; Casadio, R. I-Mutant2. 0: Predicting Stability Changes upon Mutation from the Protein Sequence or Structure. Nucleic Acids Res. 2005, 33, W306–W310. [Google Scholar] [CrossRef]
- Cheng, J.; Randall, A.Z.; Sweredoski, M.J.; Baldi, P. SCRATCH: A Protein Structure and Structural Feature Prediction Server. Nucleic Acids Res. 2005, 33, W72–W76. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An Improved Methodology to Estimate and Visualize Evolutionary Conservation in Macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. [Google Scholar] [CrossRef]
- Xu, D.; Zhang, Y. Improving the Physical Realism and Structural Accuracy of Protein Models by a Two-Step Atomic-Level Energy Minimization. Biophys. J. 2011, 101, 2525–2534. [Google Scholar] [CrossRef]
- DeLano, W.L. Pymol: An Open-Source Molecular Graphics Tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Ge, F.; Zhang, Y.; Xu, J.; Muhammad, A.; Song, J.; Yu, D.-J. Prediction of Disease-Associated NsSNPs by Integrating Multi-Scale ResNet Models with Deep Feature Fusion. Brief. Bioinform. 2022, 23, bbab530. [Google Scholar] [CrossRef]
- Slater, H.R.; Bailey, D.K.; Ren, H.; Cao, M.; Bell, K.; Nasioulas, S.; Henke, R.; Choo, K.A.; Kennedy, G.C. High-Resolution Identification of Chromosomal Abnormalities Using Oligonucleotide Arrays Containing 116,204 SNPs. Am. J. Hum. Genet. 2005, 77, 709–726. [Google Scholar] [CrossRef]
- Komar, A.A. SNPs, Silent but Not Invisible. Science 2007, 315, 466–467. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.U.; Kiani, B.H.; Abrar, M.; Jan, Z.; Zafar, I.; Ali, Y.; Alanazi, A.M.; Malik, A.; Rather, M.A.; Ahmad, A. A Comprehensive Genomic Study, Mutation Screening, Phylogenetic and Statistical Analysis of SARS-CoV-2 and Its Variant Omicron among Different Countries. J. Infect. Public Health 2022, 15, 878–891. [Google Scholar] [CrossRef]
- Khattak, S.; Rauf, M.A.; Zaman, Q.; Ali, Y.; Fatima, S.; Muhammad, P.; Li, T.; Khan, H.A.; Khan, A.A.; Ngowi, E.E. Genome-Wide Analysis of Codon Usage Patterns of SARS-CoV-2 Virus Reveals Global Heterogeneity of COVID-19. Biomolecules 2021, 11, 912. [Google Scholar] [CrossRef]
- Shah, A.A.; Amjad, M.; Hassan, J.-U.; Ullah, A.; Mahmood, A.; Deng, H.; Ali, Y.; Gul, F.; Xia, K. Molecular Insights into the Role of Pathogenic Nssnps in Grin2b Gene Provoking Neurodevelopmental Disorders. Genes 2022, 13, 1332. [Google Scholar] [CrossRef]
- Ijaz, A.; Shah, K.; Aziz, A.; Rehman, F.U.; Ali, Y.; Tareen, A.M.; Khan, K.; Ayub, M.; Wali, A. Novel Frameshift Mutations in XPC Gene Underlie Xeroderma Pigmentosum in Pakistani Families. Indian J. Dermatol. 2021, 66, 220. [Google Scholar]
- Ali, Y.; Ahmad, F.; Ullah, M.F.; Haq, N.U.; Haq, M.; Aziz, A.; Zouidi, F.; Khan, M.I.; Eldin, S.M. Structural Evaluation and Conformational Dynamics of ZNF141T474I Mutation Provoking Postaxial Polydactyly Type A. Bioengineering 2022, 9, 749. [Google Scholar] [CrossRef]
- Ahmad, S.U.; Ali, Y.; Jan, Z.; Rasheed, S.; Nazir, N.U.A.; Khan, A.; Rukh Abbas, S.; Wadood, A.; Rehman, A.U. Computational Screening and Analysis of Deleterious NsSNPs in Human p 14ARF (CDKN2A Gene) Protein Using Molecular Dynamic Simulation Approach. J. Biomol. Struct. Dyn. 2022, 41, 3964–3975. [Google Scholar] [CrossRef] [PubMed]
- Ali, Y.; Imtiaz, H.; Tahir, M.M.; Gul, F.; Saddozai, U.A.K.; ur Rehman, A.; Ren, Z.-G.; Khattak, S.; Ji, X.-Y. Fragment-Based Approaches Identified Tecovirimat-Competitive Novel Drug Candidate for Targeting the F13 Protein of the Monkeypox Virus. Viruses 2023, 15, 570. [Google Scholar] [CrossRef]
- Ajmal, A.; Ali, Y.; Khan, A.; Wadood, A.; Rehman, A.U. Identification of Novel Peptide Inhibitors for the KRas-G12C Variant to Prevent Oncogenic Signaling. J. Biomol. Struct. Dyn. 2022, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ali, Y.; Yasin, M.; Aziz, A.; Khan, A.W.; ur Rahman, S.; Haq, N.U. In-Silico Analysis of 2-Cysteine Peroxiredoxin Genes in Arabidopsis Thaliana with Possible Role in Carbon Dioxide Fixation through Carbonic Anhydrase Regulation. Pak. J. Biochem. Biotechnol. 2022, 3, 175–189. [Google Scholar] [CrossRef]
- Yue, P.; Moult, J. Identification and Analysis of Deleterious Human SNPs. J. Mol. Biol. 2006, 356, 1263–1274. [Google Scholar] [CrossRef]
- Yates, C.M.; Sternberg, M.J. The Effects of Non-Synonymous Single Nucleotide Polymorphisms (NsSNPs) on Protein–Protein Interactions. J. Mol. Biol. 2013, 425, 3949–3963. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, V.K.; Oakley, A.; Purohit, R. Mechanistic Behavior and Subtle Key Events during DNA Clamp Opening and Closing in T4 Bacteriophage. Int. J. Biol. Macromol. 2022, 208, 11–19. [Google Scholar] [CrossRef]
- Bhardwaj, V.K.; Purohit, R. A Lesson for the Maestro of the Replication Fork: Targeting the Protein-binding Interface of Proliferating Cell Nuclear Antigen for Anticancer Therapy. J. Cell. Biochem. 2022, 123, 1091–1102. [Google Scholar] [CrossRef]
- Rajendran, V.; Purohit, R.; Sethumadhavan, R. In Silico Investigation of Molecular Mechanism of Laminopathy Caused by a Point Mutation (R482W) in Lamin A/C Protein. Amino Acids 2012, 43, 603–615. [Google Scholar] [CrossRef]
- Chatterjee, C.; Sparks, D.L. Hepatic Lipase, High Density Lipoproteins, and Hypertriglyceridemia. Am. J. Pathol. 2011, 178, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Sim, N.-L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT Web Server: Predicting Effects of Amino Acid Substitutions on Proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y. A Fast Computation of Pairwise Sequence Alignment Scores between a Protein and a Set of Single-Locus Variants of Another Protein. In Proceedings of the ACM Conference on Bioinformatics, Computational Biology and Biomedicine (BCB’ 12), Orlando, FL, USA, 7–10 October 2012; pp. 414–417. [Google Scholar]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A Method and Server for Predicting Damaging Missense Mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7–20. [Google Scholar] [CrossRef]
- López-Ferrando, V.; Gazzo, A.; De La Cruz, X.; Orozco, M.; Gelpí, J.L. PMut: A Web-Based Tool for the Annotation of Pathological Variants on Proteins, 2017 Update. Nucleic Acids Res. 2017, 45, W222–W228. [Google Scholar] [CrossRef]
- Zhang, M.; Huang, C.; Wang, Z.; Lv, H.; Li, X. In Silico Analysis of Non-Synonymous Single Nucleotide Polymorphisms (NsSNPs) in the Human GJA3 Gene Associated with Congenital Cataract. BMC Mol. Cell Biol. 2020, 21, 12. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5–6. [Google Scholar] [CrossRef]
- Colovos, C.; Yeates, T.O. Verification of Protein Structures: Patterns of Nonbonded Atomic Interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Bowers, K.J.; Sacerdoti, F.D.; Salmon, J.K.; Shan, Y.; Shaw, D.E.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006. [Google Scholar]
- Maisuradze, G.G.; Liwo, A.; Scheraga, H.A. Principal Component Analysis for Protein Folding Dynamics. J. Mol. Biol. 2009, 385, 312–329. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant ID | AA Change | SIFT | Polyphen-2 | PROVEAN | SNP&GO | PhD-SNP | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Pred 1 | Scr | Effect 2 | Scr | Pred 3 | Scr | Pred 4 | RI | Pred 5 | RI | ||
| rs267607219 | W108R | Dmg | 0.00 | Pro. Dmg | 1.00 | Del | −12.81 | Disease | 8 | Disease | 9 |
| rs201249971 | C246S | Dmg | 0.00 | Pro. Dmg | 1.00 | Del | −9.36 | Disease | 7 | Disease | 9 |
| rs201868115 | H248N | Dmg | 0.00 | Pro. Dmg | 1.00 | Del | −6.56 | Disease | 7 | Disease | 9 |
| Variant ID | AA Change | PMut | I-Mutant 2.0 | ConSurf | |||
|---|---|---|---|---|---|---|---|
| Score and Percentage | Pred | Stability | DDG Value | Cons. Score | Pred | ||
| rs267607219 | W108R | (0.93) 94% | Disease | Decrease | −1.91 | 9 | Strongly conserved and prominently displayed (s) |
| rs201249971 | C246S | (0.90) 93% | Disease | Decrease | −1.55 | 9 | Strongly conserved and prominently displayed (s) |
| rs201868115 | H248N | (0.90) 93% | Disease | Decrease | −0.85 | 9 | Strongly conserved and prominently displayed (f) |
| Model | TM-Align 1 | ERRAT 2 | PROCHECK Ramachandran Plot Analysis 3 | ||||
|---|---|---|---|---|---|---|---|
| TM Score | RMSD | ERRAT Value (Overall Quality Factor) | Residues in Most Favored Regions | Residues in Additional Allowed Regions | Residues in Generously Allowed Regions | Residues in Disallowed Regions | |
| LIPHWT | Nill | Nill | 90.26% | 340 (80.3%) | 39 (9.9%) | 7 (1.8%) | 8 (2.0%) |
| LIPHW108R | 1.00 | 0.00 | 85.00% | 350 (88.8%) | 29 (7.4%) | 7 (1.8%) | 8 (2.0%) |
| LIPHC246S | 1.00 | 0.00 | 85.00% | 341 (86.5%) | 40 (10.2%) | 8 (2.0%) | 5 (1.3%) |
| LIPHH248N | 1.00 | 0.00 | 87.41% | 342 (86.8%) | 40 (10.2%) | 7 (1.8%) | 5 (1.3%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, H.A.; Asif, M.U.; Ijaz, M.K.; Alharbi, M.; Ali, Y.; Ahmad, F.; Azhar, R.; Ahmad, S.; Irfan, M.; Javed, M.; et al. In Silico Characterization and Analysis of Clinically Significant Variants of Lipase-H (LIPH Gene) Protein Associated with Hypotrichosis. Pharmaceuticals 2023, 16, 803. https://doi.org/10.3390/ph16060803
Khan HA, Asif MU, Ijaz MK, Alharbi M, Ali Y, Ahmad F, Azhar R, Ahmad S, Irfan M, Javed M, et al. In Silico Characterization and Analysis of Clinically Significant Variants of Lipase-H (LIPH Gene) Protein Associated with Hypotrichosis. Pharmaceuticals. 2023; 16(6):803. https://doi.org/10.3390/ph16060803
Chicago/Turabian StyleKhan, Hamza Ali, Muhammad Umair Asif, Muhammad Khurram Ijaz, Metab Alharbi, Yasir Ali, Faisal Ahmad, Ramsha Azhar, Sajjad Ahmad, Muhammad Irfan, Maryana Javed, and et al. 2023. "In Silico Characterization and Analysis of Clinically Significant Variants of Lipase-H (LIPH Gene) Protein Associated with Hypotrichosis" Pharmaceuticals 16, no. 6: 803. https://doi.org/10.3390/ph16060803
APA StyleKhan, H. A., Asif, M. U., Ijaz, M. K., Alharbi, M., Ali, Y., Ahmad, F., Azhar, R., Ahmad, S., Irfan, M., Javed, M., Naseer, N., & Aziz, A. (2023). In Silico Characterization and Analysis of Clinically Significant Variants of Lipase-H (LIPH Gene) Protein Associated with Hypotrichosis. Pharmaceuticals, 16(6), 803. https://doi.org/10.3390/ph16060803