

Structure-Based Design of Peptides Targeting VEGF/VEGFRs


Abstract
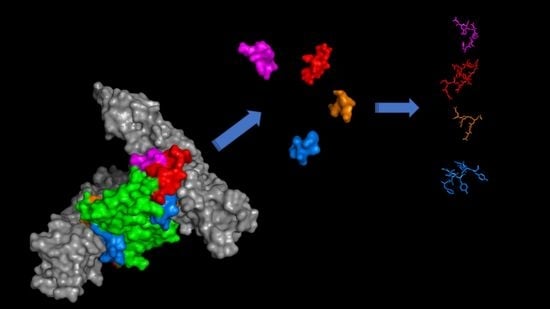
:
1. Introduction
2. The Biology of VEGF/VEGFR Molecular System
3. VEGF/VEGFR-Targeting Peptides: An Opportunity in Pharmaceutical Sciences
4. Peptides Targeting VEGF
5. Peptides Targeting VEGF Receptors
5.1. Peptide Mimetics of α1 N-Terminal Helix Region
5.2. Peptide Mimetics of Loop 1 Region
5.3. Peptide Mimetics of Loop 2 Region
5.4. Peptide Mimetics of Loop 3 Region
6. Peptide Mimetics of Discontinuous Binding Surface
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Ricard, N.; Bailly, S.; Guignabert, C.; Simons, M. The quiescent endothelium: Signalling pathways regulating organ-specific endothelial normalcy. Nat. Rev. Cardiol. 2021, 18, 565–580. [Google Scholar] [CrossRef]
- Muller, Y.A.; Li, B.; Christinger, H.W.; Wells, J.A.; Cunningham, B.C.; DeVos, A.M. Vascular endothelial growth factor: Crystal structure and functional mapping of the kinase domain receptor binding site. Proc. Natl. Acad. Sci. USA 1997, 94, 7192–7197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.A.; Jeong, M.S.; Ha, K.T.; Jang, S.B. Structure and function of vascular endothelial growth factor and its receptor system. BMB Rep. 2018, 51, 73–78. [Google Scholar] [CrossRef] [Green Version]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibuya, M. Vascular Endothelial Growth Factor (VEGF) and Its Receptor (VEGFR) Signaling in Angiogenesis: A Crucial Target for Anti- and Pro-Angiogenic Therapies. Genes Cancer 2011, 2, 1097–1105. [Google Scholar] [CrossRef]
- Shaik, F.; Cuthbert, G.A.; Homer-Vanniasinkam, S.; Muench, S.P.; Ponnambalam, S.; Harrison, M.A. Structural Basis for Vascular Endothelial Growth Factor Receptor Activation and Implications for Disease Therapy. Biomolecules 2020, 10, 1673. [Google Scholar] [CrossRef]
- Claesson-Welsh, L. VEGF receptor signal transduction—A brief update. Vasc. Pharmacol. 2016, 86, 14–17. [Google Scholar] [CrossRef]
- Davydova, N.; Harris, N.C.; Roufail, S.; Paquet-Fifield, S.; Ishaq, M.; Streltsov, V.A.; Williams, S.P.; Karnezis, T.; Stacker, S.A.; Achen, M.G. Differential Receptor Binding and Regulatory Mechanisms for the Lymphangiogenic Growth Factors Vascular Endothelial Growth Factor (VEGF)-C and -D. J. Biol. Chem. 2016, 291, 27265–27278. [Google Scholar] [CrossRef] [Green Version]
- Mabeta, P.; Steenkamp, V. The VEGF/VEGFR Axis Revisited: Implications for Cancer Therapy. Int. J. Mol. Sci. 2022, 23, 15585. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef] [PubMed]
- Daly, J.L.; Simonetti, B.; Klein, K.; Chen, K.E.; Williamson, M.K.; Anton-Plagaro, C.; Shoemark, D.K.; Simon-Gracia, L.; Bauer, M.; Hollandi, R.; et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science 2020, 370, 861–865. [Google Scholar] [CrossRef]
- Grunewald, M.; Kumar, S.; Sharife, H.; Volinsky, E.; Gileles-Hillel, A.; Licht, T.; Permyakova, A.; Hinden, L.; Azar, S.; Friedmann, Y.; et al. Counteracting age-related VEGF signaling insufficiency promotes healthy aging and extends life span. Science 2021, 373, eabc8479. [Google Scholar] [CrossRef]
- Calvo, P.M.; Hernandez, R.G.; de la Cruz, R.R.; Pastor, A.M. VEGF is an essential retrograde trophic factor for motoneurons. Proc. Natl. Acad. Sci. USA 2022, 119, e2202912119. [Google Scholar] [CrossRef] [PubMed]
- Arjunan, P.; Lin, X.C.; Tang, Z.S.; Du, Y.X.; Kumar, A.; Liu, L.X.; Yin, X.K.; Huang, L.J.; Chen, W.; Chen, Q.S.; et al. VEGF-B is a potent antioxidant. Proc. Natl. Acad. Sci. USA 2018, 115, 10351–10356. [Google Scholar] [CrossRef] [Green Version]
- Di Stasi, R.; De Rosa, L.; Romanelli, A.; D’Andrea, L.D. Peptides Interacting with Growth Factor Receptors Regulating Angiogenesis. In Frontier in Medicinal Chemistry; Atta-ur-Rahman, M., Choudhary, I., Reitz, A.B., Eds.; Bentham Science Publishers: Sharjah, Arab Emirates, 2016; Volume 9, pp. 103–160. [Google Scholar] [CrossRef] [Green Version]
- Browne, S.; Pandit, A. Engineered systems for therapeutic angiogenesis. Curr. Opin. Pharmacol. 2017, 36, 34–43. [Google Scholar] [CrossRef]
- Garcia, J.; Hurwitz, H.I.; Sandler, A.B.; Miles, D.; Coleman, R.L.; Deurloo, R.; Chinot, O.L. Bevacizumab (Avastin (R)) in cancer treatment: A review of 15 years of clinical experience and future outlook. Cancer Treat. Rev. 2020, 86, 102017. [Google Scholar] [CrossRef] [PubMed]
- Holash, J.; Davis, S.; Papadopoulos, N.; Croll, S.D.; Ho, L.; Russell, M.; Boland, P.; Leidich, R.; Hylton, D.; Burova, E.; et al. VEGF-Trap: A VEGF blocker with potent antitumor effects. Proc. Natl. Acad. Sci. USA 2002, 99, 11393–11398. [Google Scholar] [CrossRef] [Green Version]
- Kong, D.H.; Kim, M.R.; Jang, J.H.; Na, H.J.; Lee, S. A Review of Anti-Angiogenic Targets for Monoclonal Antibody Cancer Therapy. Int. J. Mol. Sci. 2017, 18, 1786. [Google Scholar] [CrossRef] [Green Version]
- Xue, L.; Gao, X.; Zhang, H.; Tang, J.; Wang, Q.; Li, F.; Li, X.; Yu, X.; Lu, Z.; Huang, Y.; et al. Antiangiogenic antibody BD0801 combined with immune checkpoint inhibitors achieves synergistic antitumor activity and affects the tumor microenvironment. BMC Cancer 2021, 21, 1134. [Google Scholar] [CrossRef]
- Musumeci, F.; Radi, M.; Brullo, C.; Schenone, S. Vascular endothelial growth factor (VEGF) receptors: Drugs and new inhibitors. J. Med. Chem. 2012, 55, 10797–10822. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Zhou, J.; Zhang, Z.; Guo, M.; Liang, J.; Zhou, F.; Long, J.; Zhang, W.; Yin, F.; Cai, H.; et al. Discovery of fruquintinib, a potent and highly selective small molecule inhibitor of VEGFR 1, 2, 3 tyrosine kinases for cancer therapy. Cancer Biol. Ther. 2014, 15, 1635–1645. [Google Scholar] [CrossRef] [Green Version]
- Henninot, A.; Collins, J.C.; Nuss, J.M. The Current State of Peptide Drug Discovery: Back to the Future? J. Med. Chem. 2018, 61, 1382–1414. [Google Scholar] [CrossRef]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic peptides: Current applications and future directions. Signal. Transduct. Target. Ther. 2022, 7, 48. [Google Scholar] [CrossRef]
- Muttenthaler, M.; King, G.F.; Adams, D.J.; Alewood, P.F. Trends in peptide drug discovery. Nat. Rev. Drug Discov. 2021, 20, 309–325. [Google Scholar] [CrossRef]
- Cabri, W.; Cantelmi, P.; Corbisiero, D.; Fantoni, T.; Ferrazzano, L.; Martelli, G.; Mattellone, A.; Tolomelli, A. Therapeutic Peptides Targeting PPI in Clinical Development: Overview, Mechanism of Action and Perspectives. Front. Mol. Biosci. 2021, 8, 697586. [Google Scholar] [CrossRef]
- Belvisi, L.; D’Andrea, L.D.; Jimenez, M.A. Editorial: Peptides Targeting Protein-Protein Interactions: Methods and Applications. Front. Mol. Biosci. 2021, 8, 780106. [Google Scholar] [CrossRef]
- Lee, A.C.; Harris, J.L.; Khanna, K.K.; Hong, J.H. A Comprehensive Review on Current Advances in Peptide Drug Development and Design. Int. J. Mol. Sci. 2019, 20, 2383. [Google Scholar] [CrossRef] [Green Version]
- Basith, S.; Manavalan, B.; Hwan Shin, T.; Lee, G. Machine intelligence in peptide therapeutics: A next-generation tool for rapid disease screening. Med. Res. Rev. 2020, 40, 1276–1314. [Google Scholar] [CrossRef] [PubMed]
- Erak, M.; Bellmann-Sickert, K.; Els-Heindl, S.; Beck-Sickinger, A.G. Peptide chemistry toolbox-Transforming natural peptides into peptide therapeutics. Bioorgan. Med. Chem. 2018, 26, 2759–2765. [Google Scholar] [CrossRef] [PubMed]
- Hoppenz, P.; Els-Heindl, S.; Beck-Sickinger, A.G. Peptide-Drug Conjugates and Their Targets in Advanced Cancer Therapies. Front. Chem. 2020, 8, 571. [Google Scholar] [CrossRef]
- Sharma, A.; Kumar, N.; Kuppermann, B.D.; Francesco, B.; Loewenstein, A. Biotherapeutics and immunogenicity: Ophthalmic perspective. Eye 2019, 33, 1359–1361. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, L.; Diana, D.; Basile, A.; Russomanno, A.; Isernia, C.; Turco, M.C.; Fattorusso, R.; D’Andrea, L.D. Design, structural and biological characterization of a VEGF inhibitor beta-hairpin-constrained peptide. Eur. J. Med. Chem. 2014, 73, 210–216. [Google Scholar] [CrossRef]
- De Rosa, L.; Capasso, D.; Diana, D.; Stefania, R.; Di Stasi, R.; Fattorusso, R.; D’Andrea, L.D. Metabolic and conformational stabilization of a VEGF-mimetic beta-hairpin peptide by click-chemistry. Eur. J. Med. Chem. 2021, 222, 113575. [Google Scholar] [CrossRef] [PubMed]
- Dubey, S.K.; Parab, S.; Dabholkar, N.; Agrawal, M.; Singhvi, G.; Alexander, A.; Bapat, R.A.; Kesharwani, P. Oral peptide delivery: Challenges and the way ahead. Drug Discov. Today 2021, 26, 931–950. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J. Advances in oral peptide therapeutics. Nat. Rev. Drug Discov. 2020, 19, 277–289. [Google Scholar] [CrossRef]
- Zizzari, A.T.; Pliatsika, D.; Gall, F.M.; Fischer, T.; Riedl, R. New perspectives in oral peptide delivery. Drug Discov. Today 2021, 26, 1097–1105. [Google Scholar] [CrossRef]
- Anselmo, A.C.; Gokarn, Y.; Mitragotri, S. Non-invasive delivery strategies for biologics. Nat. Rev. Drug Discov. 2019, 18, 19–40. [Google Scholar] [CrossRef]
- Tong, T.; Wang, L.Y.; You, X.R.; Wu, J. Nano and microscale delivery platforms for enhanced oral peptide/protein bioavailability. Biomater. Sci. 2020, 8, 5804–5823. [Google Scholar] [CrossRef]
- Cooper, B.M.; Iegre, J.; O’ Donovan, D.H.; Halvarsson, M.O.; Spring, D.R. Peptides as a platform for targeted therapeutics for cancer: Peptide-drug conjugates (PDCs). Chem. Soc. Rev. 2021, 50, 1480–1494. [Google Scholar] [CrossRef]
- Li, C.M.; Haratipour, P.; Lingeman, R.G.; Perry, J.J.P.; Gu, L.; Hickey, R.J.; Malkas, L.H. Novel Peptide Therapeutic Approaches for Cancer Treatment. Cells 2021, 10, 2908. [Google Scholar] [CrossRef] [PubMed]
- Hawala, I.; De Rosa, L.; Aime, S.; D’Andrea, L.D. An innovative approach for the synthesis of dual modality peptide imaging probes based on the native chemical ligation approach. Chem. Commun. 2020, 56, 3500–3503. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Y.; Liu, J.C.; Li, T.Q.; Wang, Y.L.; Liu, X.M.; Bai, Y.W.; Wang, C.Y.; Ju, S.G.; Huang, S.J.; Yang, C.T.; et al. A VEGFR targeting peptide-drug conjugate (PDC) suppresses tumor angiogenesis in a TACE model for hepatocellular carcinoma therapy. Cell Death Discov. 2022, 8, 411. [Google Scholar] [CrossRef] [PubMed]
- Michigami, M.; Takahashi, K.; Yamashita, H.; Ye, Z.M.; Nakase, I.; Fujii, I. A “ligand-targeting” peptide-drug conjugate: Targeted intracellular drug delivery by VEGF-binding helix-loop-helix peptides via receptor-mediated endocytosis. PLoS ONE 2021, 16, e0247045. [Google Scholar] [CrossRef] [PubMed]
- Zanjanchi, P.; Asghari, S.M.; Mohabatkar, H.; Shourian, M.; Ardestani, M.S. Conjugation of VEGFR1/R2-targeting peptide with gold nanoparticles to enhance antiangiogenic and antitumoral activity. J. Nanobiotechnol. 2022, 20, 7. [Google Scholar] [CrossRef]
- Baghban, R.; Ghasemali, S.; Farajnia, S.; Hoseinpoor, R.; Andarzi, S.; Zakariazadeh, M.; Zarredar, H. Design and In Silico Evaluation of a Novel Cyclic Disulfide-Rich anti-VEGF Peptide as a Potential Antiangiogenic Drug. Int. J. Pept. Res. Ther. 2021, 27, 2245–2256. [Google Scholar] [CrossRef]
- Checco, J.W.; Kreitler, D.F.; Thomas, N.C.; Belair, D.G.; Rettko, N.J.; Murphy, W.L.; Forest, K.T.; Gellman, S.H. Targeting diverse protein-protein interaction interfaces with alpha/beta-peptides derived from the Z-domain scaffold. Proc. Natl. Acad. Sci. USA 2015, 112, 4552–4557. [Google Scholar] [CrossRef] [Green Version]
- Karami, E.; Sabatier, J.M.; Behdani, M.; Irani, S.; Kazemi-Lomedasht, F. A nanobody-derived mimotope against VEGF inhibits cancer angiogenesis. J. Enzym. Inhib. Med. Chem. 2020, 35, 1233–1239. [Google Scholar] [CrossRef]
- Guryanov, I.; Korzhikov-Vlakh, V.; Bhattacharya, M.; Biondi, B.; Masiero, G.; Formaggio, F.; Tennikova, T.; Urtti, A. Conformationally Constrained Peptides with High Affinity to the Vascular Endothelial Growth Factor. J. Med. Chem. 2021, 64, 10900–10907. [Google Scholar] [CrossRef]
- D’Andrea, L.D.; Iaccarino, G.; Fattorusso, R.; Sorriento, D.; Carannante, C.; Capasso, D.; Trimarco, B.; Pedone, C. Targeting angiogenesis: Structural characterization and biological properties of a de novo engineered VEGF mimicking peptide. Proc. Natl. Acad. Sci. USA 2005, 102, 14215–14220. [Google Scholar] [CrossRef] [Green Version]
- Basile, A.; Del Gatto, A.; Diana, D.; Di Stasi, R.; Falco, A.; Festa, M.; Rosati, A.; Barbieri, A.; Franco, R.; Arra, C.; et al. Characterization of a Designed Vascular Endothelial Growth Factor Receptor Antagonist Helical Peptide with Antiangiogenic Activity in Vivo. J. Med. Chem. 2011, 54, 1391–1400. [Google Scholar] [CrossRef] [PubMed]
- Balsera, B.; Bonache, M.A.; Reille-Seroussi, M.; Gagey-Eilstein, N.; Vidal, M.; Gonzalez-Muniz, R.; de Vega, M.J.P. Disrupting VEGF-VEGFR1 Interaction: De Novo Designed Linear Helical Peptides to Mimic the VEGF(13-25) Fragment. Molecules 2017, 22, 1846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdulkadir, S.; Li, C.P.; Jiang, W.; Zhao, X.; Sang, P.; Wei, L.L.; Hu, Y.; Li, Q.; Cai, J.F. Modulating Angiogenesis by Proteomimetics of Vascular Endothelial Growth Factor. J. Am. Chem. Soc. 2022, 144, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Assareh, E.; Mehrnejad, F.; Mansouri, K.; Rastaghi, A.R.E.; Naderi-Manesh, H.; Asghari, S.M. A cyclic peptide reproducing the alpha 1 helix of VEGF-B binds to VEGFR-1 and VEGFR-2 and inhibits angiogenesis and tumor growth. Biochem. J. 2019, 476, 645–663. [Google Scholar] [CrossRef]
- Dmytriyeva, O.; Ajenjo, A.D.; Lundo, K.; Hertz, H.; Rasmussen, K.K.; Christiansen, A.T.; Klingelhofer, J.; Nielsen, A.L.; Hoeber, J.; Kozlova, E.; et al. Neurotrophic Effects of Vascular Endothelial Growth Factor B and Novel Mimetic Peptides on Neurons from the Central Nervous System. ACS Chem. Neurosci. 2020, 11, 1270–1282. [Google Scholar] [CrossRef]
- Garcia-Aranda, M.I.; Gonzalez-Lopez, S.; Santiveri, C.M.; Gagey-Eilstein, N.; Reille-Seroussi, M.; Martin-Martinez, M.; Inguimbert, N.; Vidal, M.; Garcia-Lopez, M.T.; Jimenez, M.A.; et al. Helical peptides from VEGF and Vammin hotspots for modulating the VEGF-VEGFR interaction. Org. Biomol. Chem. 2013, 11, 1896–1905. [Google Scholar] [CrossRef] [Green Version]
- Zanella, S.; Bocchinfuso, G.; De Zotti, M.; Arosio, D.; Marino, F.; Raniolo, S.; Pignataro, L.; Sacco, G.; Palleschi, A.; Siano, A.S.; et al. Rational Design of Antiangiogenic Helical Oligopeptides Targeting the Vascular Endothelial Growth Factor Receptors. Front. Chem. 2019, 7, 170. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zhou, L.Y.; Reille-Seroussi, M.; Gagey-Eilstein, N.; Broussy, S.; Zhang, T.Y.; Ji, L.L.; Vidal, M.; Liu, W.Q. Identification of Peptidic Antagonists of Vascular Endothelial Growth Factor Receptor 1 by Scanning the Binding Epitopes of Its Ligands. J. Med. Chem. 2017, 60, 6598–6606. [Google Scholar] [CrossRef]
- Goncalves, V.; Gautier, B.; Coric, P.; Bouaziz, S.; Lenoir, C.; Garbay, C.; Vidal, M.; Inguimbert, N. Rational design, structure, and biological evaluation of cyclic peptides mimicking the vascular endothelial growth factor. J. Med. Chem. 2007, 50, 5135–5146. [Google Scholar] [CrossRef]
- Zilberberg, L.; Shinkaruk, S.; Lequin, O.; Rousseau, B.; Hagedorn, M.; Costa, F.; Caronzolo, D.; Balke, M.; Canron, X.; Convert, O.; et al. Structure and inhibitory effects on angiogenesis and tumor development of a new vascular endothelial growth inhibitor. J. Biol. Chem. 2003, 278, 35564–35573. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Aranda, M.I.; Mirassou, Y.; Gautier, B.; Martin-Martinez, M.; Inguimbert, N.; Vidal, M.; Garcia-Lopez, M.T.; Jimenez, M.A.; Gonzalez-Muniz, R.; de Vega, M.J.P. Disulfide and amide-bridged cyclic peptide analogues of the VEGF(81-91) fragment: Synthesis, conformational analysis and biological evaluation. Bioorgan. Med. Chem. 2011, 19, 7526–7533. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Aranda, M.I.; Marrero, P.; Gautier, B.; Martin-Martinez, M.; Inguimbert, N.; Vidal, M.; Garcia-Lopez, M.T.; Jimenez, M.A.; Gonzalez-Muniz, R.; de Vega, M.J.P. Parallel solid-phase synthesis of a small library of linear and hydrocarbon-bridged analogues of VEGF(81-91): Potential biological tools for studying the VEGF/VEGFR-1 interaction. Bioorgan. Med. Chem. 2011, 19, 1978–1986. [Google Scholar] [CrossRef] [PubMed]
- Vicari, D.; Foy, K.C.; Liotta, E.M.; Kaumaya, P.T.P. Engineered Conformation-dependent VEGF Peptide Mimics Are Effective in Inhibiting VEGF Signaling Pathways. J. Biol. Chem. 2011, 286, 13612–13625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diana, D.; Basile, A.; De Rosa, L.; Di Stasi, R.; Auriemma, S.; Arra, C.; Pedone, C.; Turco, M.C.; Fattorusso, R.; D’Andrea, L.D. beta-Hairpin Peptide That Targets Vascular Endothelial Growth Factor (VEGF) Receptors DESIGN, NMR CHARACTERIZATION, AND BIOLOGICAL ACTIVITY. J. Biol. Chem. 2011, 286, 41680–41691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporale, A.; Martin, A.D.; Capasso, D.; Foca, G.; Sandomenico, A.; D’Andrea, L.D.; Grieco, P.; Ruvo, M.; Doti, N. Short PlGF-derived peptides bind VEGFR-1 and VEGFR-2 in vitro and on the surface of endothelial cells. J. Pept. Sci. 2019, 25, e3146. [Google Scholar] [CrossRef] [PubMed]
- Mirassou, Y.; Santiveri, C.M.; de Vega, M.J.P.; Gonzalez-Muniz, R.; Jimenez, M.A. Disulfide Bonds versus Trp center dot center dot center dot Trp Pairs in Irregular beta-Hairpins: NMR Structure of Vammin Loop 3-Derived Peptides as a Case Study. Chembiochem 2009, 10, 902–910. [Google Scholar] [CrossRef]
- Wang, L.; Coric, P.; Broussy, S.; Di Stasi, R.; Zhou, L.Y.; D’Andrea, L.D.; Ji, L.L.; Vidal, M.; Bouaziz, S.; Liu, W.Q. Structural studies of the binding of an antagonistic cyclic peptide to the VEGFR1 domain 2. Eur. J. Med. Chem. 2019, 169, 65–75. [Google Scholar] [CrossRef]
- Goncalves, V.; Gautier, B.; Garbay, C.; Vidal, M.; Inguimbert, N. Structure-based design of a bicyclic peptide antagonist of the vascular endothelial growth factor receptors. J. Pept. Sci. 2008, 14, 767–772. [Google Scholar] [CrossRef]
- De Rosa, L.; Finetti, F.; Diana, D.; Di Stasi, R.; Auriemma, S.; Romanelli, A.; Fattorusso, R.; Ziche, M.; Morbidelli, L.; D’Andrea, L.D. Miniaturizing VEGF: Peptides mimicking the discontinuous VEGF receptor-binding site modulate the angiogenic response. Sci. Rep. 2016, 6, 31295. [Google Scholar] [CrossRef] [Green Version]
- Sadremomtaz, A.; Mansouri, K.; Alemzadeh, G.; Safa, M.; Rastaghi, A.E.; Asghari, S.M. Dual blockade of VEGFR1 and VEGFR2 by a novel peptide abrogates VEGF-driven angiogenesis, tumor growth, and metastasis through PI3K/AKT and MAPK/ERK1/2 pathway. Bba-Gen. Subj. 2018, 1862, 2688–2700. [Google Scholar] [CrossRef]
- Behelgardi, M.F.; Zahri, S.; Mashayekhi, F.; Mansouri, K.; Asghari, S.M. A peptide mimicking the binding sites of VEGF-A and VEGF-B inhibits VEGFR-1/-2 driven angiogenesis, tumor growth and metastasis. Sci. Rep. 2018, 8, 17924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guryanov, I.; Tennikova, T.; Urtti, A. Peptide Inhibitors of Vascular Endothelial Growth Factor A: Current Situation and Perspectives. Pharmaceutics 2021, 13, 1337. [Google Scholar] [CrossRef] [PubMed]
- Brozzo, M.S.; Bjelic, S.; Kisko, K.; Schleier, T.; Leppanen, V.M.; Alitalo, K.; Winkler, F.K.; Ballmer-Hofer, K. Thermodynamic and structural description of allosterically regulated VEGFR-2 dimerization. Blood 2012, 119, 1781–1788. [Google Scholar] [CrossRef] [PubMed]
- Checco, J.W.; Gellman, S.H. Iterative Nonproteinogenic Residue Incorporation Yields alpha/beta-Peptides with a Helix-Loop-Helix Tertiary Structure and High Affinity for VEGF. Chembiochem 2017, 18, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Wiesmann, C.; Fuh, G.; Christinger, H.W.; Eigenbrot, C.; Wells, J.A.; deVos, A.M. Crystal structure at 1.7 angstrom resolution of VEGF in complex with domain 2 of the Flt-1 receptor. Cell 1997, 91, 695–704. [Google Scholar] [CrossRef] [Green Version]
- De Rosa, L.; Di Stasi, R.; D’Andrea, L.D. Pro-angiogenic peptides in biomedicine. Arch. Biochem. Biophys. 2018, 660, 72–86. [Google Scholar] [CrossRef]
- Di Stasi, R.; Diana, D.; Capasso, D.; Di Gaetano, S.; De Rosa, L.; Celentano, V.; Isernia, C.; Fattorusso, R.; D’Andrea, L.D. VEGFR Recognition Interface of a Proangiogenic VEGF-Mimetic Peptide Determined In Vitro and in the Presence of Endothelial Cells by NMR Spectroscopy. Chem.-Eur. J. 2018, 24, 11461–11466. [Google Scholar] [CrossRef]
- Ziaco, B.; Diana, D.; Capasso, D.; Palumbo, R.; Celentano, V.; Di Stasi, R.; Fattorusso, R.; D’Andrea, L.D. C-terminal truncation of Vascular Endothelial Growth Factor mimetic helical peptide preserves structural and receptor binding properties. Biochem. Biophys. Res. Comm. 2012, 424, 290–294. [Google Scholar] [CrossRef]
- De Rosa, L.; Diana, D.; Di Stasi, R.; Romanelli, A.; Sciacca, M.F.M.; Milardi, D.; Isernia, C.; Fattorusso, R.; D’Andrea, L.D. Probing the helical stability in a VEGF-mimetic peptide. Bioorg. Chem. 2021, 116, 105379. [Google Scholar] [CrossRef]
- De Rosa, L.; Diana, D.; Capasso, D.; Stefania, R.; Di Stasi, R.; Fattorusso, R.; D’Andrea, L.D. Switching the N-Capping Region from all-L to all-D Amino Acids in a VEGF Mimetic Helical Peptide. Molecules 2022, 27, 6982. [Google Scholar] [CrossRef]
- Diana, D.; Ziaco, B.; Scarabelli, G.; Pedone, C.; Colombo, G.; D’Andrea, L.D.; Fattorusso, R. Structural Analysis of a Helical Peptide Unfolding Pathway. Chem.-Eur. J. 2010, 16, 5400–5407. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, L.D.; De Rosa, L.; Vigliotti, C.; Cataldi, M. VEGF mimic peptides: Potential applications in central nervous system therapeutics. New Horiz. Transl. Med. 2017, 3, 233–251. [Google Scholar] [CrossRef] [Green Version]
- Capasso, D.; Di Gaetano, S.; Celentano, V.; Diana, D.; Festa, L.; Di Stasi, R.; De Rosa, L.; Fattorusso, R.; D’Andrea, L.D. Unveiling a VEGF-mimetic peptide sequence in the IQGAP1 protein. Mol. Biosyst. 2017, 13, 1619–1629. [Google Scholar] [CrossRef] [PubMed]
- Diana, D.; Di Stasi, R.; De Rosa, L.; Isernia, C.; D’Andrea, L.D.; Fattorusso, R. Structural investigation of the VEGF receptor interaction with a helical antagonist peptide. J. Pept. Sci. 2013, 19, 214–219. [Google Scholar] [CrossRef] [PubMed]
- She, F.Y.; Teng, P.; Peguero-Tejada, A.; Wang, M.H.; Ma, N.; Odom, T.; Zhou, M.; Gjonaj, E.; Wojtas, L.; van der Vaart, A.; et al. De Novo Left-Handed Synthetic Peptidomimetic Foldamers. Angew. Chem. Int. Edit. 2018, 57, 9916–9920. [Google Scholar] [CrossRef]
- Xue, S.Y.; Wang, L.; Cai, J.F. Sulfono-gamma-AApeptides as Protein Helical Domain Mimetics to Manipulate the Angiogenesis. Chembiochem 2022, 23, e202200298. [Google Scholar] [CrossRef]
- Assareh, E.; Mehrnejad, F.; Asghari, S.M. Structural Studies on an Anti-Angiogenic Peptide Using Molecular Modeling. Iran. J. Biotechnol. 2020, 18, e2553. [Google Scholar] [CrossRef]
- Wang, L.; Xu, M.; Hu, H.F.; Zhang, L.; Ye, F.; Jin, J.; Fang, H.M.; Chen, J.; Chen, G.Q.; Broussy, S.; et al. A Cyclic Peptide Epitope of an Under-Explored VEGF-B Loop 1 Demonstrated In Vivo Anti-Angiogenic and Anti-Tumor Activities. Front. Pharmacol. 2021, 12, 734544. [Google Scholar] [CrossRef]
- Kim, K.H.; Hur, J.; Lee, H.Y.; Lee, E.G.; Lee, S.Y. Cyclo-VEGI inhibits bronchial artery remodeling in a murine model of chronic asthma. Exp. Lung. Res. 2021, 47, 494–506. [Google Scholar] [CrossRef]
- Foy, K.C.; Liu, Z.Z.; Phillips, G.; Miller, M.; Kaumaya, P.T.P. Combination Treatment with HER-2 and VEGF Peptide Mimics Induces Potent Anti-tumor and Anti-angiogenic Responses in Vitro and in Vivo. J. Biol. Chem. 2011, 286, 13626–13637. [Google Scholar] [CrossRef] [Green Version]
- Diana, D.; Russomanno, A.; De Rosa, L.; Di Stasi, R.; Capasso, D.; Di Gaetano, S.; Romanelli, A.; Russo, L.; D’Andrea, L.D.; Fattorusso, R. Functional Binding Surface of a beta-Hairpin VEGF Receptor Targeting Peptide Determined by NMR Spectroscopy in Living Cells. Chem.-Eur. J. 2015, 21, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Diana, D.; De Rosa, L.; Palmieri, M.; Russomanno, A.; Russo, L.; La Rosa, C.; Milardi, D.; Colombo, G.; D’Andrea, L.D.; Fattorusso, R. Long range Trp-Trp interaction initiates the folding pathway of a pro-angiogenic beta-hairpin peptide. Sci. Rep. 2015, 5, 16651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celentano, V.; Diana, D.; Di Salvo, C.; De Rosa, L.; Romanelli, A.; Fattorusso, R.; D’Andrea, L.D. 1,2,3-Triazole Bridge as Conformational Constrain in -Hairpin Peptides: Analysis of Hydrogen-Bonded Positions. Chem.-Eur. J. 2016, 22, 5534–5537. [Google Scholar] [CrossRef]
- Diana, D.; Di Salvo, C.; Celentano, V.; De Rosa, L.; Romanelli, A.; Fattorusso, R.; D’Andrea, L.D. Conformational stabilization of a beta-hairpin through a triazole-tryptophan interaction. Org. Biomol. Chem. 2018, 16, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Celentano, V.; Diana, D.; De Rosa, L.; Romanelli, A.; Fattorusso, R.; D’Andrea, L.D. beta-Hairpin stabilization through an interstrand triazole bridge. Chem. Commun. 2012, 48, 762–764. [Google Scholar] [CrossRef]
- Gautier, B.; Goncalves, V.; Diana, D.; Di Stasi, R.; Teillet, F.; Lenoir, C.; Huguenot, F.; Garbay, C.; Fattorusso, R.; D’Andrea, L.D.; et al. Biochemical and Structural Analysis of the Binding Determinants of a Vascular Endothelial Growth Factor Receptor Peptidic Antagonist. J. Med. Chem. 2010, 53, 4428–4440. [Google Scholar] [CrossRef]
- Markovic-Mueller, S.; Stuttfeld, E.; Asthana, M.; Weinert, T.; Bliven, S.; Goldie, K.N.; Kisko, K.; Capitani, G.; Ballmer-Hofer, K. Structure of the Full-length VEGFR-1 Extracellular Domain in Complex with VEGF-A. Structure 2017, 25, 341–352. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Sequence a | Target | Mimicking m | Affinity | Ref. |
|---|---|---|---|---|---|
| - | NGIDFNRDKFLFL | VEGF | VEGFR-2 | - | [47] |
| α/β-VEGF-1 | V(β3-Val)NK(β3-Phe)NKE(ACPC)CN (APC)RAIE(Aib)ALDPNL NDQQFH (Aib)KIW(APC)II(APC)DC b | VEGF | Z-VEGF (VEGF affibody) | Ki = 0.11 μM | [48] |
| - | YY(Abu)AARAWSPYSSTVDAGDFRYWGQ-amide c | VEGF | Nd42 (anti-VEGF nanobody) | KA = 51 × 109 M | [49] |
| Aib2 | V(Aib)PNCDIHV(nL)WEWECFERL d | VEGF | V114* peptide | KD = 4 nM | [50] |
| Kv114* | K(Aib)KKCDIHV(nL)WEWECFERL d | VEGF | V114* peptide | KD = 540 nM | [50] |
| QK | Ac-KLTWQELYQLKYKGI-amide | VEGFR1/R2 | α1 VEGF-A | KD = 64 μM | [51] |
| MA | Ac-KLTWMELYQLAYKGI-amide | VEGFR1/R2 | α1 VEGF-A | KD = 46 μM | [52] |
| #29 | Ac-SSEEFARNWAAIN-amide | VEGFR1 | α1 VEGF-A | IC50 = 0.05 μM | [53] |
| V2 |  | VEGFR1/R2 | α1 VEGF-A | KD = 0.46 μM | [54] |
| V3 |  | VEGFR1/R2 | α1 VEGF-A | KD = 0.63 μM | [54] |
| VGB1 | CSWIDVYTRATCQPRPL | VEGFR1/R2 | α1 VEGF-B | - | [55] |
| Vefin7 | KAVSWVDVYTAAT-scaffold e | VEGFR1 | α1 VEGF-B | KD = 0.167 μM | [56] |
| #16 | Ac-EV-cycle(NHCO)3,7[EKFM(O2) K] VYQRSY-amide | VEGFR1 | α1 Vammin | IC50 = 36 μM | [57] |
| #5 | Ac-W(αMeAsp)N(αMeAsp)WR(Api)TW-amide f | VEGFR1/R2 | α1 VEGF-C | - | [58] |
| #6 | Ac-(Api)WDN (αMeAsp)WR(Api)TW-amide f | VEGFR1/R2 | α1 VEGF-C | - | [58] |
| #14 | CFQEYPDEIEYICK-amide | VEGFR1 | L1 VEGF-A | IC50 = 50.4 μM | [59] |
| #18 | Ac-CTVELMGTVAKQLVPC-amide | VEGFR1 | L1 VEGF-B | IC50 = 10.4 μM | [59] |
| #19 | CSEYPSEVEHMCS-amide | VEGFR1 | L1 PlGF | IC50 = 56.0 μM | [59] |
| #2 | Ac-CNDEGLEC-amide | VEGFR-1 | L2 VEGF-A | - | [60] |
| Cyclo-VEGI | Cycle(fPQIMRIKPHQGQHIGE) | VEGFR1/R2 | L3 VEGF-A | IC50 = 0.7 μM | [61] |
| #7 | Ac-M-cycle(CH2NHCOCH2)2,10 [GIKPHQGQG]I-amide | VEGFR1 | L3 VEGF-A | IC50 = 93.2 μM | [62] |
| #8 | Acetyl-M(aGly)GIKPHQQ(aGly)I-amide g | VEGFR1 | L3 VEGF-A | IC50 = 42.3 μM | [63] |
| VEGF-P3CYC | Acetyl-ITMQCGIHQGQHPKIRMIC EMSF-amide | VEGFR2 | L3 VEGF-A | KD = 11 nM | [64] |
| HPLW | KQLLWIRSGDRPWYYTS | VEGFR1/R2 | L3 PlGF | KD = 32 μM | [65] |
| HPLC | KQCLWIRSGDRPWYCTS | VEGFR1/R2 | L3 PlGF | - | [34] |
| HPLW2 | Ac-KQLL(Bpg)IRSGDRP(OrnN3) YWTS-amide h | VEGFR1/R2 | L3 PlGF | - | [35] |
| PLGF2 | QLLKIRSHDRPSYVE-amide | VEGFR1 | L3 PlGF | KD = 86 μM | [66] |
| C1C12W3W10 | CRWNPRTQSWKC | VEGFR2 | L3 Vammin | - | [67] |
| #7 | c[YYDEGLEE]-amide | VEGFR1 | L2/α1 VEGF-A | IC50 = 39.9 μM | [60] |
| #3 | c[YKDEGLEE]-NHCH2CH2Ph(3,4-diOH) | VEGFR1 | L2/α1 VEGF-A | IC50 = 196 μM | [68] |
| VG3F | KFMDVYQRSY(Ahx)elGedncs(Ahx)ECRPK-amide i | VEGFR1 | α1/L2/L3 VEGF-A | IC50 = 52 μM | [69] |
| EP6 | Ac-E(C-7Ahp-QIMRIKPHQGQHIGET S)KFMDVYQLKYKGI-amide l | VEGFR1/R2 | α1/L3 VEGF-A | KD = 139 μM | [70] |
| VGB | CIKPHQGQHICNDE | VEGFR1/R2 | L2/L3 VEGF-A | - | [71] |
| VGB4 | KQLVIKPHGQILMIRYPSSQLEM | VEGFR1/R2 | L1/L2 VEGF-B + L3 VEGF-A | - | [72] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Stasi, R.; De Rosa, L.; D’Andrea, L.D. Structure-Based Design of Peptides Targeting VEGF/VEGFRs. Pharmaceuticals 2023, 16, 851. https://doi.org/10.3390/ph16060851
Di Stasi R, De Rosa L, D’Andrea LD. Structure-Based Design of Peptides Targeting VEGF/VEGFRs. Pharmaceuticals. 2023; 16(6):851. https://doi.org/10.3390/ph16060851
Chicago/Turabian StyleDi Stasi, Rossella, Lucia De Rosa, and Luca Domenico D’Andrea. 2023. "Structure-Based Design of Peptides Targeting VEGF/VEGFRs" Pharmaceuticals 16, no. 6: 851. https://doi.org/10.3390/ph16060851
APA StyleDi Stasi, R., De Rosa, L., & D’Andrea, L. D. (2023). Structure-Based Design of Peptides Targeting VEGF/VEGFRs. Pharmaceuticals, 16(6), 851. https://doi.org/10.3390/ph16060851