


On the Road towards Small-Molecule Programmed Cell Death 1 Ligand 1 Positron Emission Tomography Tracers: A Ligand-Based Drug Design Approach

 , , , , and
, , , , and 
Abstract
:1. Introduction
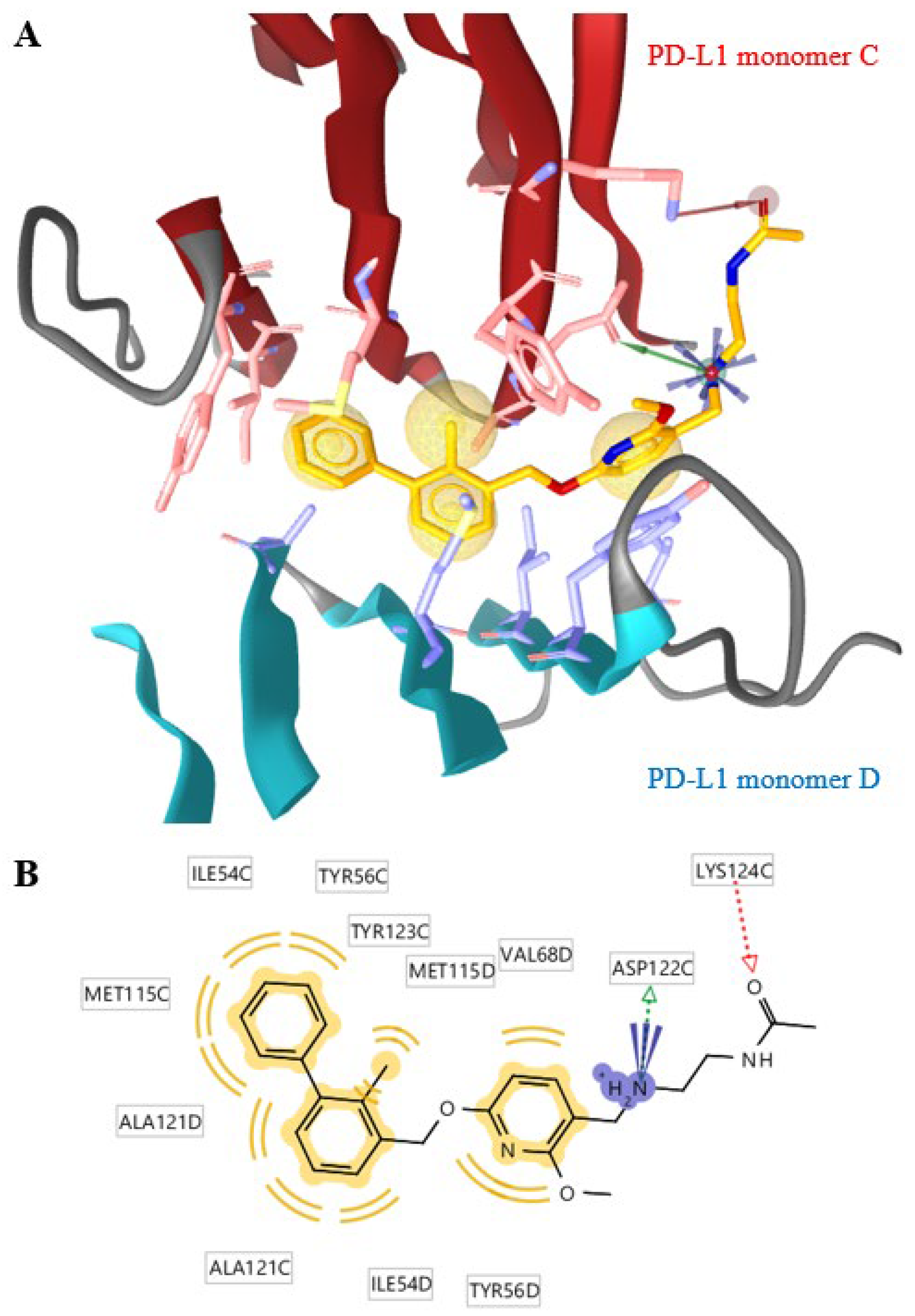
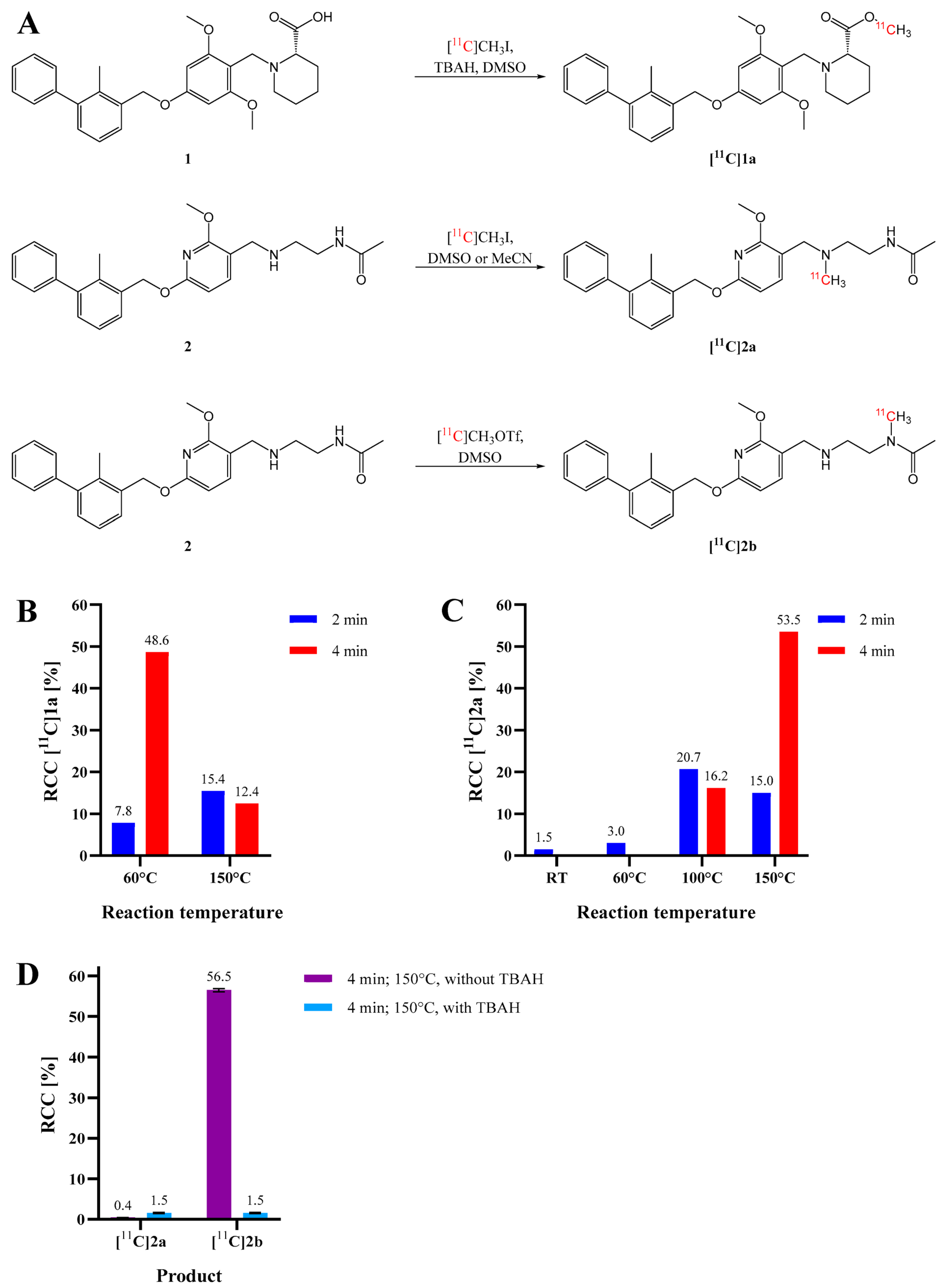
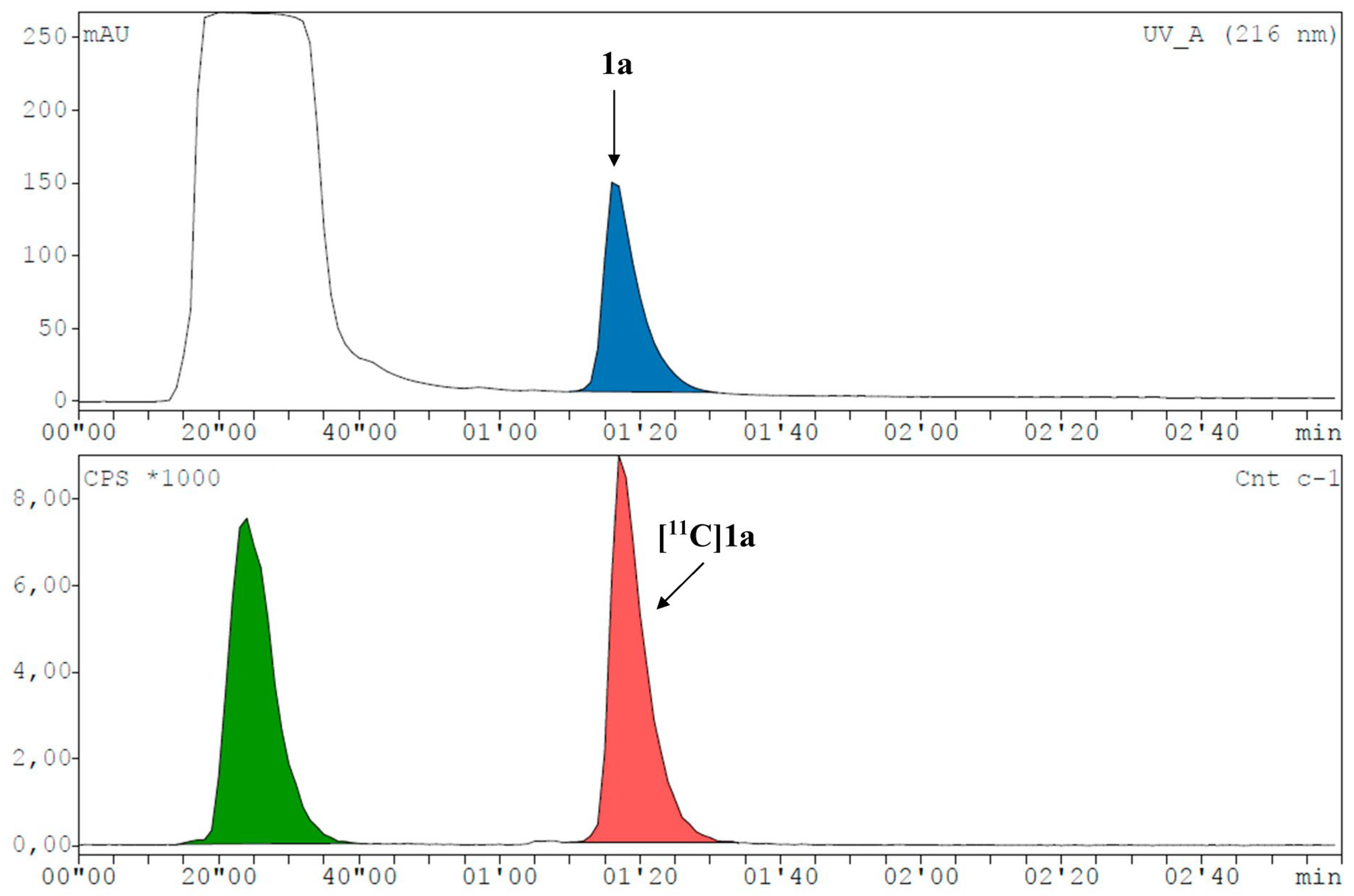
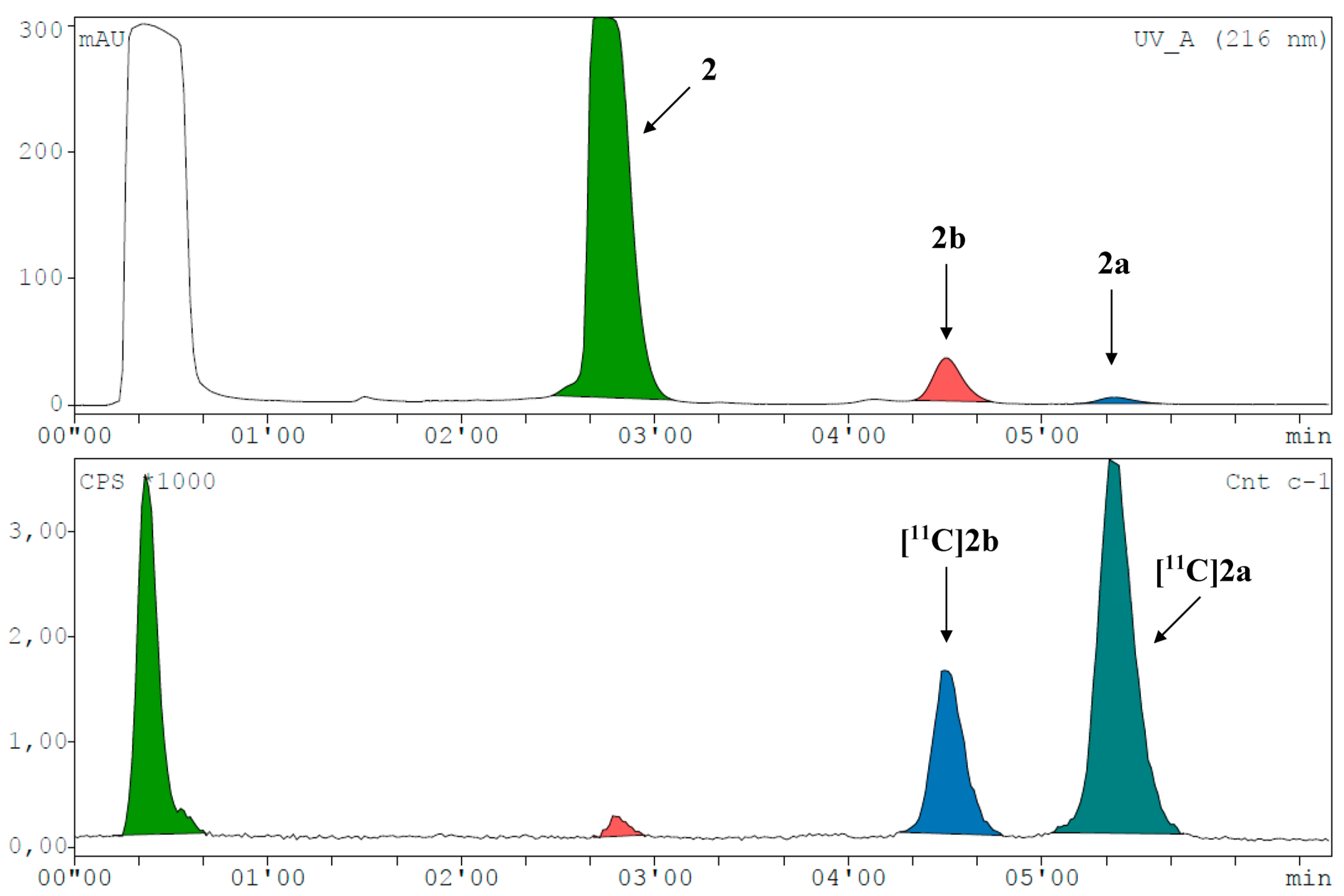
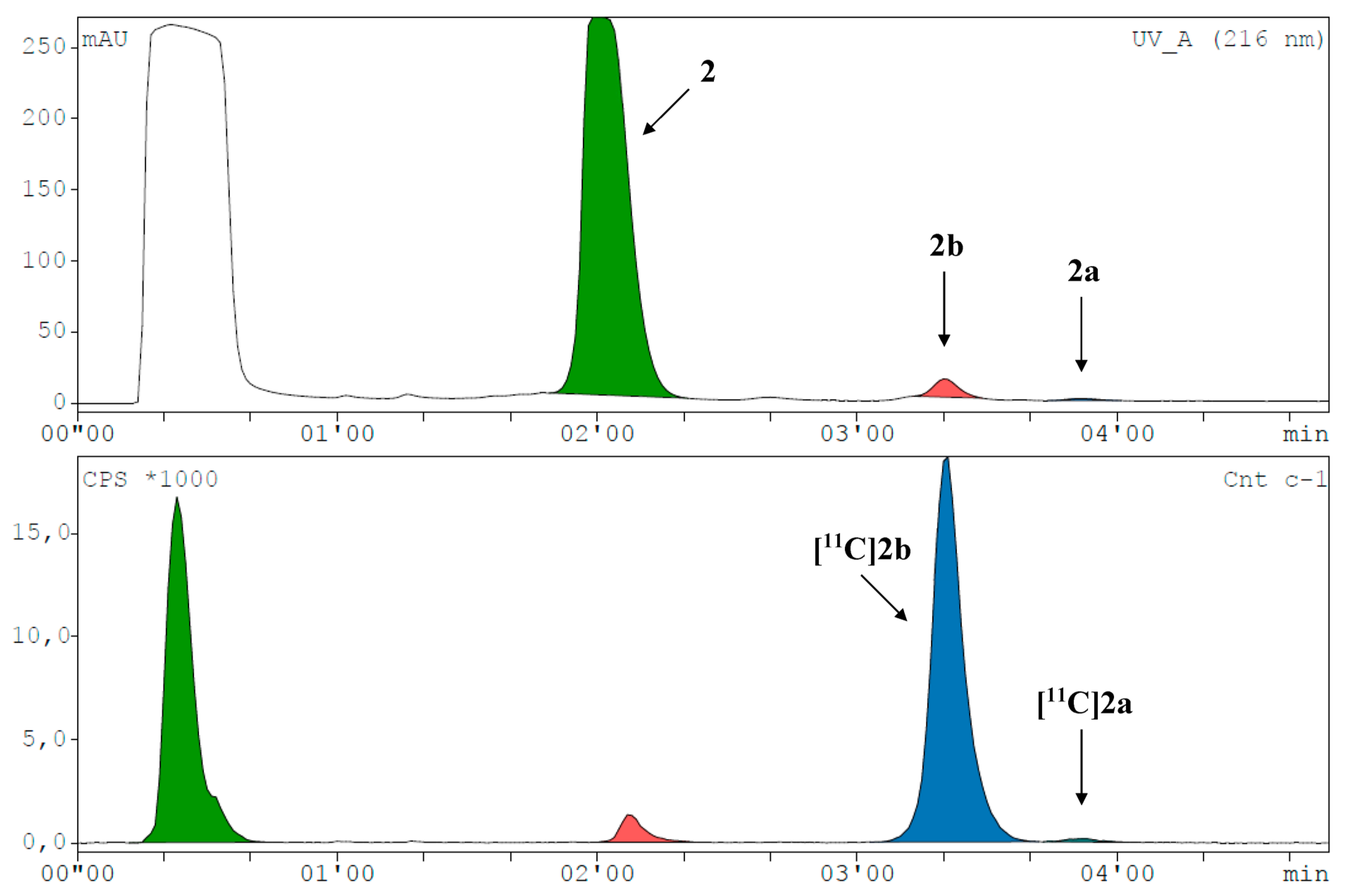
2. Results and Discussion
3. Experiments
3.1. General Information
3.2. Methods
3.2.1. High-Performance Liquid Chromatography
3.2.2. Characterization
3.3. Ligand Docking Experiments
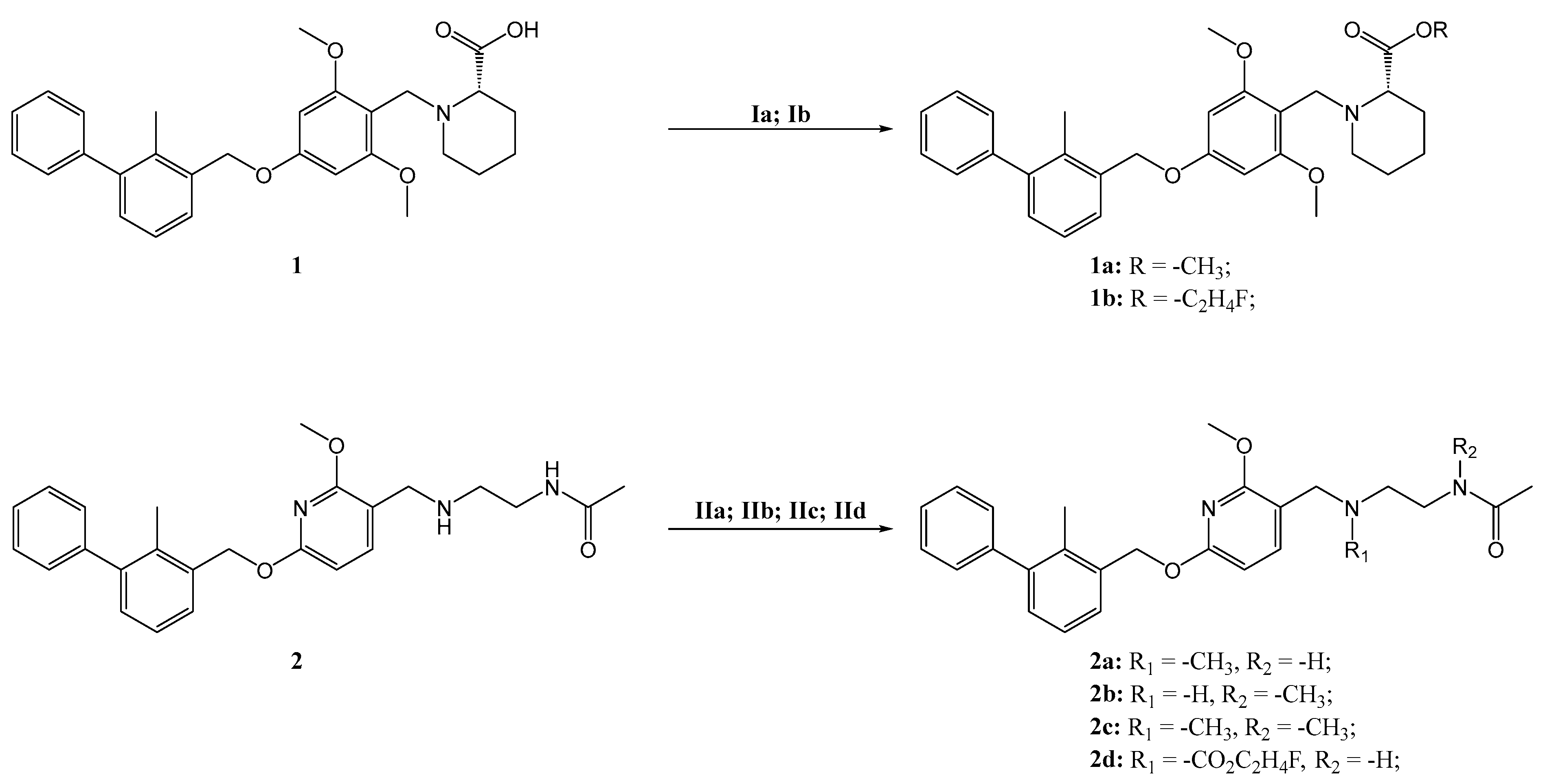
3.4. Syntheses
3.4.1. Synthesis of Methyl (S)-1-(2,6-Dimethoxy-4-((2-methyl-[1,1′-biphenyl]-3-yl)methoxy)benzyl)piperidine-2-carboxylate (1a)
3.4.2. Synthesis of 2-Fluoroethyl (S)-1-(2,6-Dimethoxy-4-((2-methyl-[1,1′-biphenyl]-3-yl)methoxy)benzyl)piperidine-2-carboxylate (1b)
3.4.3. Synthesis of N-(2-(((2-Methoxy-6-((2-methyl-[1,1′-biphenyl]-3-yl)methoxy)pyridin-3-yl)methyl)(methyl)amino)ethyl)acetamide (2a)
3.4.4. Synthesis of N-(2-(((2-Methoxy-6-((2-methyl-[1,1′-biphenyl]-3-yl)methoxy)pyridin-3-yl)methyl)amino)ethyl)-N-methylacetamide (2b)
3.4.5. Synthesis of N-(2-(((2-Methoxy-6-((2-methyl-[1,1′-biphenyl]-3-yl)methoxy)pyridin-3-yl)methyl)(methyl)amino)ethyl)-N-methylacetamide (2c)
3.4.6. Synthesis of 2-Fluoroethyl (2-Acetamidoethyl)((2-methoxy-6-((2-methyl-[1,1′-biphenyl]-3-yl)methoxy)pyridin-3-yl)methyl)carbamate (2d)
3.5. In-Solution Stability Measurements
3.6. Lipophilicity and Calculated Physicochemical Properties
3.7. Binding Affinity Measurements
3.8. Radiosyntheses with Carbon-11
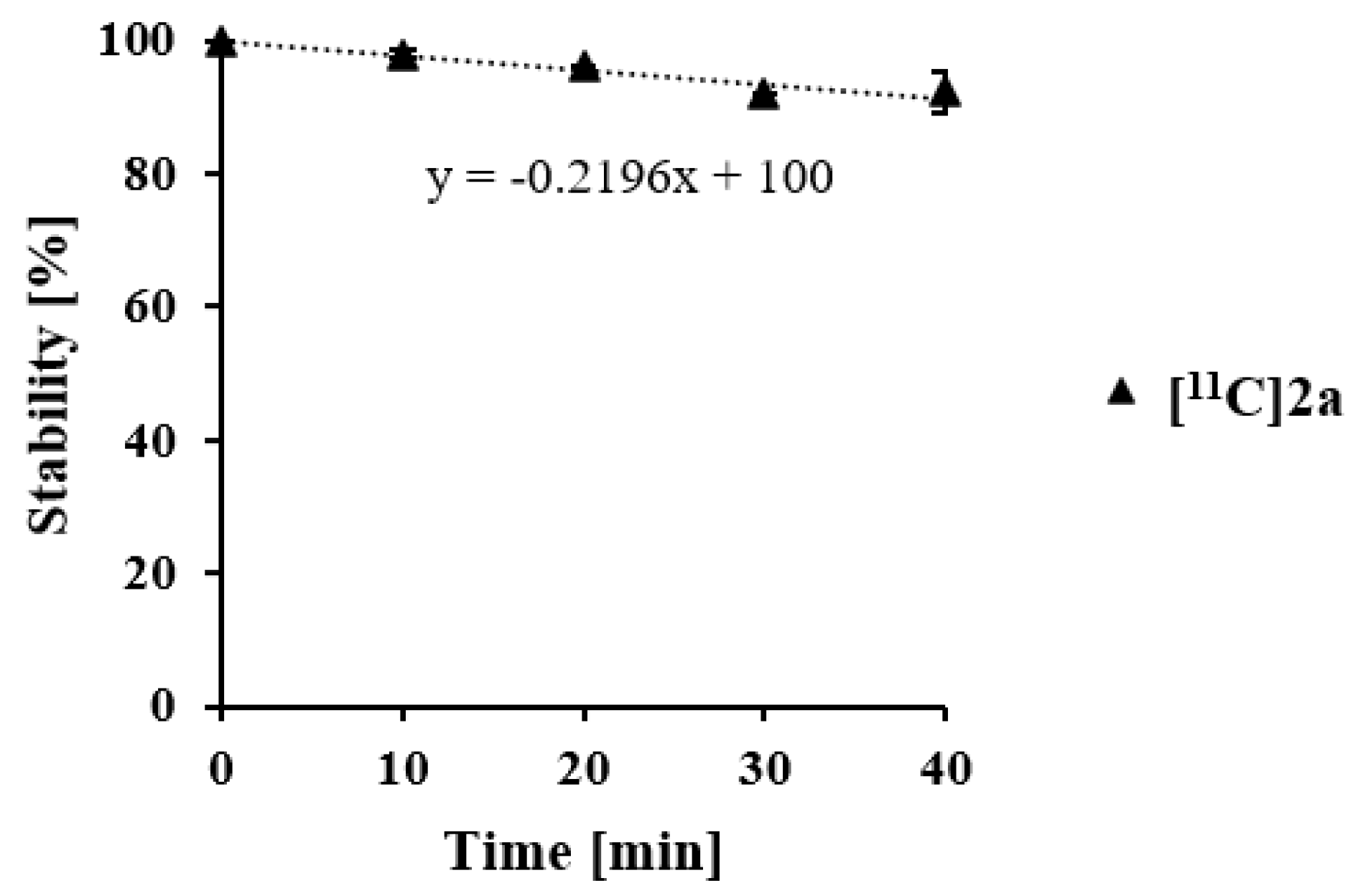
3.9. Metabolic Stability
3.10. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nobel Prize Outreach AB 2023. The Nobel Prize in Physiology or Medicine. 2018. Available online: https://www.nobelprize.org/prizes/medicine/2018/summary/ (accessed on 28 April 2023).
- Zhang, Z.; Liu, S.; Zhang, B.; Qiao, L.; Zhang, Y.; Zhang, Y. T Cell Dysfunction and Exhaustion in Cancer. Front. Cell Dev. Biol. 2020, 8, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of indications of FDA-approved immune checkpoint inhibitors per NCCN guidelines with the level of evidence. Cancers 2020, 12, 738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hettich, M.; Braun, F.; Bartholomä, M.D.; Schirmbeck, R.; Niedermann, G. High-Resolution PET Imaging with Therapeutic Antibody-based PD-1/PD-L1 Checkpoint Tracers. Theranostics 2016, 6, 1629–1640. [Google Scholar] [CrossRef] [PubMed]
- Bensch, F.; van der Veen, E.L.; Lub-de Hooge, M.N.; Jorritsma-Smit, A.; Boellaard, R.; Kok, I.C.; Oosting, S.F.; Schröder, C.P.; Hiltermann, T.J.N.; van der Wekken, A.J.; et al. 89Zr-atezolizumab imaging as a non-invasive approach to assess clinical response to PD-L1 blockade in cancer. Nat. Med. 2018, 24, 1852–1858. [Google Scholar] [CrossRef] [PubMed]
- Griessinger, C.M.; Olafsen, T.; Mascioni, A.; Jiang, Z.K.; Zamilpa, C.; Jia, F.; Torgov, M.; Romero, J.M.; Marchioni, F.; Satpayev, D.; et al. The PET-Tracer [89]Zr-Df-IAB22M2C Enables Monitoring of Intratumoral CD8 T-cell Infiltrates in Tumor-Bearing Humanized Mice after T-cell Bispecific Antibody Treatment. Cancer Res. 2020, 80, 2903–2913. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Jiang, L.; Li, K.; Li, H.; Lv, G.; Lin, J.; Qiu, L. Immuno-PET imaging of 68Ga-labeled nanobody Nb109 for dynamic monitoring the PD-L1 expression in cancers. Cancer Immunol. Immunother. 2021, 70, 1721–1733. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, D.J.; Smith, R.A.; Morin, P.; Lipovšek, D.; Gokemeijer, J.; Cohen, D.; Lafont, V.; Tran, T.; Cole, E.L.; Wright, M.; et al. Synthesis and Biologic Evaluation of a Novel 18F-Labeled Adnectin as a PET Radioligand for Imaging PD-L1 Expression. J. Nucl. Med. 2018, 59, 529–535. [Google Scholar] [CrossRef] [Green Version]
- Trotter, D.E.G.; Meng, X.; McQuade, P.; Rubins, D.; Klimas, M.; Zeng, Z.; Connolly, B.M.; Miller, P.J.; O’Malley, S.S.; Lin, S.-A.; et al. In Vivo Imaging of the Programmed Death Ligand 1 by 18F PET. J. Nucl. Med. 2017, 58, 1852–1857. [Google Scholar] [CrossRef] [Green Version]
- Krutzek, F.; Kopka, K.; Stadlbauer, S. Development of Radiotracers for Imaging of the PD-1/PD-L1 Axis. Pharmaceuticals 2022, 15, 747. [Google Scholar] [CrossRef]
- Chupak, L.S.; Zheng, X. Compounds Useful as Immunomodulators. WO 2015/034820 A1, 12 March 2015. [Google Scholar]
- Zak, K.M.; Grudnik, P.; Guzik, K.; Zieba, B.J.; Musielak, B.; Dömling, A.; Dubin, G.; Holak, T.A. Structural basis for small molecule targeting of the programmed death ligand 1 (PD-L1). Oncotarget 2016, 7, 30323–30335. [Google Scholar] [CrossRef] [Green Version]
- Shi, D.; An, X.; Bai, Q.; Bing, Z.; Zhou, S.; Liu, H.; Yao, X. Computational Insight Into the Small Molecule Intervening PD-L1 Dimerization and the Potential Structure-Activity Relationship. Front. Chem. 2019, 7, 764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Jin, Y.; Wang, B.; Liu, B. Molecular Mechanism of Small-Molecule Inhibitors in Blocking the PD-1/PD-L1 Pathway through PD-L1 Dimerization. Int. J. Mol. Sci. 2021, 22, 4766. [Google Scholar] [CrossRef]
- Hevener, K.; Zhao, W.; Ball, M.D.; Babaoglu, K.; Qi, J.; White, S.; Lee, R. Validation of Molecular Docking Programs for Virtual Screening against Dihydropteroate Synthase. J. Chem. Inf. Model. 2009, 49, 444–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCann, N.; Phan, D.; Attalla, M.; Puxty, G.; Fernandes, D.; Conway, W.; Wang, X.; Burns, R.; van Altena, I.; Lawrance, G.; et al. Molecular interactions between amine and carbonate species in aqueous solution—Kinetics and thermodynamics. Energy Procedia 2009, 1, 995–1002. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; He, J.; Sun, R.C. Carbamate synthesis from amines and dialkyl carbonate over inexpensive and clean acidic catalyst-Sulfamic acid. Chin. Chem. Lett. 2010, 21, 794–797. [Google Scholar] [CrossRef]
- Mcomie, J.F.W.; Watts, M.L.; West, D.E. Demethylation of aryl methyl ethers by boron tribromide. Tetrahedron 1968, 24, 2289–2292. [Google Scholar] [CrossRef]
- Pajouhesh, H.; Lenz, G.R. Medicinal chemical properties of successful central nervous system drugs. NeuroRx 2005, 2, 541–553. [Google Scholar] [CrossRef] [Green Version]
- Hitchcock, S.A.; Pennington, L.D. Structure−Brain Exposure Relationships. J. Med. Chem. 2006, 49, 7559–7583. [Google Scholar] [CrossRef]
- Jouini, N.; Cardinale, J.; Mindt, T.L. Evaluation of a Radiolabeled Macrocyclic Peptide as Potential PET Imaging Probe for PD−L1. ChemMedChem 2022, 17, e202200091. [Google Scholar] [CrossRef]
- Arnott, J.A.; Kumar, R.; Planey, S.L. Lipophilicity Indices for Drug Development. J. Appl. Biopharm. Pharmacokinet. 2013, 1, 31–36. [Google Scholar] [CrossRef] [Green Version]
- Shultz, M.D. Setting expectations in molecular optimizations: Strengths and limitations of commonly used composite parameters. Bioorg. Med. Chem. Lett. 2013, 23, 5980–5991. [Google Scholar] [CrossRef]
- Donovan, S.F.; Pescatore, M.C. Method for measuring the logarithm of the octanol–water partition coefficient by using short octadecyl–poly(vinyl alcohol) high-performance liquid chromatography columns. J. Chromatogr. A 2002, 952, 47–61. [Google Scholar] [CrossRef]
- Vraka, C.; Nics, L.; Wagner, K.H.; Hacker, M.; Wadsak, W.; Mitterhauser, M. LogP, a yesterday’s value? Nucl. Med. Biol. 2017, 50, 1–10. [Google Scholar] [CrossRef]
- Pichler, V.; Ozenil, M.; Bamminger, K.; Vraka, C.; Hacker, M.; Langer, O.; Wadsak, W. Pitfalls and solutions of the fully-automated radiosynthesis of [11C]metoclopramide. EJNMMI Radiopharm. Chem. 2019, 4, 31. [Google Scholar] [CrossRef]
- Dancey, C.P.; Reidy, J. Statistics without Maths for Psychology: Using SPSS for Windows, 3rd ed.; Prentice Hall: Hoboken, NJ, USA, 2004. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substance | Binding Affinity Score | Affinity (kcal/mol) |
|---|---|---|
| 1 | −37.35 ± 1.21 | −9.93 ± 0.99 |
| 1a | −30.70 ± 0.20 | −9.57 ± 0.17 |
| 1b | −37.68 ± 3.52 | −10.3 ± 0.06 |
| 2 | −37.96 ± 2.02 | −10.2 ± 0.21 |
| 2a | −30.85 ± 2.19 | −9.80 ± 0.20 |
| 2b | −31.12 ± 3.67 | −9.73 ± 0.25 |
| 2c | −33.44 ± 1.86 | −9.63 ± 0.76 |
| 2d | −35.52 ± 1.51 | −10.2 ± 0.06 |
| Substance | clogP | clogD | tPSA (Å2) | |
|---|---|---|---|---|
| 1 | 4.60 | 2.73 | 68.23 | 3.16 ± 0.16 |
| 1a | 6.93 | 6.01 | 57.23 | 4.90 ± 0.27 |
| 1b | 7.18 | 6.21 | 57.23 | 5.02 ± 0.28 |
| 2 | 4.41 | 3.64 | 71.95 | 3.88 ± 0.12 |
| 2a | 4.95 | 4.42 | 63.16 | 4.13 ± 0.16 |
| 2b | 4.81 | 3.83 | 63.16 | 3.97 ± 0.13 |
| 2c | 5.35 | 4.64 | 54.37 | 4.28 ± 0.18 |
| 2d | 5.37 | 4.52 | 89.46 | 4.23 ± 0.17 |
| IC50 (nM) | |||
|---|---|---|---|
| Substance | BMS [11] | Cisbio [22] | This Work |
| Atezolizumab | 3.84 | 4.07 ± 0.42 | |
| 1 | 6–100 | 177 | 202 ± 27 |
| 1a | 5760 ± 613 | ||
| 1b | 1440 ± 144 | ||
| 2 | 18 | 101 ± 10 | |
| 2a | 430 ± 62 | ||
| 2b | 524 ± 67 | ||
| 2c | 1310 ± 185 | ||
| 2d | 9880 ± 1390 * | ||
| Inhibitor 3 | 146 | 113 ± 45 | |
| Substance | LE (kcal/mol/Heavy Atom) | BEI | LLE | LELP |
|---|---|---|---|---|
| 1 | 0.27 | 14.1 | 3.5 | 11.8 |
| 1a | 0.20 | 10.7 | 0.3 | 24.0 |
| 1b | 0.22 | 11.4 | 0.9 | 22.9 |
| 2 | 0.32 | 16.7 | 3.1 | 12.3 |
| 2a | 0.28 | 14.7 | 2.2 | 14.8 |
| 2b | 0.27 | 14.5 | 2.3 | 14.4 |
| 2c | 0.25 | 13.1 | 1.6 | 17.1 |
| 2d | 0.19 | 9.9 | 0.8 | 22.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bamminger, K.; Pichler, V.; Vraka, C.; Nehring, T.; Pallitsch, K.; Lieder, B.; Hacker, M.; Wadsak, W. On the Road towards Small-Molecule Programmed Cell Death 1 Ligand 1 Positron Emission Tomography Tracers: A Ligand-Based Drug Design Approach. Pharmaceuticals 2023, 16, 1051. https://doi.org/10.3390/ph16071051
Bamminger K, Pichler V, Vraka C, Nehring T, Pallitsch K, Lieder B, Hacker M, Wadsak W. On the Road towards Small-Molecule Programmed Cell Death 1 Ligand 1 Positron Emission Tomography Tracers: A Ligand-Based Drug Design Approach. Pharmaceuticals. 2023; 16(7):1051. https://doi.org/10.3390/ph16071051
Chicago/Turabian StyleBamminger, Karsten, Verena Pichler, Chrysoula Vraka, Tina Nehring, Katharina Pallitsch, Barbara Lieder, Marcus Hacker, and Wolfgang Wadsak. 2023. "On the Road towards Small-Molecule Programmed Cell Death 1 Ligand 1 Positron Emission Tomography Tracers: A Ligand-Based Drug Design Approach" Pharmaceuticals 16, no. 7: 1051. https://doi.org/10.3390/ph16071051
APA StyleBamminger, K., Pichler, V., Vraka, C., Nehring, T., Pallitsch, K., Lieder, B., Hacker, M., & Wadsak, W. (2023). On the Road towards Small-Molecule Programmed Cell Death 1 Ligand 1 Positron Emission Tomography Tracers: A Ligand-Based Drug Design Approach. Pharmaceuticals, 16(7), 1051. https://doi.org/10.3390/ph16071051