
Exploring Proteus mirabilis Methionine tRNA Synthetase Active Site: Homology Model Construction, Molecular Dynamics, Pharmacophore and Docking Validation


Abstract
:1. Introduction
2. Results and Discussion
2.1. Homology Model and Validation
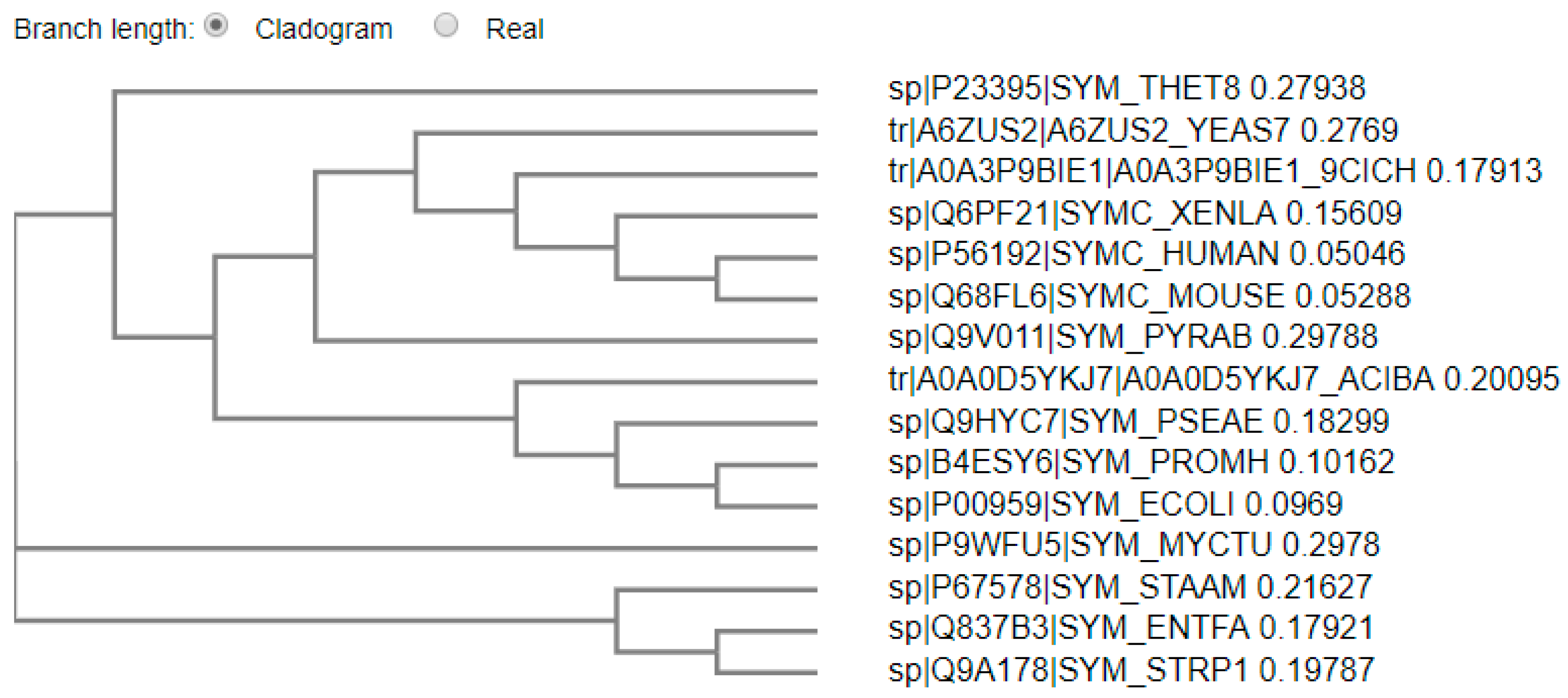
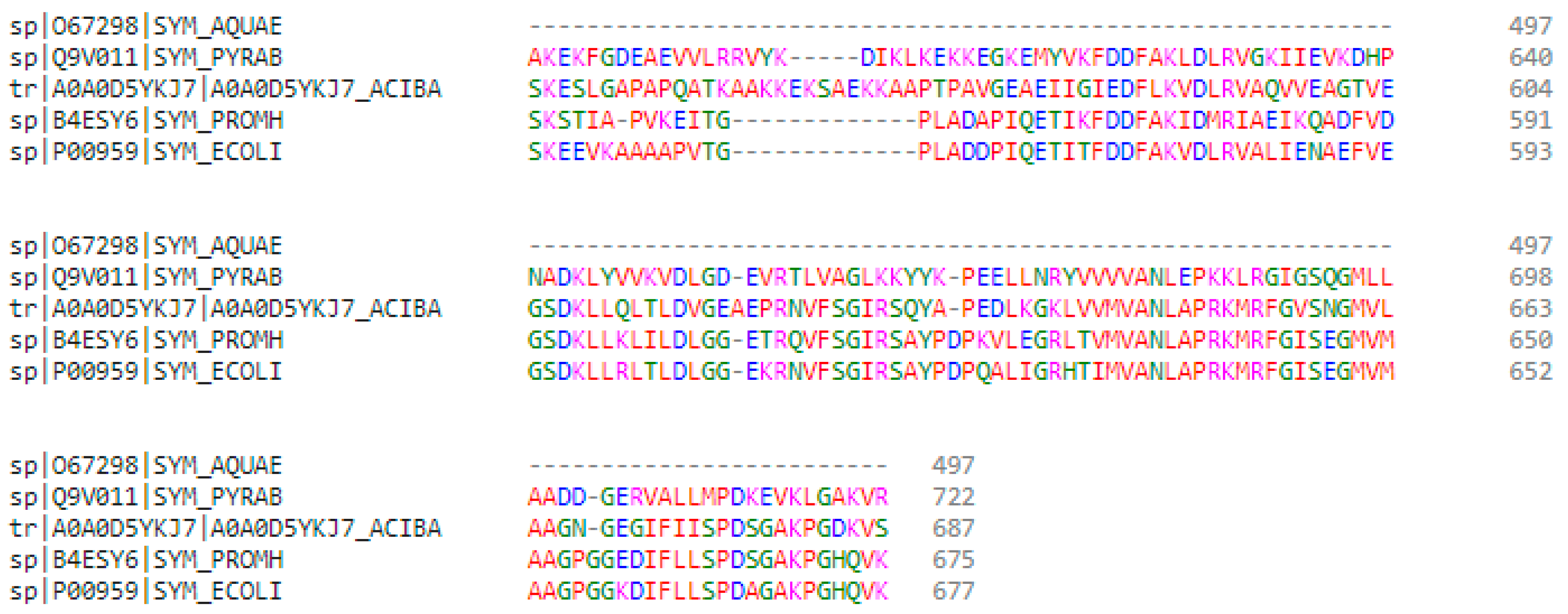
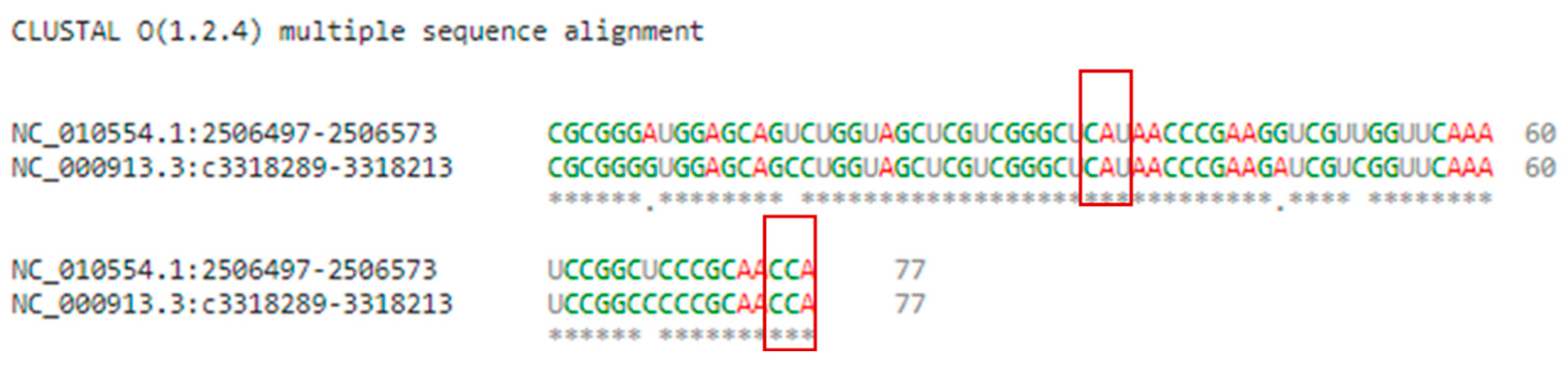
2.2. Multiple Sequence and Structural Alignments
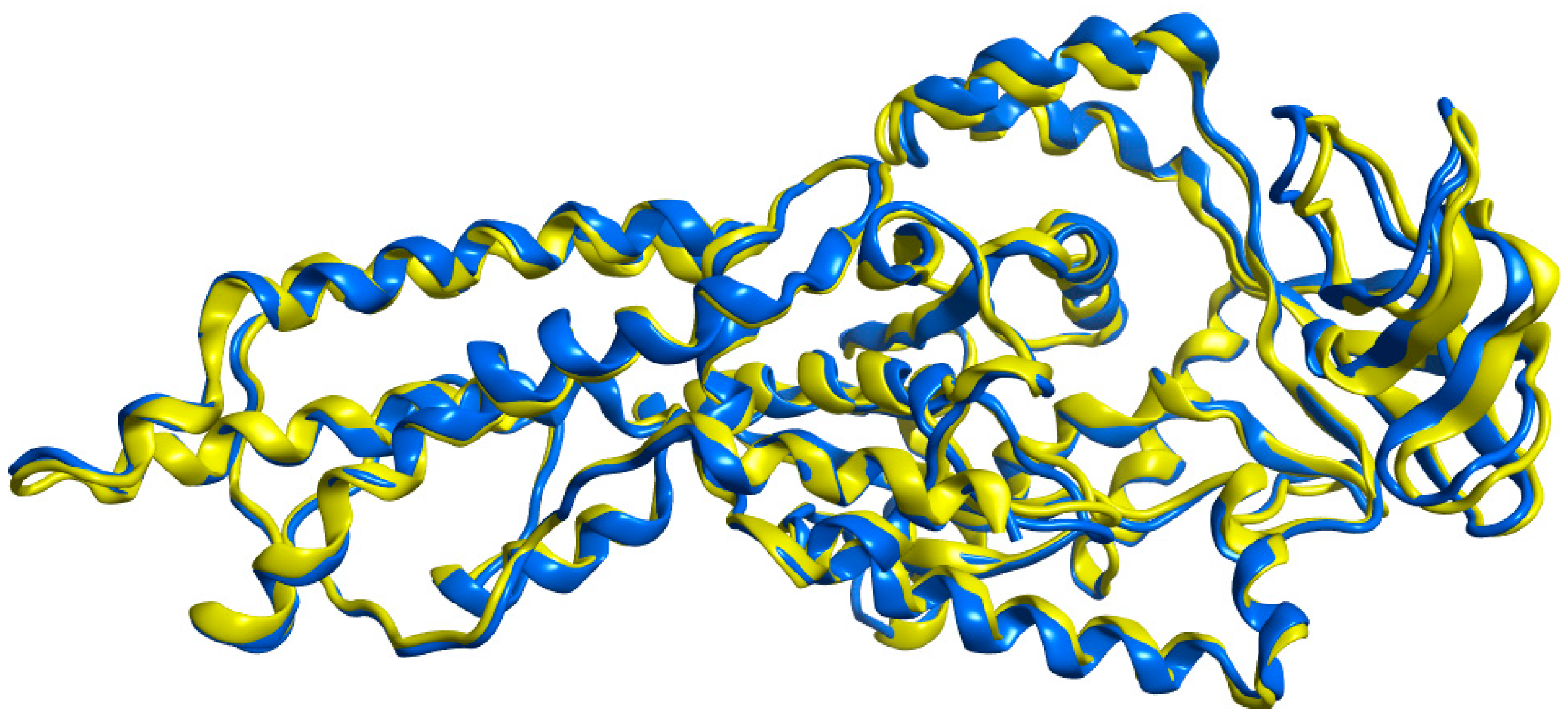
2.3. 3D Homology Model
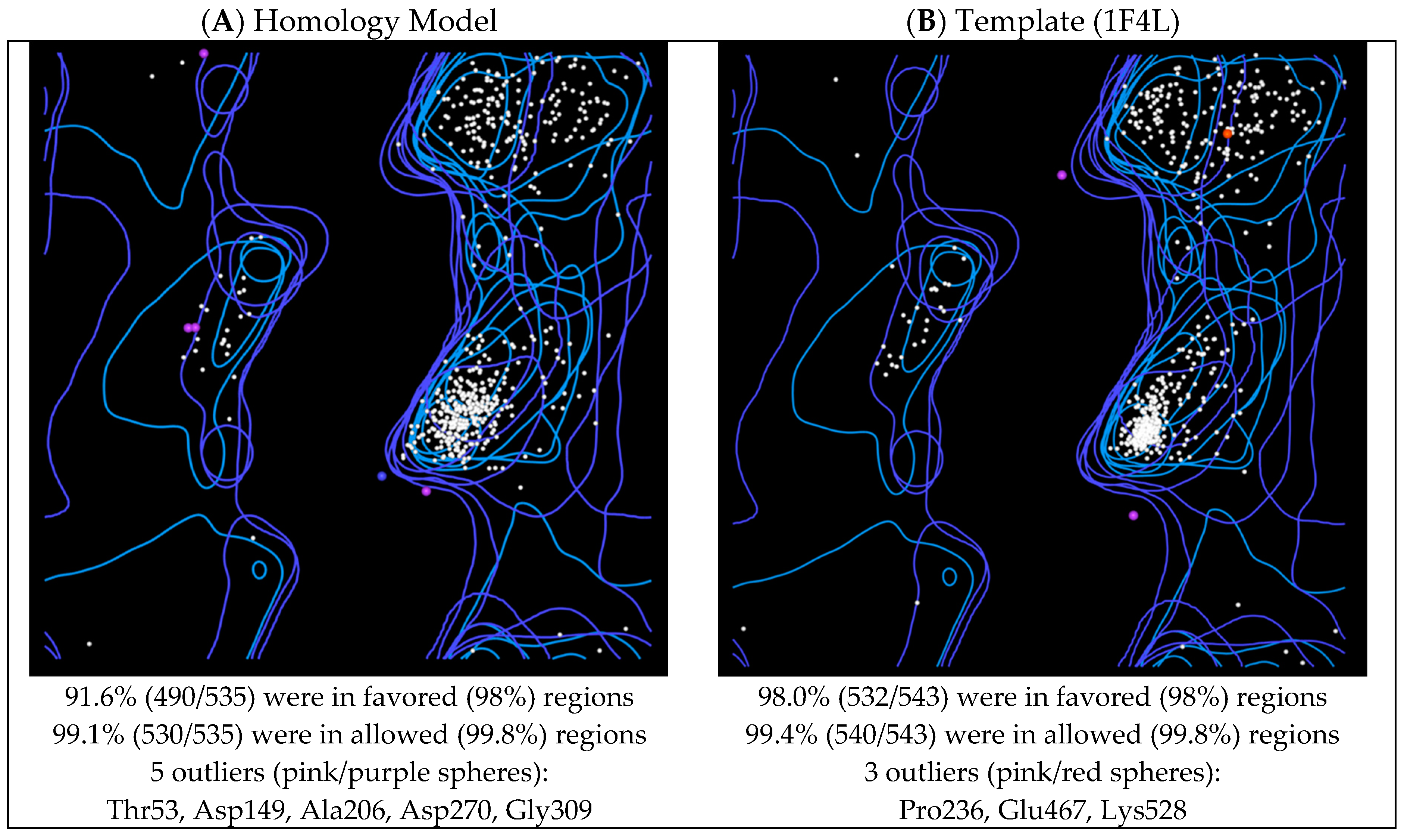
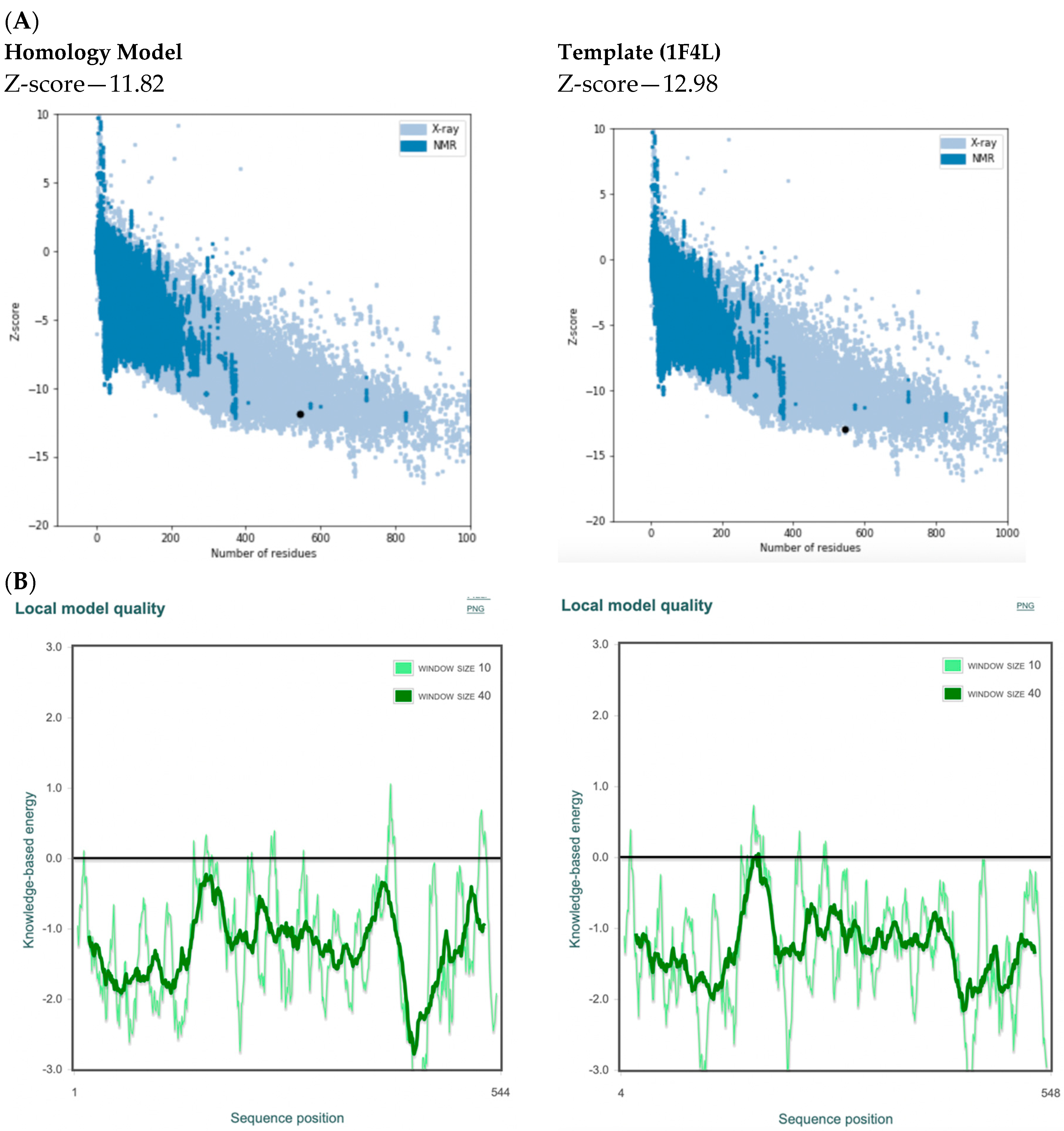
2.4. Model Evaluation/Validation
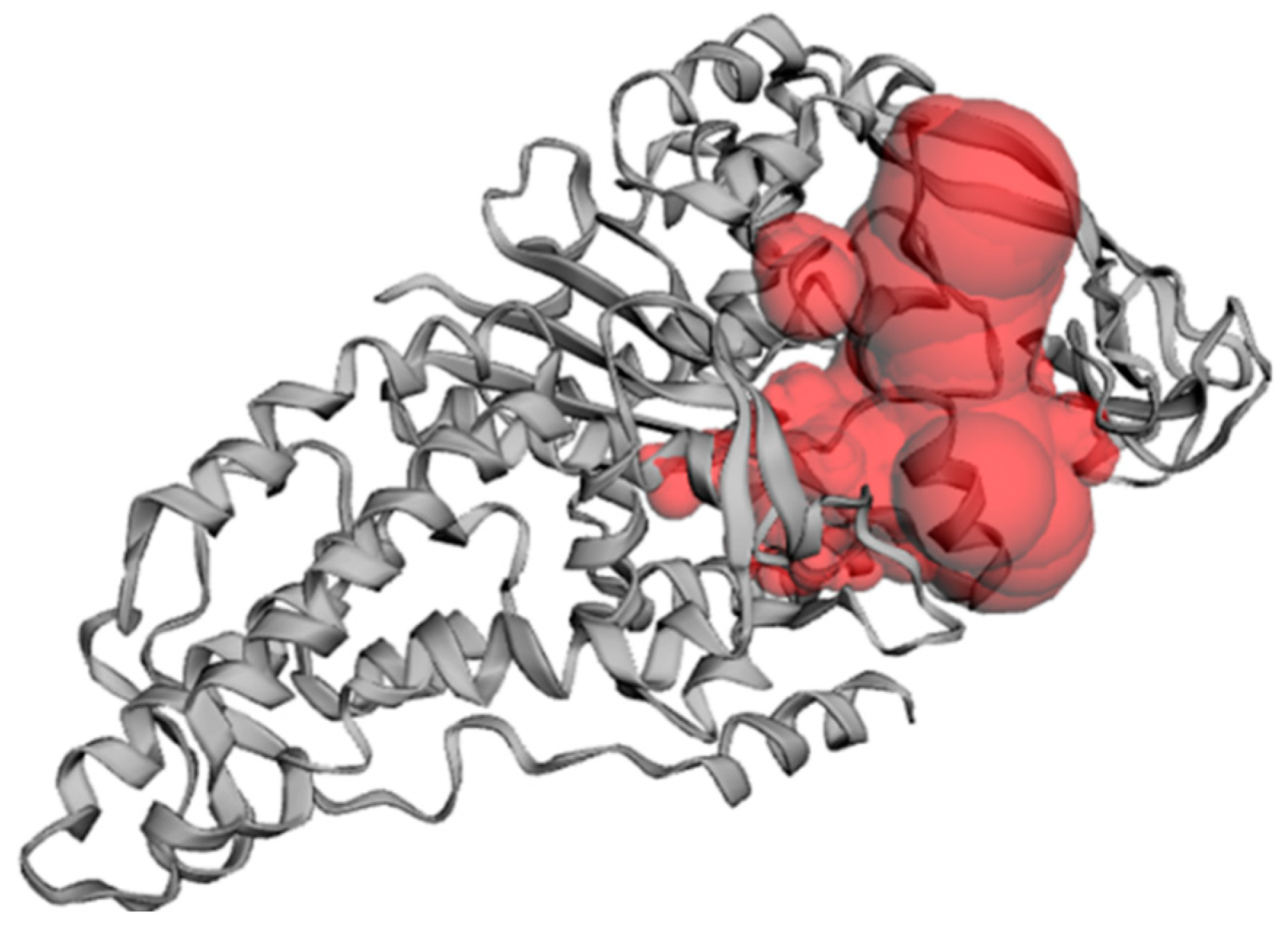
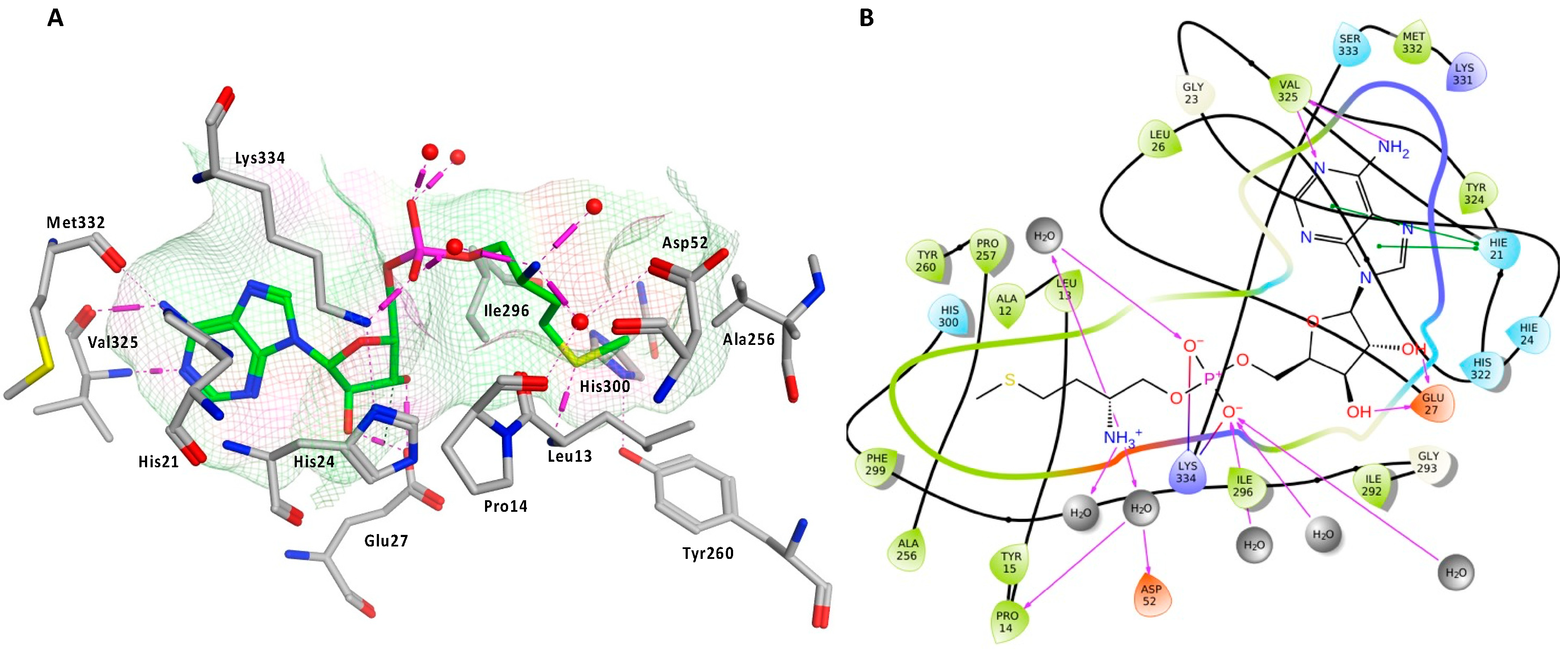
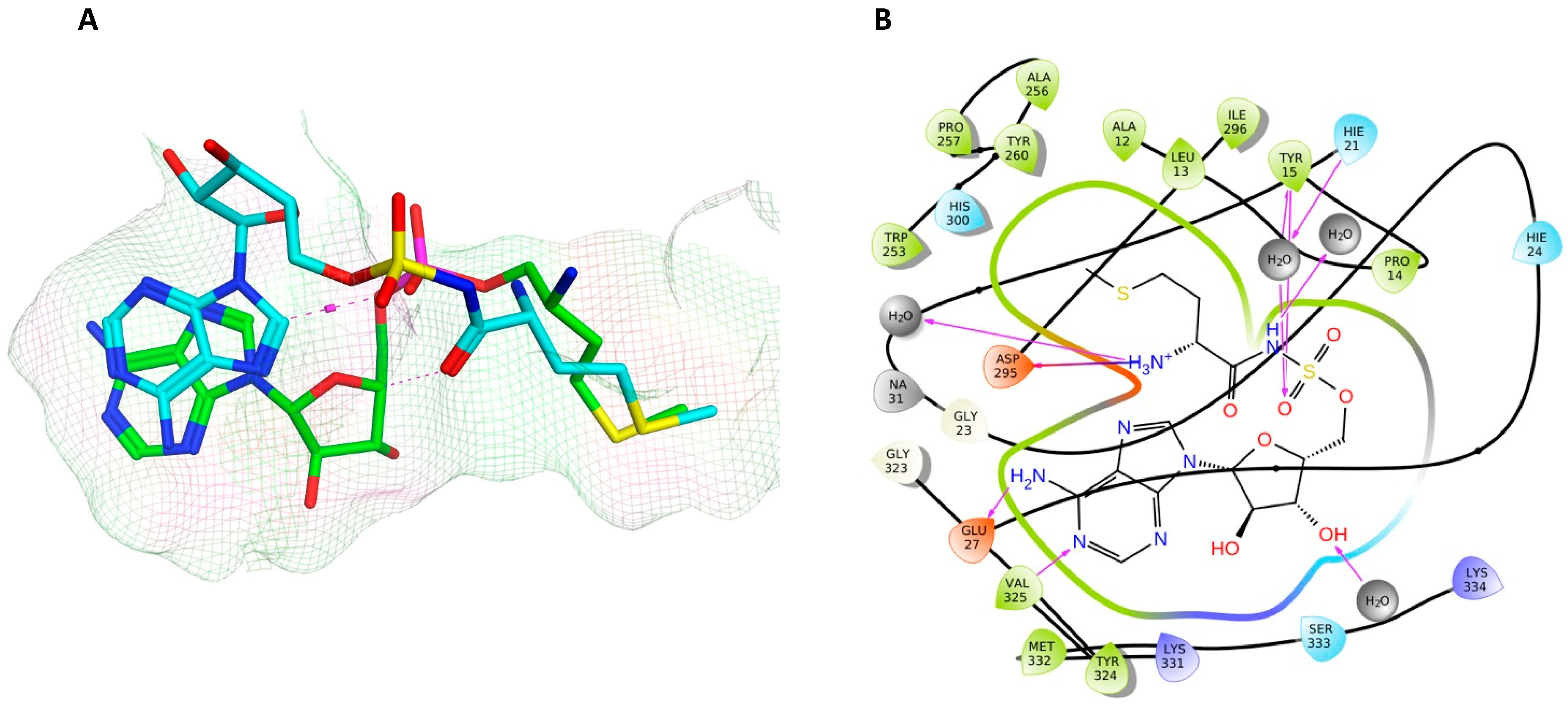
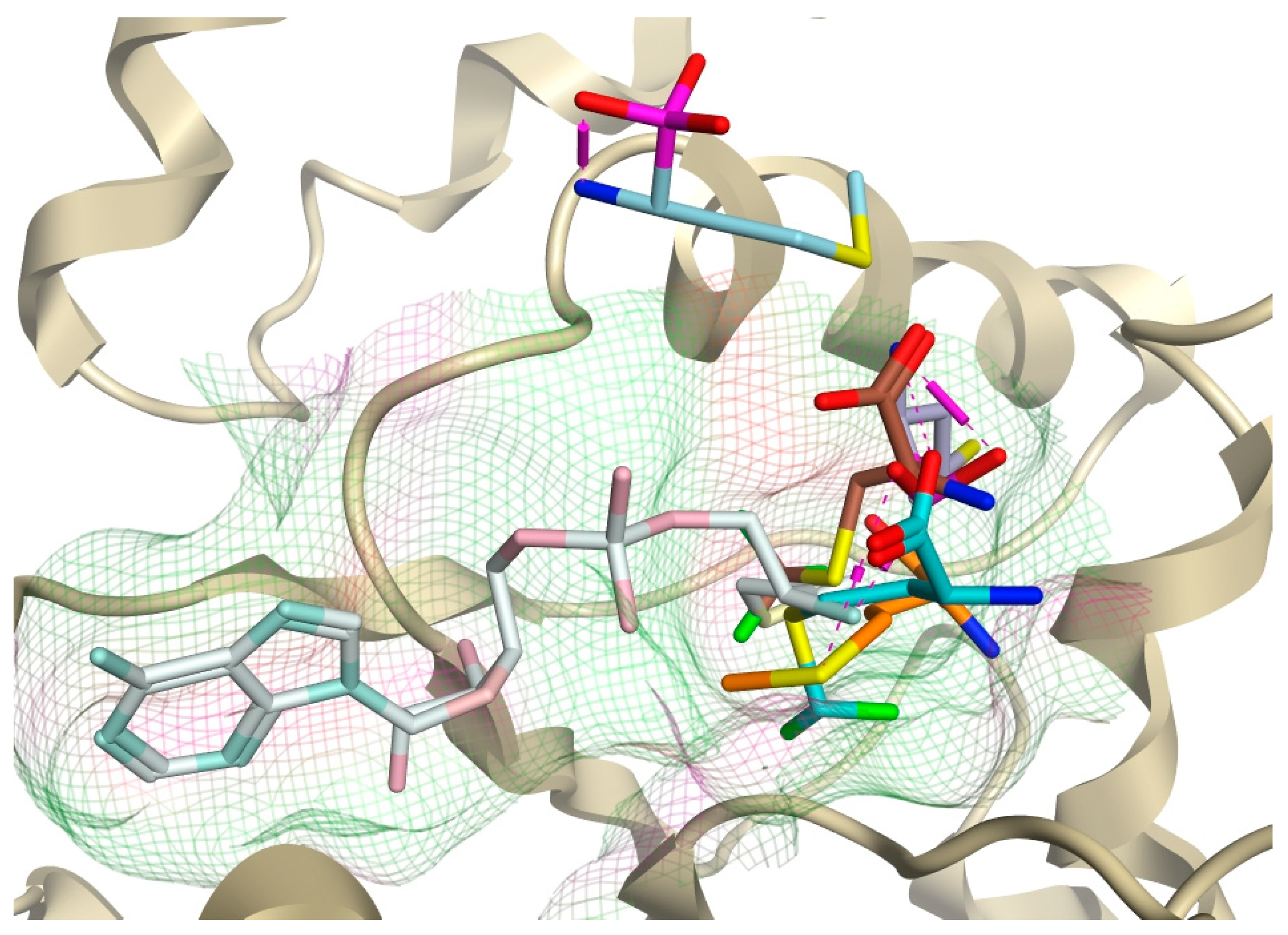
2.5. Active Site Validation and Docking
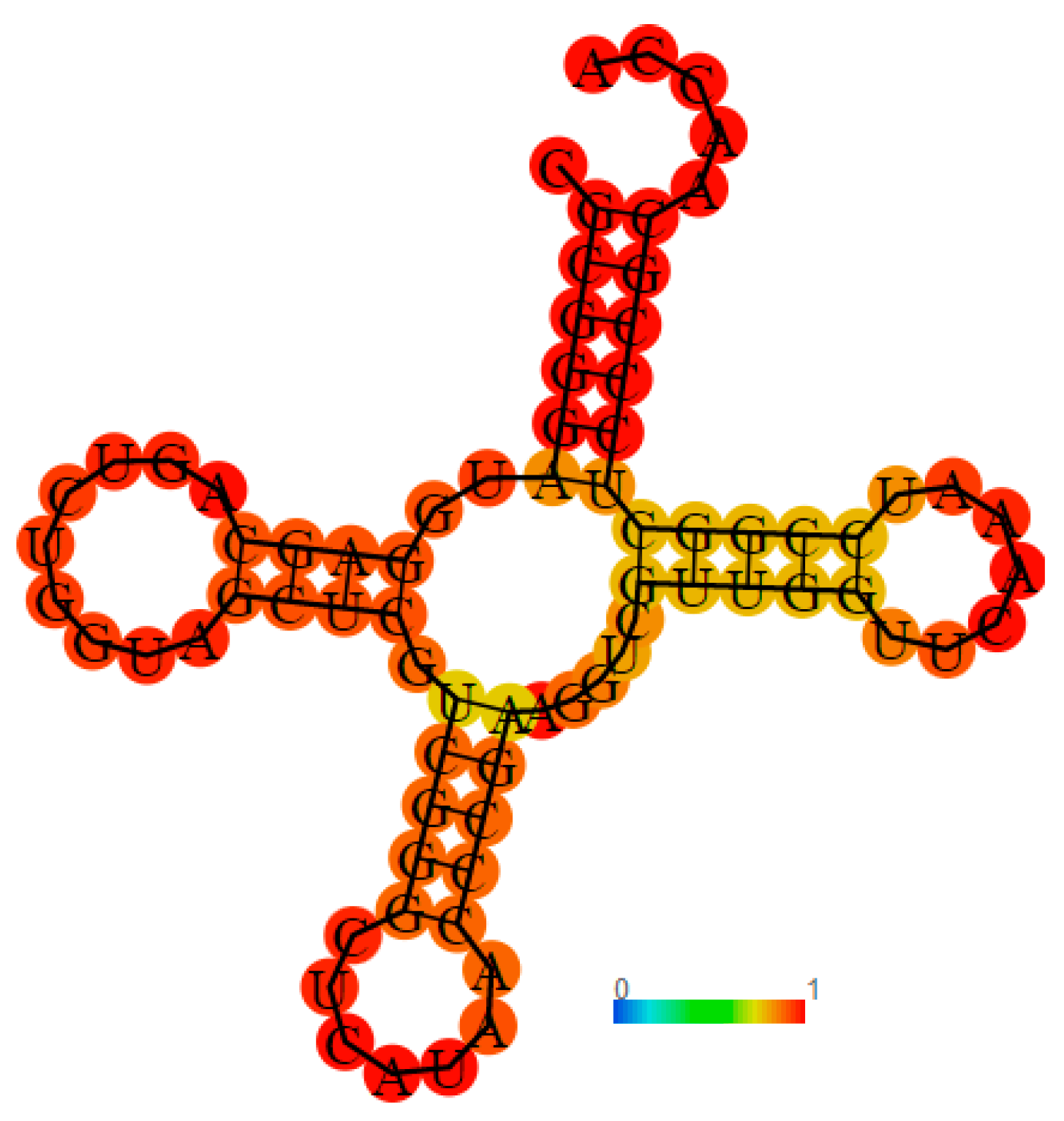
2.6. Recognition and Binding of P. mirabilis MetRS with Cognate tRNAmet
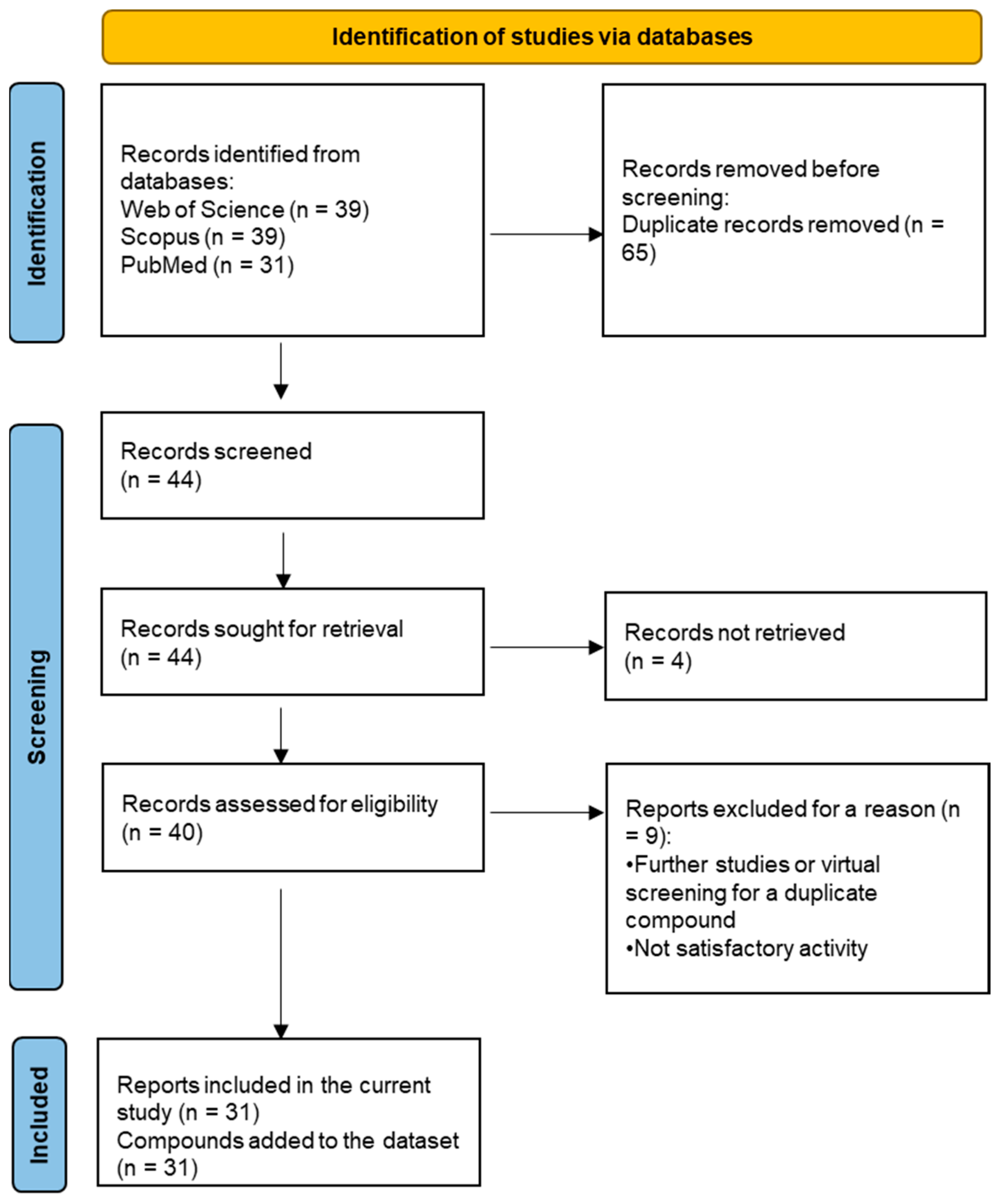
2.7. Systematic Dataset Preparation
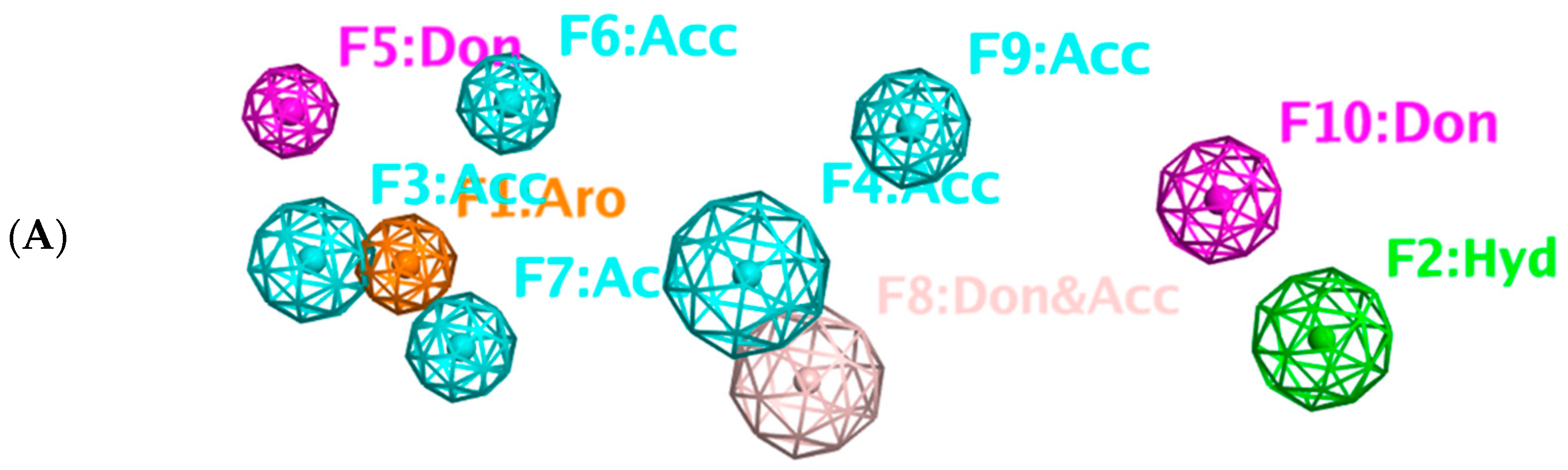
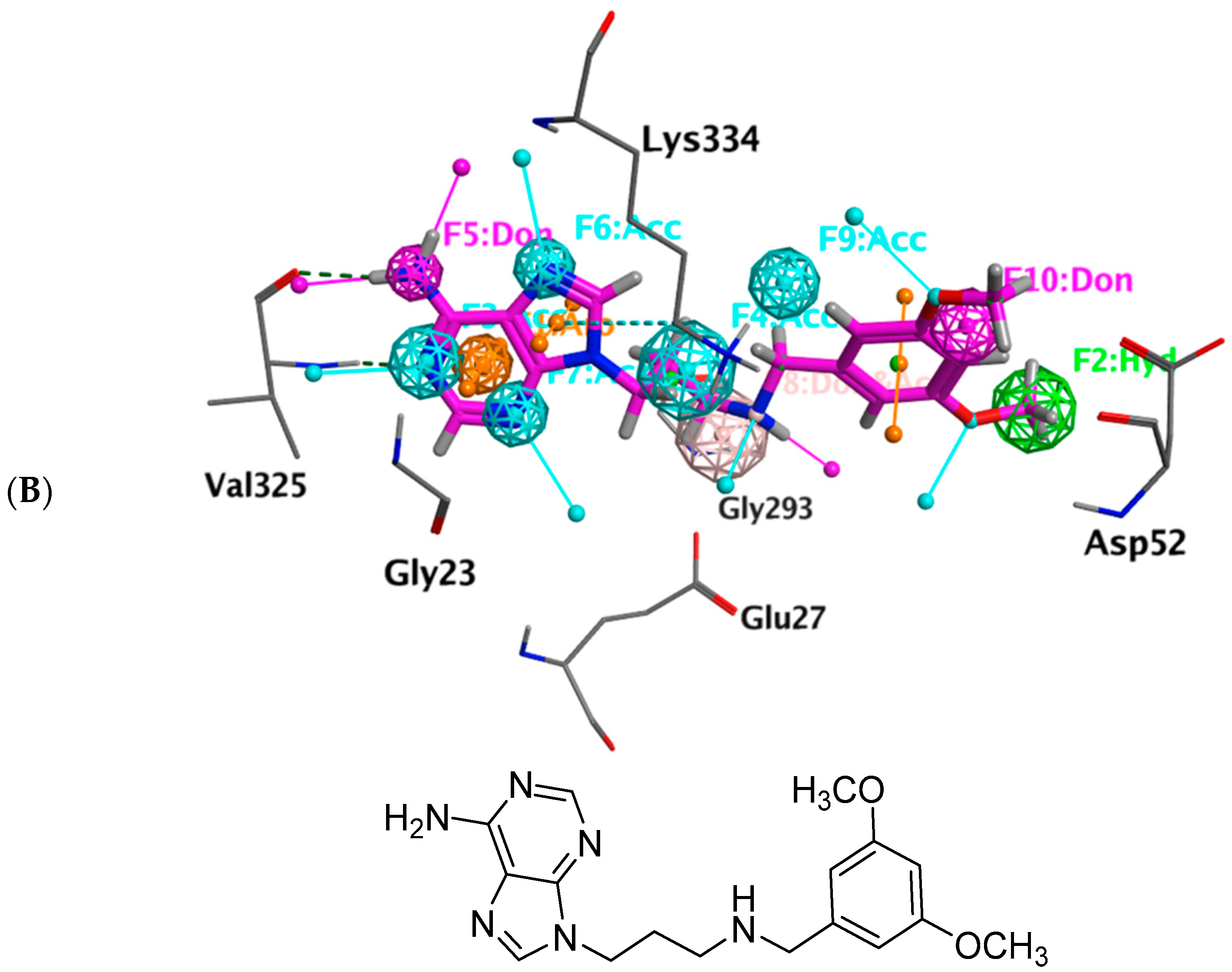
2.8. Pharmacophore-Based Virtual Screening
3. Materials and Methods
3.1. Homology Search
3.2. Multiple Sequence and Structure Alignment
3.3. 3D Model Building
3.4. Model Validation
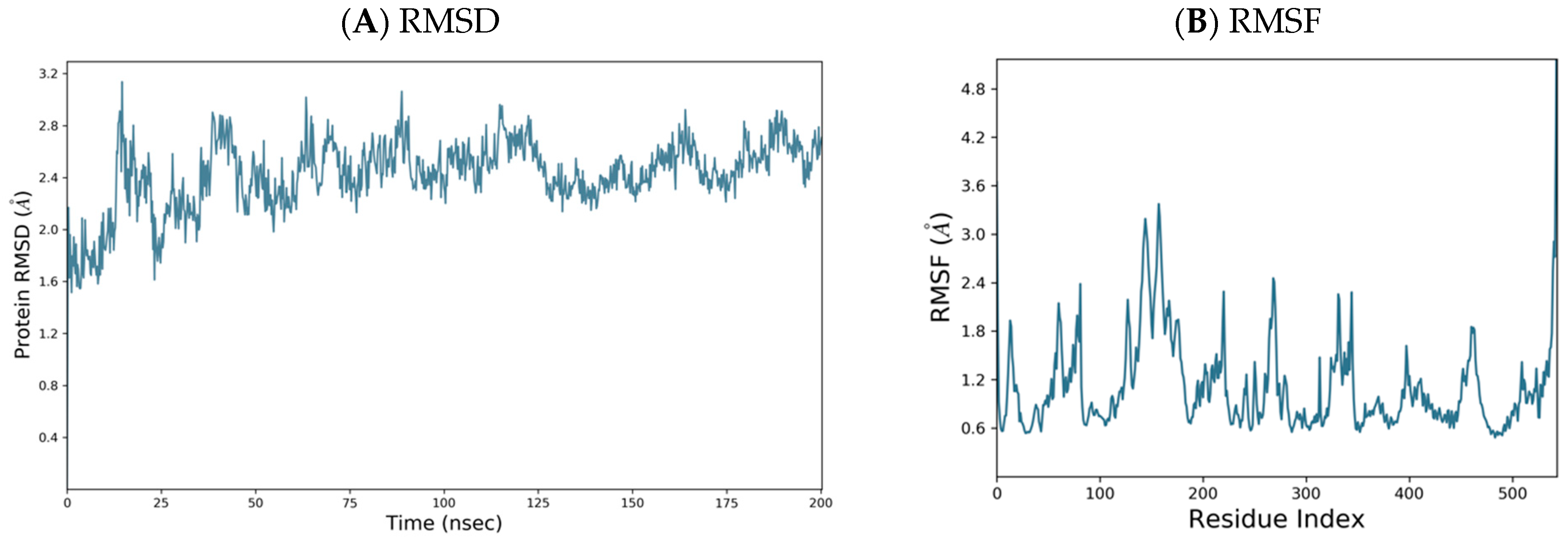
3.5. Molecular Dynamics and Docking Studies
3.6. Systemic Dataset Preparation
3.7. Pharmacophore Query
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Matthews, S.J.; Lancaster, J.W. Urinary Tract Infections in the Elderly Population. Am. J. Geriatr. Pharmacother. 2011, 9, 286–309. [Google Scholar] [CrossRef]
- Papazafiropoulou, A.; Daniil, I.; Sotiropoulos, A.; Balampani, E.; Kokolaki, A.; Bousboulas, S.; Konstantopoulou, S.; Skliros, E.; Petropoulou, D.; Pappas, S. Prevalence of Asymptomatic Bacteriuria in Type 2 Diabetic Subjects with and without Microalbuminuria. BMC Res. Notes 2010, 3, 169. [Google Scholar] [CrossRef]
- Adams-Sapper, S.; Sergeevna-Selezneva, J.; Tartof, S.; Raphael, E.; An Diep, B.; Perdreau-Remington, F.; Riley, L.W. Globally Dispersed Mobile Drug-Resistance Genes in Gram-Negative Bacterial Isolates from Patients with Bloodstream Infections in a US Urban General Hospital. J. Med. Microbiol 2012, 61, 968–974. [Google Scholar] [CrossRef]
- Lubart, E.; Segal, R.; Haimov, E.; Dan, M.; Baumoehl, Y.; Leibovitz, A. Bacteremia in a Multilevel Geriatric Hospital. J. Am. Med. Dir. Assoc. 2011, 12, 204–207. [Google Scholar] [CrossRef]
- O’Hara, C.M.; Brenner, F.W.; Miller, J.M. Classification, Identification, and Clinical Significance of Proteus, Providencia, and Morganella. Clin. Microbiol. Rev. 2000, 13, 534–546. [Google Scholar] [CrossRef]
- Jacobsen, S.M.; Stickler, D.J.; Mobley, H.L.T.; Shirtliff, M.E. Complicated Catheter-Associated Urinary Tract Infections Due to Escherichia coli and Proteus mirabilis. Clin. Microbiol. Rev. 2008, 21, 26–59. [Google Scholar] [CrossRef]
- Karlowsky, J.A.; Lagacé-Wiens, P.R.S.; Simner, P.J.; DeCorby, M.R.; Adam, H.J.; Walkty, A.; Hoban, D.J.; Zhanel, G.G. Antimicrobial Resistance in Urinary Tract Pathogens in Canada from 2007 to 2009: CANWARD Surveillance Study. Antimicrob. Agents Chemother. 2011, 55, 3169–3175. [Google Scholar] [CrossRef]
- Gupta, K.; Hooton, T.M.; Naber, K.G.; Wullt, B.; Colgan, R.; Miller, L.G.; Moran, G.J.; Nicolle, L.E.; Raz, R.; Schaeffer, A.J.; et al. International Clinical Practice Guidelines for the Treatment of Acute Uncomplicated Cystitis and Pyelonephritis in Women: A 2010 Update by the Infectious Diseases Society of America and the European Society for Microbiology and Infectious Diseases. Clin. Infect. Dis. 2011, 52, e103–e120. [Google Scholar] [CrossRef]
- Ma, K.L.; Wang, C.X. Analysis of the Spectrum and Antibiotic Resistance of Uropathogens in Vitro: Results Based on a Retrospective Study from a Tertiary Hospital. Am. J. Infect. Control. 2013, 41, 601–606. [Google Scholar] [CrossRef]
- Schito, G.C.; Naber, K.G.; Botto, H.; Palou, J.; Mazzei, T.; Gualco, L.; Marchese, A. The ARESC Study: An International Survey on the Antimicrobial Resistance of Pathogens Involved in Uncomplicated Urinary Tract Infections. Int. J. Antimicrob. Agents 2009, 34, 407–413. [Google Scholar] [CrossRef]
- Adamus-Bialek, W.; Zajac, E.; Parniewski, P.; Kaca, W. Comparison of Antibiotic Resistance Patterns in Collections of Escherichia coli and Proteus mirabilis Uropathogenic Strains. Mol. Biol. Rep. 2013, 40, 3429–3435. [Google Scholar] [CrossRef]
- McCoy, A.J.; Liu, H.; Falla, T.J.; Gunn, J.S. Identification of Proteus mirabilis Mutants with Increased Sensitivity to Antimicrobial Peptides. Antimicrob. Agents Chemother. 2001, 45, 2030–2037. [Google Scholar] [CrossRef] [PubMed]
- Belas, R.; Manos, J.; Suvanasuthi, R. Proteus mirabilis ZapA Metalloprotease Degrades a Broad Spectrum of Substrates, Including Antimicrobial Peptides. Infect. Immun. 2004, 72, 5159–5167. [Google Scholar] [CrossRef] [PubMed]
- Eriani, G.; Delarue, M.; Poch, O.; Gangloff, J.; Moras, D. Partition of TRNA Synthetases into Two Classes Based on Mutually Exclusive Sets of Sequence Motifs. Nature 1990, 347, 203–206. [Google Scholar] [CrossRef]
- Cassels, R.; Oliva, B.; Knowles, D. Occurrence of the Regulatory Nucleotides PpGpp and PppGpp Following Induction of the Stringent Response in Staphylococci. J. Bacteriol. 1995, 177, 5161–5165. [Google Scholar] [CrossRef] [PubMed]
- Cusack, S.; Härtlein, M.; Leberman, R. Sequence, Structural and Evolutionary Relationships between Class 2 Aminoacyl-TRNA Synthetases. Nucleic Acids Res. 1991, 19, 3489–3498. [Google Scholar] [CrossRef]
- Deniziak, M.A.; Barciszewski, J. Methionyl-TRNA Synthetase. Acta Biochim. Pol. 2001, 48, 337–350. [Google Scholar] [CrossRef]
- Sugiura, I.; Nureki, O.; Ugaji-Yoshikawa, Y.; Kuwabara, S.; Shimada, A.; Tateno, M.; Lorber, B.; Giegé, R.; Moras, D.; Yokoyama, S.; et al. The 2.0 Å Crystal Structure of Thermus Thermophilus Methionyl-tRNA Synthetase Reveals Two RNA-Binding Modules. Structure 2000, 8, 197–208. [Google Scholar] [CrossRef]
- Meng, X.-Y.; Zhang, H.-X.; Mezei, M.; Cui, M. Molecular Docking: A Powerful Approach for Structure-Based Drug Discovery. Curr. Comput. Aided-Drug Des. 2012, 7, 146–157. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Seidel, T.; Ibis, G.; Bendix, F.; Wolber, G. Strategies for 3D Pharmacophore-Based Virtual Screening. Drug Discov. Today Technol. 2010, 7, e221–e228. [Google Scholar] [CrossRef]
- Altschul, S. Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E. ExPASy: The Proteomics Server for in-Depth Protein Knowledge and Analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef] [PubMed]
- Serre, L.; Verdon, G.; Choinowski, T.; Hervouet, N.; Risler, J.L.; Zelwer, C. How Methionyl-tRNA Synthetase Creates Its Amino Acid Recognition Pocket upon L-Methionine Binding. J. Mol. Biol. 2001, 306, 863–876. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Cowley, A.; Uludag, M.; Gur, T.; McWilliam, H.; Squizzato, S.; Park, Y.M.; Buso, N.; Lopez, R. The EMBL-EBI Bioinformatics Web and Programmatic Tools Framework. Nucleic Acids Res. 2015, 43, W580–W584. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, Scalable Generation of High-quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Jones, D.T. Protein Secondary Structure Prediction Based on Position-Specific Scoring Matrices. J. Mol. Biol. 1999, 292, 195–202. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE 2019.0102); Chemical Computing Group Inc.: Montreal, QC, Canada, 2019; Available online: http://www.Chemcomp.Com (accessed on 2 January 2019).
- Desmond Schrödinger Release 2020-1. Available online: https://www.schrodinger.com/products/desmond (accessed on 1 March 2023).
- Bowers, K.J.; Sacerdoti, F.D.; Salmon, J.K.; Shan, Y.; Shaw, D.E.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; et al. Molecular Dynamics—Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing-SC ’06, Tampa, FL, USA, 11–17 November 2006; ACM Press: New York, NY, USA, 2006; p. 84. [Google Scholar]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and Better Reference Data for Improved All-Atom Structure Validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef]
- Crepin, T.; Schmitt, E.; Mechulam, Y.; Sampson, P.B.; Vaughan, M.D.; Honek, J.F.; Blanquet, S. Use of Analogues of Methionine and Methionyl Adenylate to Sample Conformational Changes during Catalysis in Escherichia coli Methionyl-tRNA Synthetase. J. Mol. Biol. 2003, 332, 59–72. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-Web: Interactive Web Service for the Recognition of Errors in Three-Dimensional Structures of Proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed Atlas of Surface Topography of Proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef] [PubMed]
- Arnez, J.G.; Moras, D. Structural and Functional Considerations of the Aminoacylation Reaction. Trends Biochem. Sci. 1997, 22, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, G.; Pelka, H.; Schulman, L.H. Identification of the TRNA Anticodon Recognition Site of Escherichia coli Methionyl-tRNA Synthetase. Biochemistry 1990, 29, 2220–2225. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, K.; Ogiso, Y.; Nakama, T.; Fukai, S.; Nureki, O. Structural Basis for Anticodon Recognition by Methionyl-TRNA Synthetase. Nat. Struct. Mol. Biol. 2005, 12, 931–932. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M.; Stiegler, P. Optimal Computer Folding of Large RNA Sequences Using Thermodynamics and Auxiliary Information. Nucleic Acids Res. 1981, 9, 133–148. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 Statement: An Updated Guideline for Reporting Systematic Reviews. Int. J. Surg. 2021, 88, 105906. [Google Scholar] [CrossRef]
- Zhang, Z.; Barros-Álvarez, X.; Gillespie, J.R.; Ranade, R.M.; Huang, W.; Shibata, S.; Molasky, N.M.R.; Faghih, O.; Mushtaq, A.; Choy, R.K.M.; et al. Structure-Guided Discovery of Selective Methionyl-tRNA Synthetase Inhibitors with Potent Activity against: Trypanosoma brucei. RSC Med. Chem. 2020, 11, 885–895. [Google Scholar] [CrossRef]
- Michaels, S.A.; Shih, H.-W.; Zhang, B.; Navaluna, E.D.; Zhang, Z.; Ranade, R.M.; Gillespie, J.R.; Merritt, E.A.; Fan, E.; Buckner, F.S.; et al. Methionyl-tRNA Synthetase Inhibitor Has Potent In Vivo Activity in a Novel Giardia lamblia Luciferase Murine Infection Model. J. Antimicrob. Chemother. 2020, 75, 1218–1227. [Google Scholar] [CrossRef]
- Faghih, O.; Zhang, Z.; Ranade, R.M.; Gillespie, J.R.; Creason, S.A.; Huang, W.; Shibata, S.; Barros-Álvarez, X.; Verlinde, C.L.M.J.; Hol, W.G.J.; et al. Development of Methionyl-tRNA Synthetase Inhibitors as Antibiotics for Gram-Positive Bacterial Infections. Antimicrob. Agents Chemother. 2017, 61, 10–1128. [Google Scholar] [CrossRef]
- Buckner, F.S.; Ranade, R.M.; Gillespie, J.R.; Shibata, S.; Hulverson, M.A.; Zhang, Z.; Huang, W.; Choi, R.; Verlinde, C.L.M.J.; Hol, W.G.J.; et al. Optimization of Methionyl tRNA-Synthetase Inhibitors for Treatment of Cryptosporidium Infection. Antimicrob. Agents Chemother. 2019, 63, 10–1128. [Google Scholar] [CrossRef]
- Nayak, S.U.; Griffiss, J.M.; Blumer, J.; O’Riordan, M.A.; Gray, W.; McKenzie, R.; Jurao, R.A.; An, A.T.; Le, M.; Bell, S.J.; et al. Safety, Tolerability, Systemic Exposure, and Metabolism of CRS3123, a Methionyl-tRNA Synthetase Inhibitor Developed for Treatment of Clostridium difficile, in a Phase 1 Study. Antimicrob. Agents Chemother. 2017, 61, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.Y.; Kim, J.E.; Wetzel, A.B.; de van der Schueren, W.J.; Shibata, S.; Ranade, R.M.; Liu, J.; Zhang, Z.; Gillespie, J.R.; Buckner, F.S.; et al. Structures of Trypanosoma brucei Methionyl-tRNA Synthetase with Urea-Based Inhibitors Provide Guidance for Drug Design against Sleeping Sickness. PLoS Negl. Trop. Dis. 2014, 8, e2775. [Google Scholar] [CrossRef] [PubMed]
- Shibata, S.; Gillespie, J.R.; Ranade, R.M.; Koh, C.Y.; Kim, J.E.; Laydbak, J.U.; Zucker, F.H.; Hol, W.G.J.; Verlinde, C.L.M.J.; Buckner, F.S.; et al. Urea-Based Inhibitors of Trypanosoma brucei Methionyl-tRNA Synthetase: Selectivity and in Vivo Characterization. J. Med. Chem. 2012, 55, 6342–6351. [Google Scholar] [CrossRef]
- Ranade, R.M.; Gillespie, J.R.; Shibata, S.; Verlinde, C.L.M.J.; Fan, E.; Hol, W.G.J.; Buckner, F.S. Induced Resistance to Methionyl-tRNA Synthetase Inhibitors in Trypanosoma bruceibrucei is Due to Overexpression of the Target. Antimicrob. Agents Chemother. 2013, 57, 3021–3028. [Google Scholar] [CrossRef] [PubMed]
- Shibata, S.; Gillespie, J.R.; Kelley, A.M.; Napuli, A.J.; Zhang, Z.; Kovzun, K.V.; Pefley, R.M.; Lam, J.; Zucker, F.H.; van Voorhis, W.C.; et al. Selective Inhibitors of Methionyl-tRNA Synthetase Have Potent Activity against Trypanosoma brucei Infection in Mice. Antimicrob. Agents Chemother. 2011, 55, 1982–1989. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; He, G.; Jiang, Q.L.; Han, B.; Peng, C. Novel Hybrid Virtual Screening Protocol Based on Molecular Docking and Structure-Based Pharmacophore for Discovery of Methionyl-tRNA Synthetase Inhibitors as Antibacterial Agents. Int. J. Mol. Sci. 2013, 14, 14225–14239. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.Y.; Kim, J.E.; Shibata, S.; Ranade, R.M.; Yu, M.; Liu, J.; Gillespie, J.R.; Buckner, F.S.; Verlinde, C.L.M.J.; Fan, E.; et al. Distinct States of Methionyl-tRNA Synthetase Indicate Inhibitor Binding by Conformational Selection. Structure 2012, 20, 1681–1691. [Google Scholar] [CrossRef]
- Farhanullah; Kang, T.; Yoon, E.-J.; Choi, E.-C.; Kim, S.; Lee, J. 2-[2-Substituted-3-(3,4-Dichlorobenzylamino)Propylamino]-1H-Quinolin-4-Ones as Staphylococcus aureus Methionyl-tRNA Synthetase Inhibitors. Eur. J. Med. Chem. 2009, 44, 239–250. [Google Scholar] [CrossRef]
- Finn, J.; Stidham, M.; Hilgers, M.; Kedar, G.C. Identification of Novel Inhibitors of Methionyl-tRNA Synthetase (MetRS) by Virtual Screening. Bioorg. Med. Chem. Lett. 2008, 18, 3932–3937. [Google Scholar] [CrossRef]
- Farhanullah; Kim, S.Y.; Yoon, E.-J.; Choi, E.-C.; Kim, S.; Kang, T.; Samrin, F.; Puri, S.; Lee, J. Design and Synthesis of Quinolinones as Methionyl-tRNA Synthetase Inhibitors. Bioorg. Med. Chem. 2006, 14, 7154–7159. [Google Scholar] [CrossRef]
- Vaughan, M.; Sampson, P.; Daub, E.; Honek, J. Investigation of Bioisosteric Effects on the Interaction of Substrates/ Inhibitors with the Methionyl-tRNA Synthetase from Escherichia coli. Med. Chem. 2005, 1, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.E.; Kim, S.Y.; Kim, S.; Kang, T.; Lee, J. Deoxyribosyl Analogues of Methionyl and Isoleucyl Sulfamate Adenylates as Inhibitors of Methionyl-tRNA and Isoleucyl-tRNA Synthetases. Bioorg Med. Chem. Lett. 2005, 15, 3389–3393. [Google Scholar] [CrossRef]
- Lee, J.; Kim, S.E.; Lee, J.Y.; Kim, S.Y.; Kang, S.U.; Seo, S.H.; Chun, M.W.; Kang, T.; Choi, S.Y.; Kim, H.O. N-Alkoxysulfamide, N-Hydroxysulfamide, and Sulfamate Analogues of Methionyl and Isoleucyl Adenylates as Inhibitors of Methionyl-tRNA and Isoleucyl-tRNA Synthetases. Bioorg. Med. Chem. Lett. 2003, 13, 1087–1092. [Google Scholar] [CrossRef] [PubMed]
- Ochsner, U.A.; Young, C.L.; Stone, K.C.; Dean, F.B.; Janjic, N.; Critchley, I.A. Mode of Action and Biochemical Characterization of REP8839, a Novel Inhibitor of Methionyl-tRNA Synthetase. Antimicrob. Agents Chemother. 2005, 49, 4253–4262. [Google Scholar] [CrossRef] [PubMed]
- Tandon, M.; Coffen, D.L.; Gallant, P.; Keith, D.; Ashwell, M.A. Potent and Selective Inhibitors of Bacterial Methionyl tRNA Synthetase Derived from an Oxazolone–Dipeptide Scaffold. Bioorg. Med. Chem. Lett. 2004, 14, 1909–1911. [Google Scholar] [CrossRef] [PubMed]
- Jarvest, R.L.; Armstrong, S.A.; Berge, J.M.; Brown, P.; Elder, J.S.; Brown, M.J.; Copley, R.C.B.; Forrest, A.K.; Hamprecht, D.W.; O’Hanlon, P.J.; et al. Definition of the Heterocyclic Pharmacophore of Bacterial Methionyl tRNA Synthetase Inhibitors: Potent Antibacterially Active Non-Quinolone Analogues. Bioorg. Med. Chem. Lett. 2004, 14, 3937–3941. [Google Scholar] [CrossRef]
- Kim, S.Y.; Lee, J. 3-D-QSAR Study and Molecular Docking of Methionyl-tRNA Synthetase Inhibitors. Bioorg. Med. Chem. 2003, 11, 5325–5331. [Google Scholar] [CrossRef]
- Jarvest, R.L.; Berge, J.M.; Brown, M.J.; Brown, P.; Elder, J.S.; Forrest, A.K.; Houge-Frydrych, C.S.V.; O’Hanlon, P.J.; McNair, D.J.; Rittenhouse, S.; et al. Optimisation of Aryl Substitution Leading to Potent Methionyl tRNA Synthetase Inhibitors with Excellent Gram-Positive Antibacterial Activity. Bioorg. Med. Chem. Lett. 2003, 13, 665–668. [Google Scholar] [CrossRef]
- Jarvest, R.L.; Berge, J.M.; Brown, P.; Houge-Frydrych, C.S.V.; O’Hanlon, P.J.; McNair, D.J.; Pope, A.J.; Rittenhouse, S. Conformational Restriction of Methionyl tRNA Synthetase Inhibitors Leading to Analogues with Potent Inhibition and Excellent Gram-Positive Antibacterial Activity. Bioorg. Med. Chem. Lett. 2003, 13, 1265–1268. [Google Scholar] [CrossRef]
- Lee, J.; Kang, S.U.; Kim, S.Y.; Kim, S.E.; Jo, Y.J.; Kim, S. Vanilloid and Isovanilloid Analogues as Inhibitors of Methionyl-tRNA and Isoleucyl-tRNA Synthetases. Bioorg. Med. Chem. Lett. 2001, 11, 965–968. [Google Scholar] [CrossRef]
- Lee, J.; Kang, S.U.; Kim, S.Y.; Kim, S.E.; Kang, M.K.; Jo, Y.J.; Kim, S. Ester and Hydroxamate Analogues of Methionyl and Isoleucyl Adenylates as Inhibitors of Methionyl-tRNA and Isoleucyl-tRNA Synthetases. Bioorg. Med. Chem. Lett. 2001, 11, 961–964. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kang, S.U.; Kang, M.K.; Chun, M.W.; Jo, Y.J.; Kkwak, J.H.; Kim, S. Methionyl Adenylate Analogues as Inhibitors of Methionyl-tRNA Synthetase. Bioorg. Med. Chem. Lett. 1999, 9, 1365–1370. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Zhang, Z.; Ranade, R.M.; Gillespie, J.R.; Barros-Álvarez, X.; Creason, S.A.; Shibata, S.; Verlinde, C.L.M.J.; Hol, W.G.J.; Buckner, F.S.; et al. Optimization of a Binding Fragment Targeting the “Enlarged Methionine Pocket” Leads to Potent Trypanosoma brucei Methionyl-tRNA Synthetase Inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 2702–2707. [Google Scholar] [CrossRef]
- Eissa, A.G.; Blaxland, J.A.; Williams, R.O.; Metwally, K.A.; El-Adl, S.M.; Lashine, E.-S.M.E.-S.M.; Baillie, L.W.J.; Simons, C. Targeting Methionyl tRNA Synthetase: Design, Synthesis and Antibacterial Activity against Clostridium difficile of Novel 3-Biaryl-N-Benzylpropan-1-Amine Derivatives. J. Enzym. Inhib. Med. Chem. 2016, 31, 1694–1697. [Google Scholar] [CrossRef]
- Huang, W.; Zhang, Z.; Barros-Álvarez, X.; Koh, C.Y.; Ranade, R.M.; Gillespie, J.R.; Creason, S.A.; Shibata, S.; Verlinde, C.L.M.J.; Hol, W.G.J.; et al. Structure-Guided Design of Novel Trypanosoma brucei Methionyl-tRNA Synthetase Inhibitors. Eur. J. Med. Chem. 2016, 124, 1081–1092. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Koh, C.Y.; Ranade, R.M.; Shibata, S.; Gillespie, J.R.; Hulverson, M.A.; Huang, W.; Nguyen, J.; Pendem, N.; Gelb, M.H.; et al. 5-Fluoroimidazo [4,5-b]Pyridine Is a Privileged Fragment That Conveys Bioavailability to Potent Trypanosomal Methionyl-tRNA Synthetase Inhibitors. ACS Infect. Dis. 2016, 2, 399–404. [Google Scholar] [CrossRef]
- Ranade, R.M.; Zhang, Z.; Gillespie, J.R.; Shibata, S.; Verlinde, C.L.M.J.; Hol, W.G.J.; Fan, E.; Buckner, F.S. Inhibitors of Methionyl-tRNA Synthetase Have Potent Activity against Giardia Intestinalis Trophozoites. Antimicrob. Agents Chemother. 2015, 59, 7128–7131. [Google Scholar] [CrossRef]
- Pearson, M.M.; Sebaihia, M.; Churcher, C.; Quail, M.A.; Seshasayee, A.S.; Luscombe, N.M.; Abdellah, Z.; Arrosmith, C.; Atkin, B.; Chillingworth, T.; et al. Complete Genome Sequence of Uropathogenic Proteus mirabilis, a Master of Both Adherence and Motility. J. Bacteriol. 2008, 190, 4027–4037. [Google Scholar] [CrossRef]
- Schaffer, A.A. Improving the Accuracy of PSI-BLAST Protein Database Searches with Composition-Based Statistics and Other Refinements. Nucleic. Acids Res. 2001, 29, 2994–3005. [Google Scholar] [CrossRef]
- RCSB Protein Data Bank (PDB). Available online: http://www.rcsb.org (accessed on 1 March 2023).
- Weiner, S.J.; Kollman, P.A.; Nguyen, D.T.; Case, D.A. An All Atom Force Field for Simulations of Proteins and Nucleic Acids. J. Comput. Chem. 1986, 7, 230–252. [Google Scholar] [CrossRef]
- Kishk, S.M.; Kishk, R.M.; Yassen, A.S.A.; Nafie, M.S.; Nemr, N.A.; ElMasry, G.; Al-Rejaie, S.; Simons, C. Molecular Insights into Human Transmembrane Protease Serine-2 (TMPS2) Inhibitors against SARS-CoV2: Homology Modelling, Molecular Dynamics, and Docking Studies. Molecules 2020, 25, 5007. [Google Scholar] [CrossRef] [PubMed]
- Noureldin, N.A.; Richards, J.; Kothayer, H.; Baraka, M.M.; Eladl, S.M.; Wootton, M.; Simons, C. Phenylalanyl tRNA Synthetase (PheRS) Substrate Mimics: Design, Synthesis, Molecular Dynamics and Antimicrobial Evaluation. RSC Adv. 2022, 12, 2511–2524. [Google Scholar] [CrossRef] [PubMed]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organism | PDB Code | BLAST a Score | Sequence Identity b | Sequence Identity % | Positive % | Chain Length | E-Value |
|---|---|---|---|---|---|---|---|
| E. coli | 1F4L | 964 | 443/551 | 80 | 91 | 551 | 0.0 |
| A. baumannii | 5URB | 697 | 319/544 | 59 | 77 | 567 | 0.0 |
| P. abyss | 1RQG | 380 | 243/727 | 33 | 51 | 722 | 2 × 10−120 |
| PDB | HIGH Region Motif | KMSKS Motif | Zinc Binding Residues | ATP Binding |
|---|---|---|---|---|
| 1F4L | 15–25 | 333–337 | 145, 148, 158 and 161 | 336 |
| Ligand | Structure | P/L RMSD (Å) 0 ns | P/L RMSD (Å) 200 ns |
|---|---|---|---|
| Methionine |  | 2.14/1.50 | 3.74/6.80 |
| MPH |  | 1.46/1.18 | 4.00/6.67 |
| MPJ |  | 2.44/1.97 | 2.84/6.49 |
| 2FM |  | 1.59/1.79 | 2.57/5.29 |
| MF3 |  | 1.36/2.72 | 4.08/3.17 |
| MSP |  | 1.15/2.52 | 2.65/7.56 |
| Methionyl adenylate |  | 1.92/3.63 | 2.59/4.36 |
| A. aeolicus | E. coli | P. mirabilis |
|---|---|---|
| Asn356 | Asn391 | Asn390 |
| Arg360 | Arg395 | Arg394 |
| Trp430 | Trp461 | Trp460 |
| Compound | Target Organism | Ref. |
|---|---|---|
 | Trypanosoma brucei | [40] |
 | Giardia lamblia | [41,42] |
 | Cryptosporidium | [43] |
 | Clostridium difficile | [44] |
 | Trypanosoma brucei | [45,46] |
 | Trypanosoma brucei | [47,48] |
 | MRSA/Staphylococcus aureus | [49] |
 | Pseudomonas aeruginosa | [49] |
 | Trypanosoma brucei | [50] |
 | Staphylococcus aureus | [51] |
 | Staphylococcus aureus | [51] |
 | Staphylococcus aureus | [52] |
 | Staphylococcus aureus | [53] |
 | Escherichia coli | [54,55,56] |
 | Staphylococcus aureus | [57] |
 | Staphylococcus aureus | [58] |
 | Staphylococcus aureus | [59] |
 | Staphylococcus aureus | [60] |
 | Staphylococcus aureus | [60] |
 | Staphylococcus aureus | [61] |
 | Staphylococcus aureus | [61] |
 | Staphylococcus aureus | [61] |
 | Staphylococcus aureus | [62] |
 | Escherichia coli | [63] |
 | Escherichia coli | [64] |
 | Escherichia coli | [65] |
 | Trypanosoma brucei | [66] |
 | Clostridium difficile | [67] |
 | Trypanosoma brucei | [68] |
 | Trypanosoma brucei | [69] |
 | Trypanosoma brucei | [70] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elbaramawi, S.S.; Eissa, A.G.; Noureldin, N.A.; Simons, C. Exploring Proteus mirabilis Methionine tRNA Synthetase Active Site: Homology Model Construction, Molecular Dynamics, Pharmacophore and Docking Validation. Pharmaceuticals 2023, 16, 1263. https://doi.org/10.3390/ph16091263
Elbaramawi SS, Eissa AG, Noureldin NA, Simons C. Exploring Proteus mirabilis Methionine tRNA Synthetase Active Site: Homology Model Construction, Molecular Dynamics, Pharmacophore and Docking Validation. Pharmaceuticals. 2023; 16(9):1263. https://doi.org/10.3390/ph16091263
Chicago/Turabian StyleElbaramawi, Samar S., Ahmed G. Eissa, Nada A. Noureldin, and Claire Simons. 2023. "Exploring Proteus mirabilis Methionine tRNA Synthetase Active Site: Homology Model Construction, Molecular Dynamics, Pharmacophore and Docking Validation" Pharmaceuticals 16, no. 9: 1263. https://doi.org/10.3390/ph16091263
APA StyleElbaramawi, S. S., Eissa, A. G., Noureldin, N. A., & Simons, C. (2023). Exploring Proteus mirabilis Methionine tRNA Synthetase Active Site: Homology Model Construction, Molecular Dynamics, Pharmacophore and Docking Validation. Pharmaceuticals, 16(9), 1263. https://doi.org/10.3390/ph16091263