

Protective Effect of Peptide Calcium Channel Blocker Omega-Hexatoxin-Hv1a on Epithelial Cell during Ischemia–Reperfusion Injury
 ,
,
Abstract
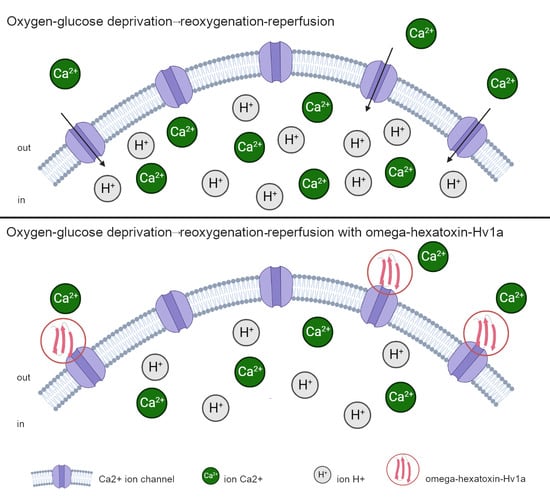
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
3. Discussion
4. Materials and Methods
- Peptide synthesis, quality control and formation of the secondary structure of toxin
- 2.
- Cell culture and experiment condition
- 3.
- Measuring the level of apoptosis, necrosis, mitochondrial membrane potential, concentration of ROS, NO, ATP, GSH, calcium, sodium, potassium ions, pH and cell index
- 4.
- ATP analysis
- 5.
- Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, Y.; Zhang, H.; Li, Z.; Yan, X.; Li, Y.; Liu, S. microRNA-130a-5p suppresses myocardial ischemia reperfusion injury by downregulating the HMGB2/NF-κB axis. BMC Musculoskelet. Disord. 2021, 21, 121. [Google Scholar] [CrossRef]
- Wu, Q.; Mao, Z.; Liu, J.; Huang, J.; Wang, N. Ligustilide Attenuates Ischemia Reperfusion-Induced Hippocampal Neuronal Apoptosis via Activating the PI3K/Akt Pathway. Front. Pharmacol. 2020, 11, 979. [Google Scholar] [CrossRef]
- El Kady, A.H.; Elkafoury, B.M.; Saad, D.A.; el-Wahed, D.M.; Baher, W.; Ahmed, M.A. Hepatic ischemia reperfusion injury: Effect of moderate intensity exercise and oxytocin compared to L-arginine in a rat model. Egypt. Liver J. 2021, 11, 45. [Google Scholar] [CrossRef]
- Shang, Y.; Madduma Hewage, S.; Wijerathne, C.U.B.; Siow, Y.L.; Isaak, C.K.; Karmin, O. Kidney Ischemia-Reperfusion Elicits Acute Liver Injury and Inflammatory Response. Front. Med. 2020, 7, 201. [Google Scholar] [CrossRef]
- Capuzzimati, M.; Hough, O.; Liu, M. Cell death and ischemia-reperfusion injury in lung transplantation. J. Heart Lung Transplant. 2022, 41, 1003–1013. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.-Y.; Yiang, G.-T.; Liao, W.-T.; Tsai, A.P.Y.; Cheng, Y.-L.; Cheng, P.-W.; Li, C.-Y.; Li, C.J. Current Mechanistic Concepts in Ischemia and Reperfusion Injury. Cell Physiol. Biochem. 2018, 46, 1650–1667. [Google Scholar] [CrossRef]
- He, J.; Liu, D.; Zhao, L.; Zhou, D.; Rong, J.; Zhang, L.; Xia, Z. Myocardial ischemia/reperfusion injury: Mechanisms of injury and implications for management. Exp. Ther. Med. 2022, 23, 430. [Google Scholar] [CrossRef]
- Wang, W.Z.; Fang, X.-H.; Stephenson, L.L.; Khiabani, K.T.; Zamboni, W.A. Ischemia/reperfusion-induced necrosis and apoptosis in the cells isolated from rat skeletal muscle. J. Orthop. Res. 2008, 26, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Neblina, F.; Toledo, A.H.; Toledo-Pereyra, L.H. Molecular biology of apoptosis in ischemia and reperfusion. J. Investig. Surg. 2015, 18, 335–350. [Google Scholar] [CrossRef] [PubMed]
- Menezes-Rodrigues, F.S.; Tavares, J.G.P.; Vasques, E.R.; Errante, P.R.; Araújo, E.A.; Pires-Oliveira, M.; Scorza, C.A.; Scorza, F.A.; Taha, M.O.; Caricati-Neto, A. Cardioprotective effects of pharmacological blockade of the mitochondrial calcium uniporter on myocardial ischemia-reperfusion injury. Acta Cir. Bras. 2020, 35, e202000306. [Google Scholar] [CrossRef]
- Rizzuto, R.; Pinton, P.; Ferrari, D.; Chami, M.; Szabadkai, G.; Magalhães, P.J.; Di Virgilio, F.; Pozzan, T. Calcium and apoptosis: Facts and hypotheses. Oncogene 2003, 22, 8619–8627. [Google Scholar] [CrossRef] [PubMed]
- Zhivotovsky, B.; Orrenius, S. Calcium and cell death mechanisms: A perspective from the cell death community. Cell Calcium. 2011, 50, 211–221. [Google Scholar] [CrossRef]
- Sadrhaghighi, G.; Abbaszadeh, S.; Babataheri, S.; Garjani, A.; Soraya, H. Effects of pre-treatment with metoprolol and diltiazem on cerebral ischemia/reperfusion-induced injuries. Braz. J. Pharm. Sci. 2022, 58, e21086. [Google Scholar] [CrossRef]
- Bao, M.; Huang, W.; Zhao, Y.; Fang, X.; Zhang, Y.; Gao, F.; Huang, D.; Wang, B.; Shi, G. Verapamil Alleviates Myocardial Ischemia/Reperfusion Injury by Attenuating Oxidative Stress via Activation of SIRT1. Front. Pharmacol. 2022, 13, 822640. [Google Scholar] [CrossRef]
- Søfteland, J.M.; Oltean, M. Intestinal Ischemia-Reperfusion Injury and Calcium Channel Blockers: Getting to the Core of the Problem. J. Investig. Surg. 2021, 34, 808–809. [Google Scholar] [CrossRef] [PubMed]
- Godfraind, T. Discovery and Development of Calcium Channel Blockers. Front. Pharmacol. 2017, 8, 286. [Google Scholar] [CrossRef]
- Fernandez, R.C.; Konate, K.; Josse, E.; Nargeot, J.; Barrère-Lemaire, S.; Boisguérin, P. Therapeutic Peptides to Treat Myocardial Ischemia-Reperfusion Injury. Front. Cardiovasc. Med. 2022, 9, 792885. [Google Scholar] [CrossRef]
- Oliveira, K.M.; Lavor, M.S.L.; Silva, O.C.M.; Fukushima, F.B.; Rosado, I.R.; Silva, J.F.; Martins, B.C.; Guimarães, L.B.; Gomez, M.V.; Melo, M.M.; et al. Omega-conotoxin MVIIC attenuates neuronal apoptosis in vitro and improves significant recovery after spinal cord injury in vivo in rats. Int. J. Clin. Exp. Pathol. 2014, 7, 3524–3536. [Google Scholar]
- Lubbers, N.L.; Campbell, T.J.; Polakowski, J.S.; Bulaj, G.; Layer, R.T.; Moore, J.; Gross, G.J.; Cox, B.F. Postischemic administration of CGX-1051, a peptide from cone snail venom, reduces infarct size in both rat and dog models of myocardial ischemia and reperfusion. J. Cardiovasc. Pharmacol. 2005, 46, 141–146. [Google Scholar] [CrossRef]
- Chassagnon, I.R.; McCarthy, C.A.; Chin, Y.K.-Y.; Pineda, S.S.; Keramidas, A.; Mobli, M.; Pham, V.; De Silva, T.M.; Lynch, J.W.; Widdop, R.E.; et al. Potent neuroprotection after stroke afforded by a double-knot spider-venom peptide that inhibits acid-sensing ion channel 1a. Proc. Natl. Acad. Sci. USA 2017, 114, 3750–3755. [Google Scholar] [CrossRef]
- Xu, X.; Lai, Y.; Hua, Z.-C. Apoptosis and apoptotic body: Disease message and therapeutic target potentials. Biosci. Rep. 2019, 29, BSR20180992. [Google Scholar] [CrossRef]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of cell death: The calcium-apoptosis link. Nat. Rev. Mol. Cell Biol. 2003, 4, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Soaresm, R.O.S.; Losadam, D.M.; Jordanim, M.C.; Évoram, P.; Castro-E-Silvam, O. Ischemia/Reperfusion Injury Revisited: An Overview of the Latest Pharmacological Strategies. Int. J. Mol. Sci. 2019, 20, 5034. [Google Scholar] [CrossRef] [PubMed]
- Chong, Y.; Hayes, J.L.; Sollod, B. The omega-atracotoxins: Selective blockers of insect M-LVA and HVA calcium channels. Biochem. Pharmacol. 2007, 74, 623–638. [Google Scholar] [CrossRef] [PubMed]
- Ponticelli, C. Ischaemia-reperfusion injury: A major protagonist in kidney transplantation. Nephrol. Dial. Transplant. 2014, 29, 1134–1140. [Google Scholar] [CrossRef]
- Saren, G.; Wong, A.; Lu, Y.-B.; Baciu, C.; Zhou, W.; Zamel, R.; Soltanieh, S.; Sugihara, J.; Liu, M. Ischemia-Reperfusion Injury in a Simulated Lung Transplant Setting Differentially Regulates Transcriptomic Profiles between Human Lung Endothelial and Epithelial Cells. Cells 2021, 10, 2713. [Google Scholar] [CrossRef]
- Skryma, R.; Prevarskaya, N.; Vacher, P.; Dufy, B. Voltage-dependent Ca2+ channels in Chinese hamster ovary (CHO) cells. FEBS Lett. 1994, 349, 289–294. [Google Scholar] [CrossRef]
- Iurova, E.; Beloborodov, E.; Tazintseva, E.; Fomin, A.; Shutov, A.; Slesarev, S.; Saenko, Y.; Saenko, Y. Arthropod toxins inhibiting Ca2+ and Na+ channels prevent AC-1001 H3 peptide-induced apoptosis. J. Pept. Sci. 2020, 27, e3288. [Google Scholar] [CrossRef]
- Inserte, J.; Barba, I.; Hernando, V.; Abellán, A.; Ruiz-Meana, M.; Rodríguez-Sinovas, A.; Garcia-Dorado, D. Effect of acidic reperfusion on prolongation of intracellular acidosis and myocardial salvage. Cardiovasc. Res. 2008, 77, 782–790. [Google Scholar] [CrossRef]
- Pike, M.M.; Kitakaze, M.; Marban, E. 23Na-NMR measurements of intracellular sodium in intact perfused ferret hearts during ischemia and reperfusion. Am. J. Physiol. 1990, 259, H1767–H1773. [Google Scholar] [CrossRef]
- Gao, F.; Gong, B.; Christopher, T.A.; Lopez, B.L.; Karasawa, A.; Ma, X.L. Anti-apoptotic effect of benidipine, a long-lasting vasodilating calcium antagonist, in ischaemic/reperfused myocardial cells. Br. J. Pharmacol. 2001, 132, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Cimen, O.; Eken, H.; Cimen, F.K.; Bilgin, A.O.; Pehlivanoglu, K.; Kurnaz, E.; Gülaboglu, M.; Suleyman, B.; Suleyman, H. Benidipine can prevent liver ischemia reperfusion injury in rats: A biochemical and histopathological evaluation. Biotechnol. Biotechnol. Equip. 2019, 33, 1645–1652. [Google Scholar] [CrossRef]
- Zhang, A.; Fu, S.; Chen, L.; Ren, L.; Qu, S.; Zhang, Y.; Yao, L.; Yang, S. Lacidipine attenuates apoptosis via a caspase-3 dependent pathway in human kidney cells. Cell Physiol. Biochem. 2013, 32, 1040–1049. [Google Scholar] [CrossRef]
- Jia, Z.; Chen, Q.; Qin, H. Ischemia-induced apoptosis of intestinal epithelial cells correlates with altered integrin distribution and disassembly of F-actin triggered by calcium overload. J. Biomed. Biotechnol. 2012, 2012, 617539. [Google Scholar] [CrossRef] [PubMed]
- Lemasters, J.J.; Bond, J.M.; Chacon, E.; Harper, I.S.; Kaplan, S.H.; Ohata, H.; Trollinger, D.R.; Herman, B.; Cascio, W.E. The pH paradox in ischemia-reperfusion injury to cardiac myocytes. EXS 1996, 76, 99–114. [Google Scholar] [CrossRef]
- Cohen, M.V.; Yang, X.-M.; Downey, J.M. The pH hypothesis of postconditioning: Staccato reperfusion reintroduces oxygen and perpetuates myocardial acidosis. Circulation 2007, 115, 1895–1903. [Google Scholar] [CrossRef] [PubMed]
- Iurova, E.; Beloborodov, E.; Rastorgueva, E. Peptide Sodium Channels Modulator Mu-Agatoxin-Aa1a Prevents Ischemia-Reperfusion Injury of Cells. Molecules 2023, 28, 3174. [Google Scholar] [CrossRef]
- Arita, Y.; Fukui, T.; Ogasawara, N. Slow-flow phenomenon after drug-coated balloon angioplasty for lower-extremity arteries is associated with lack of prescribing of calcium channel blockers. Indian Heart J. 2023, 75, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Kranzhöfer, A.F.; Weingärtner, O.; Oberhoff, M.; Karsch, K.R. Effect of a dihydropyridine-type calcium channel blocker on vascular remodeling after experimental balloon angioplasty. Cardiovasc. Hematol. Agents Med. Chem. 2011, 9, 1–6. [Google Scholar] [CrossRef]
- Moore, S.J.; Leung, C.L.; Norton, H.K.; Cochran, J.R. Engineering agatoxin, a cystine-knot peptide from spider venom, as a molecular probe for in vivo tumor imaging. PLoS ONE 2013, 8, e60498. [Google Scholar] [CrossRef]
- Wenger, R.H.; Kurtcuoglu, V.; Scholz, C.C.; Marti, H.H.; Hoogewijs, D. Frequently asked questions in hypoxia research. Hypoxia 2015, 3, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Bolaños, J.M.G.; Morán, Á.M.; Balao da Silva, C.M. Autophagy and apoptosis have a role in the survival or death of stallion spermatozoa during conservation in refrigeration. PLoS ONE 2012, 7, e30688. [Google Scholar] [CrossRef]
- Saenko, Y.V.; Glushchenko, E.S.; Zolotovskii, I.O.; Sholokhov, E.; Kurkov, A. Mitochondrial dependent oxidative stress in cell culture induced by laser radiation at 1265 nm. Lasers Med. Sci. 2016, 31, 405–413. [Google Scholar] [CrossRef]
- Zeidler, D.; Zähringer, U.; Gerber, I.; Dubery, I.; Hartung, T.; Bors, W.; Hutzler, P.; Durner, J. Innate immunity in Arabidopsis thaliana: Lipopolysaccharides activate nitric oxide synthase (NOS) and induce defense genes. Proc. Natl. Acad. Sci. USA 2004, 101, 15811–15816. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Noack, H.; Possel, H.; Keilhoff, G.; Wolf, G. Glutathione levels in primary glial cultures: Monochlorobimane provides evidence of cell type-specific distribution. Glia 1999, 27, 152–161. [Google Scholar] [CrossRef]
- Fonteriz, R.I.; de la Fuente, S.; Moreno, A.; Lobatón, C.D.; Montero, M.; Alvarez, J. Monitoring mitochondrial [Ca2+] dynamics with rhod-2, ratiometric pericam and aequorin. Cell Calcium. 2010, 48, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Tay, B.; Stewart, T.A.; Davis, F.M.; Deuis, J.R.; Vetter, I. Development of a high-throughput fluorescent no-wash sodium influx assay. PLoS ONE 2019, 14, e0213751. [Google Scholar] [CrossRef]
- Camilli, G.; Bohm, M.; Piffer, A.C.; Lavenir, R.; Williams, D.L.; Neven, B.; Grateau, G.; Georgin-Lavialle, S.; Quintin, J. β-Glucan-induced reprogramming of human macrophages inhibits NLRP3 inflammasome activation in cryopyrinopathies. J. Clin. Investig. 2020, 130, 4561–4573. [Google Scholar] [CrossRef]
- Alvarez-Leefmans, F.J.; Herrera-Pérez, J.J.; Márquez, M.S.; Blanco, V.M. Simultaneous measurement of water volume and pH in single cells using BCECF and fluorescence imaging microscopy. Biophys. J. 2006, 90, 608–618. [Google Scholar] [CrossRef]
- Ke, N.; Wang, X.; Xu, X.; Abassi, Y.A. The xCELLigence system for real-time and label-free monitoring of cell viability. Methods Mol. Biol. 2011, 740, 33–43. [Google Scholar] [CrossRef]
- Liu, H.; Jiang, Y.; Luo, Y.; Jiang, W. A simple and rapid determination of ATP, ADP and AMP concentrations in pericarp tissue of litchi fruit by high performance liquid chromatography. Food Technol. Biotechnol. 2006, 44, 531–534. [Google Scholar]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iurova, E.; Rastorgueva, E.; Beloborodov, E.; Pogodina, E.; Fomin, A.; Sugak, D.; Viktorov, D.; Tumozov, I.; Saenko, Y. Protective Effect of Peptide Calcium Channel Blocker Omega-Hexatoxin-Hv1a on Epithelial Cell during Ischemia–Reperfusion Injury. Pharmaceuticals 2023, 16, 1314. https://doi.org/10.3390/ph16091314
Iurova E, Rastorgueva E, Beloborodov E, Pogodina E, Fomin A, Sugak D, Viktorov D, Tumozov I, Saenko Y. Protective Effect of Peptide Calcium Channel Blocker Omega-Hexatoxin-Hv1a on Epithelial Cell during Ischemia–Reperfusion Injury. Pharmaceuticals. 2023; 16(9):1314. https://doi.org/10.3390/ph16091314
Chicago/Turabian StyleIurova, Elena, Eugenia Rastorgueva, Evgenii Beloborodov, Evgeniya Pogodina, Aleksandr Fomin, Dmitrii Sugak, Denis Viktorov, Ivan Tumozov, and Yury Saenko. 2023. "Protective Effect of Peptide Calcium Channel Blocker Omega-Hexatoxin-Hv1a on Epithelial Cell during Ischemia–Reperfusion Injury" Pharmaceuticals 16, no. 9: 1314. https://doi.org/10.3390/ph16091314
APA StyleIurova, E., Rastorgueva, E., Beloborodov, E., Pogodina, E., Fomin, A., Sugak, D., Viktorov, D., Tumozov, I., & Saenko, Y. (2023). Protective Effect of Peptide Calcium Channel Blocker Omega-Hexatoxin-Hv1a on Epithelial Cell during Ischemia–Reperfusion Injury. Pharmaceuticals, 16(9), 1314. https://doi.org/10.3390/ph16091314