
Hitting the Target! Challenges and Opportunities for TGF-β Inhibition for the Treatment of Cardiac fibrosis


{kind=link}
{kind=link}
Abstract
:1. The Need for Anti-Fibrotic Therapies in Heart Failure Management
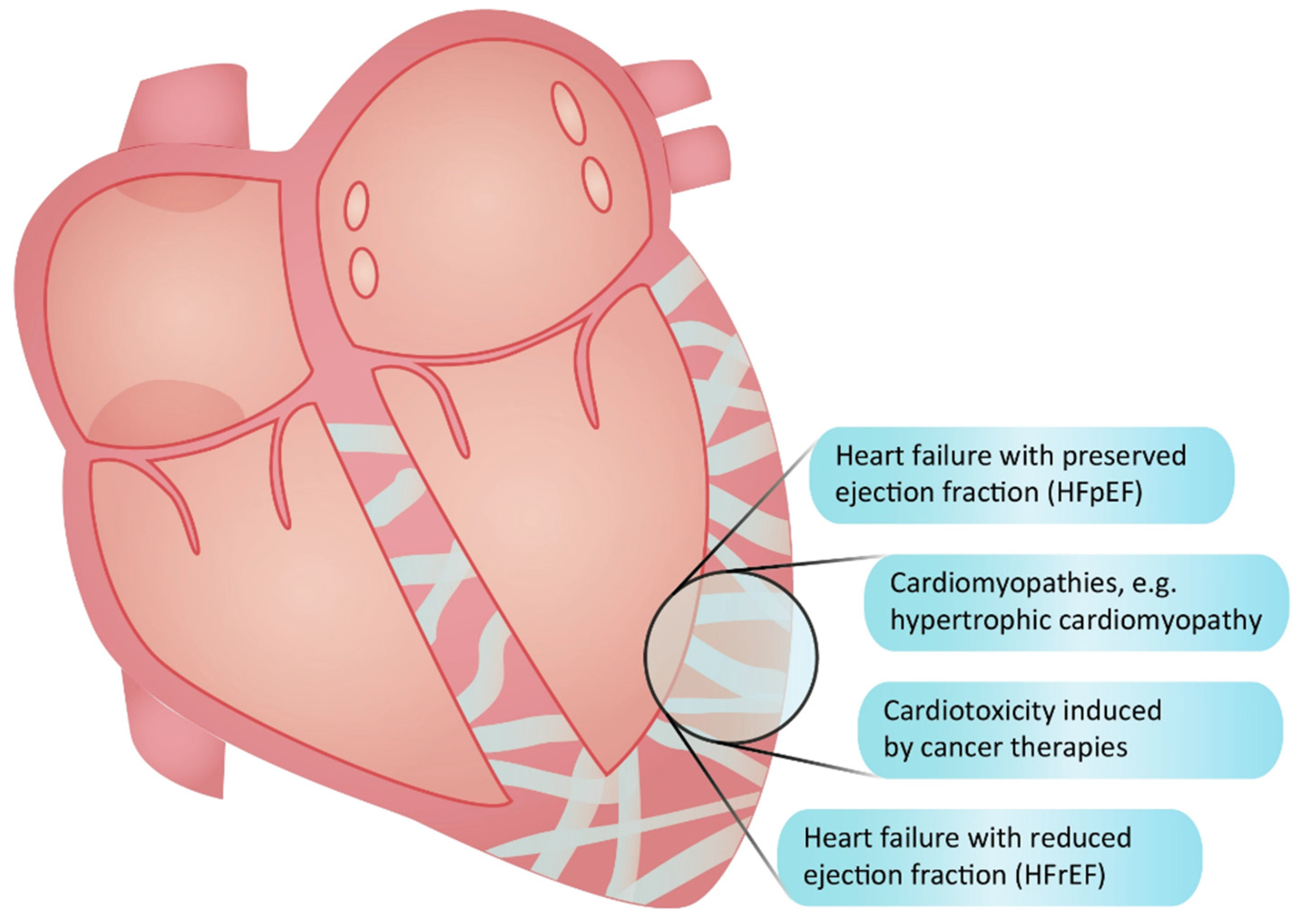
2. The Significance of Fibrosis as a Therapeutic Target in Cardiomyopathies and Cardiotoxicities

3. The Dual Nature of Fibrosis and TGF-β in the Heart: Adaptive and Maladaptive Roles
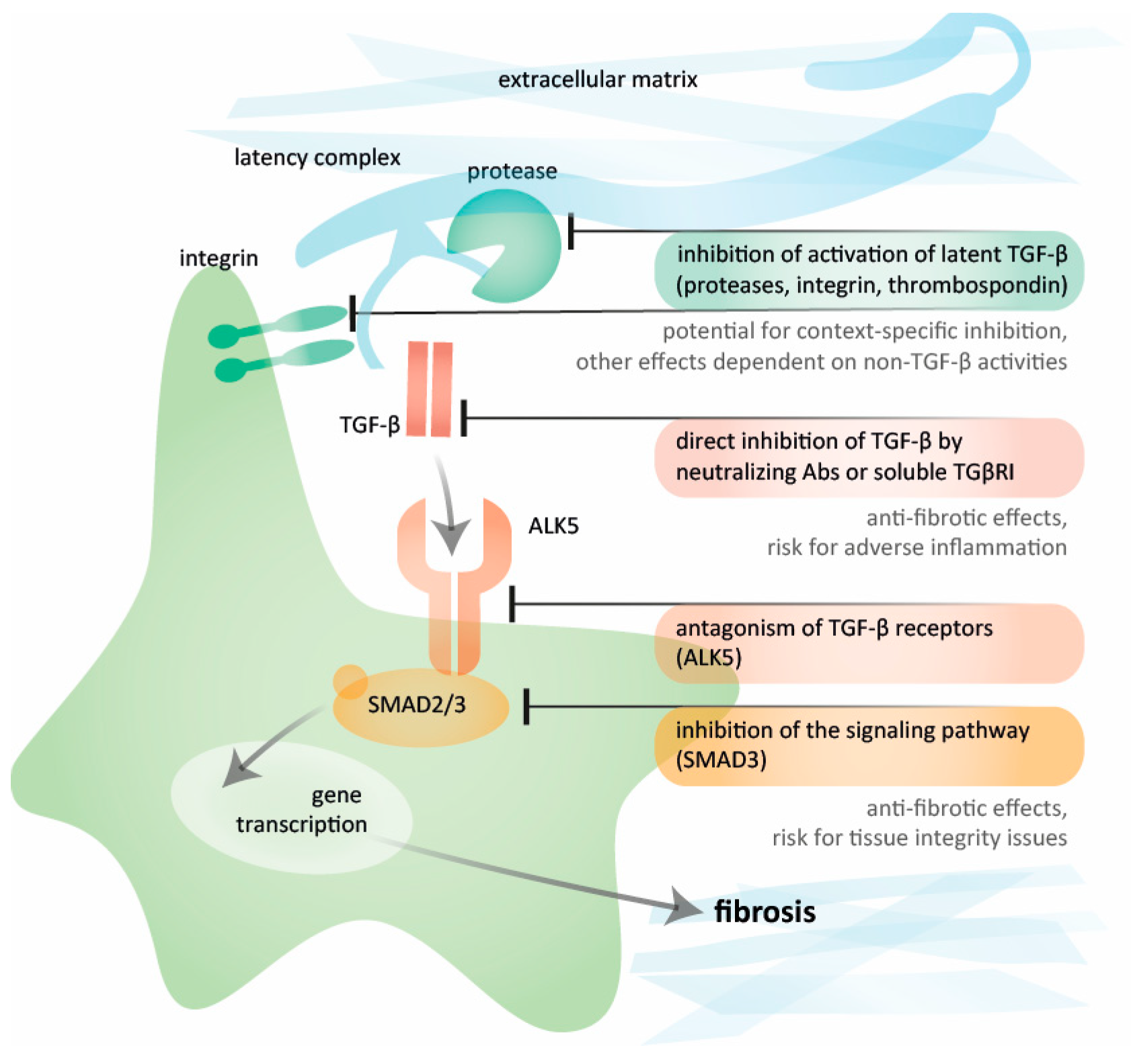
4. Targeting Activators of Latent TGF-β for Selective Inhibition
5. Inhibiting TGF-β Directly: Targets in the Signaling Pathway

6. Direct Blockade of TGF-β: Timing Matters!
7. Balancing Efficacy and Adverse Effects of Inhibitors of TGF-β Receptors
8. SMAD Deficiency and Loss of Structural Integrity
9. The Challenges of Targeting MMPs in Heart Disease
10. Enhancing Precision in Integrin Antagonism for the Prevention of Latent TGF-β Activation
11. Targeted Inhibition through Other Activators of Latent TGF-β
12. TGF-β-Suppressing Agents with Unknown Mechanisms of Action
13. Through the Backdoor: TGF-β Modulation via the Renin–Angiotensin–Aldosterone System
14. Modest Anti-Fibrotic Effects of Other Heart Failure Drugs
15. Challenges in Translating Anti-Fibrotic Strategies from Other Organs to the Heart
16. Conclusions and Future Directions
Funding
Conflicts of Interest
References
- Henderson, N.C.; Rieder, F.; Wynn, T.A. Fibrosis: From mechanisms to medicines. Nature 2020, 587, 555–566. [Google Scholar] [CrossRef]
- Kato, S.; Saito, N.; Kirigaya, H.; Gyotoku, D.; Iinuma, N.; Kusakawa, Y.; Iguchi, K.; Nakachi, T.; Fukui, K.; Futaki, M.; et al. Prognostic significance of quantitative assessment of focal myocardial fibrosis in patients with heart failure with preserved ejection fraction. Int. J. Cardiol. 2015, 191, 314–319. [Google Scholar] [CrossRef]
- Gulati, A.; Jabbour, A.; Ismail, T.F.; Guha, K.; Khwaja, J.; Raza, S.; Morarji, K.; Brown, T.D.H.; Ismail, N.A.; Dweck, M.R.; et al. Association of Fibrosis With Mortality and Sudden Cardiac Death in Patients With Nonischemic Dilated Cardiomyopathy. J. Am. Med. Assoc. 2013, 309, 896–908. [Google Scholar] [CrossRef]
- Moreo, A.; Ambrosio, G.; De Chiara, B.; Pu, M.; Tran, T.; Mauri, F.; Raman, S.V. Influence of myocardial fibrosis on left ventricular diastolic function: Noninvasive assessment by cardiac magnetic resonance and echo. Circ. Cardiovasc. Imaging 2009, 2, 437–443. [Google Scholar] [CrossRef]
- Murtha, L.A.; Morten, M.; Schuliga, M.J.; Mabotuwana, N.S.; Hardy, S.A.; Waters, D.W.; Burgess, J.K.; Ngo, D.T.; Sverdlov, A.L.; Knight, D.A.; et al. The Role of Pathological Aging in Cardiac and Pulmonary Fibrosis. Aging Dis. 2019, 10, 419–428. [Google Scholar] [CrossRef]
- López, B.; Ravassa, S.; Moreno, M.U.; José, G.S.; Beaumont, J.; González, A.; Díez, J. Diffuse myocardial fibrosis: Mechanisms, diagnosis and therapeutic approaches. Nat. Rev. Cardiol. 2021, 18, 479–498. [Google Scholar] [CrossRef]
- Sweeney, M.; Corden, B.; Cook, S.A. Targeting cardiac fibrosis in heart failure with preserved ejection fraction: Mirage or miracle? EMBO Mol. Med. 2020, 12, e10865. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2023 Focused Update of the 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2023, 44, 3627–3639. [Google Scholar] [CrossRef]
- Díez, J.; de Boer, R.A. Management of cardiac fibrosis is the largest unmet medical need in heart failure. Cardiovasc. Res. 2021, 118, e20–e22. [Google Scholar] [CrossRef]
- Heerebeek Lv Borbély, A.; Niessen, H.W.M.; Bronzwaer, G.F.; Velden, J.v.d.; Stienen, G.J.M.; Linke, W.A.; Laarman, G.J.; Paulus, W.J. Myocardial Structure and Function Differ in Systolic and Diastolic Heart Failure. Circulation 2006, 113, 1966–1973. [Google Scholar] [CrossRef]
- Dai, Z.; Aoki, T.; Fukumoto, Y.; Shimokawa, H. Coronary perivascular fibrosis is associated with impairment of coronary blood flow in patients with non-ischemic heart failure. J. Cardiol. 2012, 60, 416–421. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef]
- Simmonds, S.J.; Cuijpers, I.; Heymans, S.; Jones, E.A.V. Cellular and Molecular Differences between HFpEF and HFrEF: A Step Ahead in an Improved Pathological Understanding. Cells 2020, 9, 242. [Google Scholar] [CrossRef]
- Schlittler, M.; Pramstaller, P.P.; Rossini, A.; De Bortoli, M. Myocardial Fibrosis in Hypertrophic Cardiomyopathy: A Perspective from Fibroblasts. Int. J. Mol. Sci. 2023, 24, 14845. [Google Scholar] [CrossRef]
- O’Hanlon, R.; Grasso, A.; Roughton, M.; Moon, J.C.; Clark, S.; Wage, R.; Webb, J.; Kulkarni, M.; Dawson, D.; Sulaibeekh, L.; et al. Prognostic significance of myocardial fibrosis in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2010, 56, 867–874. [Google Scholar] [CrossRef]
- Ellims, A.H.; Iles, L.M.; Ling, L.H.; Hare, J.L.; Kaye, D.M.; Taylor, A.J. Diffuse myocardial fibrosis in hypertrophic cardiomyopathy can be identified by Cardiovascular magnetic resonance, and is associated with left ventricular diastolic dysfunction. J. Cardiovasc. Magn. Reson. 2012, 14, 76. [Google Scholar] [CrossRef]
- McLellan, A.J.A.; Ellims, A.H.; Prabhu, S.; Voskoboinik, A.; Iles, L.M.; Hare, J.L.; Kaye, D.M.; Macciocca, I.; Mariani, J.A.; Kalman, J.M.; et al. Diffuse Ventricular Fibrosis on Cardiac Magnetic Resonance Imaging Associates With Ventricular Tachycardia in Patients With Hypertrophic Cardiomyopathy. J. Cardiovasc. Electrophysiol. 2016, 27, 571–580. [Google Scholar] [CrossRef]
- Todiere, G.; Aquaro, G.D.; Piaggi, P.; Formisano, F.; Barison, A.; Masci, P.G.; Strata, E.; Bacigalupo, L.; Marzilli, M.; Pingitore, A.; et al. Progression of myocardial fibrosis assessed with cardiac magnetic resonance in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2012, 60, 922–929. [Google Scholar] [CrossRef]
- Green, J.J.; Berger, J.S.; Kramer, C.M.; Salerno, M. Prognostic value of late gadolinium enhancement in clinical outcomes for hypertrophic cardiomyopathy. JACC Cardiovasc. Imaging 2012, 5, 370–377. [Google Scholar] [CrossRef]
- Raman, B.; Ariga, R.; Spartera, M.; Sivalokanathan, S.; Chan, K.; Dass, S.; Petersen, S.E.; Daniels, M.J.; Francis, J.; Smillie, R.; et al. Progression of myocardial fibrosis in hypertrophic cardiomyopathy: Mechanisms and clinical implications. Eur. Heart J.Cardiovasc. Imaging 2018, 20, 157–167. [Google Scholar] [CrossRef]
- Marian, A.J. Role of the Extracellular Matrix in the Pathogenesis of Hypertrophic Cardiomyopathy. JACC Basic Transl. Sci. 2019, 4, 506–508. [Google Scholar] [CrossRef]
- Bittencourt, M.I.; Cader, S.A.; Araújo, D.V.; Salles, A.L.F.; Albuquerque, F.N.; Spineti, P.P.M.; Albuquerque, D.C.; Mourilhe-Rocha, R. Role of Myocardial Fibrosis in Hypertrophic Cardiomyopathy: A Systematic Review and Updated Meta-Analysis of Risk Markers for Sudden Death. Arq. Bras. Cardiol. 2019, 112, 281–289. [Google Scholar] [CrossRef]
- Weng, Z.; Yao, J.; Chan, R.H.; He, J.; Yang, X.; Zhou, Y.; He, Y. Prognostic Value of LGE-CMR in HCM: A Meta-Analysis. JACC Cardiovasc. Imaging 2016, 9, 1392–1402. [Google Scholar] [CrossRef]
- Olivotto, I.; Oreziak, A.; Barriales-Villa, R.; Abraham, T.P.; Masri, A.; Garcia-Pavia, P.; Saberi, S.; Lakdawala, N.K.; Wheeler, M.T.; Owens, A.; et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020, 396, 759–769. [Google Scholar] [CrossRef]
- Green, E.M.; Wakimoto, H.; Anderson, R.L.; Evanchik, M.J.; Gorham, J.M.; Harrison, B.C.; Henze, M.; Kawas, R.; Oslob, J.D.; Rodriguez, H.M.; et al. A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science 2016, 351, 617–621. [Google Scholar] [CrossRef]
- Maron, B.J.; Shirani, J.; Poliac, L.C.; Mathenge, R.; Roberts, W.C.; Mueller, F.O. Sudden death in young competitive athletes. Clinical, demographic, and pathological profiles. J. Am. Med. Assoc. 1996, 276, 199–204. [Google Scholar] [CrossRef]
- Herrmann, J. Adverse cardiac effects of cancer therapies: Cardiotoxicity and arrhythmia. Nat. Rev. Cardiol. 2020, 17, 474–502. [Google Scholar] [CrossRef]
- Packard, R.R.S. Cardiac fibrosis in oncologic therapies. Curr. Opin. Physiol. 2022, 29, 100575. [Google Scholar] [CrossRef]
- Hinz, B.; Lagares, D. Evasion of apoptosis by myofibroblasts: A hallmark of fibrotic diseases. Nat. Rev. Rheumatol. 2020, 16, 11–31. [Google Scholar] [CrossRef]
- Khalil, H.; Kanisicak, O.; Prasad, V.; Correll, R.N.; Fu, X.; Schips, T.; Vagnozzi, R.J.; Liu, R.; Huynh, T.; Lee, S.J.; et al. Fibroblast-specific TGF-β–Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Investig. 2017, 127, 3770–3783. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Transforming growth factor-β in myocardial disease. Nat. Rev. Cardiol. 2022, 19, 435–455. [Google Scholar] [CrossRef]
- Meng, X.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Dobaczewski, M.; Chen, W.; Frangogiannis, N.G. Transforming growth factor (TGF)-beta signaling in cardiac remodeling. J. Mol. Cell. Cardiol. 2011, 51, 600–606. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Transforming growth factor–β in tissue fibrosis. J. Exp. Med. 2020, 217, e20190103. [Google Scholar] [CrossRef]
- Massagué, J.; Sheppard, D. TGF-beta signaling in health and disease. Cell 2023, 186, 4007–4037. [Google Scholar] [CrossRef]
- Kulkarni, A.B.; Karlsson, S. Transforming growth factor-beta 1 knockout mice. A mutation in one cytokine gene causes a dramatic inflammatory disease. Am. J. Pathol. 1993, 143, 3–9. [Google Scholar]
- Loeys, B.L.; Schwarze, U.; Holm, T.; Callewaert, B.L.; Thomas, G.H.; Pannu, H.; De Backer, J.F.; Oswald, G.L.; Symoens, S.; Manouvrier, S.; et al. Aneurysm Syndromes Caused by Mutations in the TGF-β Receptor. N. Engl. J. Med. 2006, 355, 788–798. [Google Scholar] [CrossRef]
- Annes, J.P.; Munger, J.S.; Rifkin, D.B. Making sense of latent TGFbeta activation. J. Cell. Sci. 2003, 116 Pt 2, 217–224. [Google Scholar] [CrossRef]
- Vistnes, M.; Erusappan, P.M.; Sasi, A.; Nordén, E.S.; Bergo, K.; Romaine, A.; Lunde, I.G.; Zhang, L.; Olsen, M.B.; Øgaard, J.; et al. Inhibition of the extracellular enzyme ADAMTS4 prevents cardiac fibrosis and dysfunction. Cardiovasc. Res. 2023, 119, 1915–1927. [Google Scholar] [CrossRef]
- Budi, E.H.; Schaub, J.R.; Decaris, M.; Turner, S.; Derynck, R. TGF-β as a driver of fibrosis: Physiological roles and therapeutic opportunities. J. Pathol. 2021, 254, 358–373. [Google Scholar] [CrossRef]
- Rifkin, D.B.; Rifkin, W.J.; Zilberberg, L. LTBPs in biology and medicine: LTBP diseases. Matrix Biol. 2018, 71–72, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Robertson, I.B.; Rifkin, D.B. Regulation of the Bioavailability of TGF-β and TGF-β-Related Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a021907. [Google Scholar] [CrossRef]
- Massam-Wu, T.; Chiu, M.; Choudhury, R.; Choudhury, R.; Chaudhry, S.S.; Baldwin, A.K.; McGovern, A.; Baldock, C.; Shuttleworth, C.A.; Kielty, C.M. Assembly of fibrillin microfibrils governs extracellular deposition of latent TGFβ. J. Cell. Sci. 2010, 123, 3006–3018. [Google Scholar] [CrossRef] [PubMed]
- Dallas, S.L.; Sivakumar, P.; Jones, C.J.P.; Chen, Q.; Peters, D.M.; Mosher, D.F.; Humphries, M.J.; Kielty, C.M. Fibronectin regulates latent transforming growth factor-β (TGFβ) by controlling matrix assembly of latent TGFβ-binding protein-1. J. Biol. Chem. 2005, 280, 18871–18880. [Google Scholar] [CrossRef]
- Klingberg, F.; Chau, G.; Walraven, M.; Boo, S.; Koehler, A.; Chow, M.L.; Olsen, A.L.; Im, M.; Lodyga, M.; Wells, R.G.; et al. The fibronectin ED-A domain enhances recruitment of latent TGF-beta-binding protein-1 to the fibroblast matrix. J. Cell. Sci. 2018, 131, jcs201293. [Google Scholar] [CrossRef]
- Hinz, B. The extracellular matrix and transforming growth factor-β1: Tale of a strained relationship. Matrix Biol. 2015, 47, 54–65. [Google Scholar] [CrossRef]
- Taipale, J.; Lohi, J.; Saarinen, J.; Kovanen, P.T.; Keski-Oja, J. Human mast cell chymase and leukocyte elastase release latent transforming growth factor-β1 from the extracellular matrix of cultured human epithelial and endothelial cells. J. Biol. Chem. 1995, 270, 4689–4696. [Google Scholar] [CrossRef]
- Yu, Q.; Stamenkovic, I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000, 14, 163–176. [Google Scholar] [CrossRef]
- Crawford, S.E.; Stellmach, V.; Murphy-Ullrich, J.E.; Ribeiro, S.M.F.; Lawler, J.; Hynes, R.O.; Boivin, G.P.; Bouck, N. Thrombospondin-1 Is a Major Activator of TGF-β1 In Vivo. Cell 1998, 93, 1159–1170. [Google Scholar] [CrossRef]
- Stockis, J.; Dedobbeleer, O.; Lucas, S. Role of GARP in the activation of latent TGF-β1. Mol. Biosyst. 2017, 13, 1925–1935. [Google Scholar] [CrossRef]
- Vistnes, M.; Aronsen, J.M.; Lunde, I.G.; Sjaastad, I.; Carlson, C.R.; Christensen, G. Pentosan polysulfate decreases myocardial expression of the extracellular matrix enzyme ADAMTS4 and improves cardiac function in vivo in rats subjected to pressure overload by aortic banding. PLoS ONE 2014, 9, e89621. [Google Scholar] [CrossRef]
- Massagué, J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef]
- Hill, C.S. Transcriptional Control by the SMADs. Cold Spring Harb. Perspect. Biol. 2016, 8, a022079. [Google Scholar] [CrossRef] [PubMed]
- Tecalco-Cruz, A.C.; Ríos-López, D.G.; Vázquez-Victorio, G.; Rosales-Alvarez, R.E.; Macías-Silva, M. Transcriptional cofactors Ski and SnoN are major regulators of the TGF-β/Smad signaling pathway in health and disease. Signal Transduct. Target. Ther. 2018, 3, 15. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-Smad pathways in TGF-β signaling. Cell. Res. 2009, 19, 128–139. [Google Scholar] [CrossRef]
- Heldin, C.-H.; Moustakas, A. Signaling Receptors for TGF-β Family Members. Cold Spring Harb. Perspect. Biol. 2016, 8, a022053. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.; Fu, M.; Wang, M.; Wei, Y.; Wei, X. Targeting TGF-β signal transduction for fibrosis and cancer therapy. Mol. Cancer 2022, 21, 104. [Google Scholar] [CrossRef] [PubMed]
- Mitra, M.S.; Lancaster, K.; Adedeji, A.O.; Palanisamy, G.S.; Dave, R.A.; Zhong, F.; Holdren, M.S.; Turley, S.J.; Liang, W.C.; Wu, Y.; et al. A Potent Pan-TGFβ Neutralizing Monoclonal Antibody Elicits Cardiovasc.ular Toxicity in Mice and Cynomolgus Monkeys. Toxicol. Sci. 2020, 175, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Ikeuchi, M.; Tsutsui, H.; Shiomi, T.; Matsusaka, H.; Matsushima, S.; Wen, J.; Kubota, T.; Takeshita, A. Inhibition of TGF-beta signaling exacerbates early cardiac dysfunction but prevents late remodeling after infarction. Cardiovasc. Res. 2004, 64, 526–535. [Google Scholar] [CrossRef]
- Frantz, S.; Hu, K.; Adamek, A.; Wolf, J.; Sallam, A.; Maier, S.K.G.; Lonning, S.; Ling, H.; Ertl, G.; Bauersachs, J. Transforming growth factor beta inhibition increases mortality and left ventricular dilatation after myocardial infarction. Basic Res Cardiol. 2008, 103, 485–492. [Google Scholar] [CrossRef]
- Okada, H.; Takemura, G.; Kosai, K.I.; Li, Y.; Takahashi, T.; Esaki, M.; Yuge, K.; Miyata, S.; Maruyama, R.; Mikami, A.; et al. Postinfarction Gene Therapy Against Transforming Growth Factor-β Signal Modulates Infarct Tissue Dynamics and Attenuates Left Ventricular Remodeling and Heart Failure. Circulation 2005, 111, 2430–2437. [Google Scholar] [CrossRef]
- Kuwahara, F.; Kai, H.; Tokuda, K.; Kai, M.; Takeshita, A.; Egashira, K.; Imaizumi, T. Transforming Growth Factor-β Function Blocking Prevents Myocardial Fibrosis and Diastolic Dysfunction in Pressure-Overloaded Rats. Circulation 2002, 106, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Song, X.; Liu, Y.; Wu, Y.; Shi, J.; Zhang, F.; Pan, Y.; Cao, Z.; Zhang, K.; Liu, J.; et al. Application of recombinant TGF-β1 inhibitory peptide to alleviate isoproterenol-induced cardiac fibrosis. Appl. Microbiol. Biotechnol. 2023, 107, 6251–6262. [Google Scholar] [CrossRef]
- Lacouture, M.E.; Morris, J.C.; Lawrence, D.P.; Tan, A.R.; Olencki, T.E.; Shapiro, G.I.; Dezube, B.J.; Berzofsky, J.A.; Hsu, F.J.; Guitart, J. Cutaneous keratoacanthomas/squamous cell carcinomas associated with neutralization of transforming growth factor β by the monoclonal antibody fresolimumab (GC1008). Cancer Immunol. Immunother. 2015, 64, 437–446. [Google Scholar] [CrossRef]
- Petersen, M.; Thorikay, M.; Deckers, M.; van Dinther, M.; Grygielko, E.T.; Gellibert, F.; de Gouville, A.C.; Huet, S.; ten Dijke, P.; Laping, N.J. Oral administration of GW788388, an inhibitor of TGF-β type I and II receptor kinases, decreases renal fibrosis. Kidney Int. 2008, 73, 705–715. [Google Scholar] [CrossRef]
- Hammad, S.; Cavalcanti, E.; Werle, J.; Caruso, M.L.; Dropmann, A.; Ignazzi, A.; Ebert, M.P.; Dooley, S.; Giannelli, G. Galunisertib modifies the liver fibrotic composition in the Abcb4Ko mouse model. Arch. Toxicol. 2018, 92, 2297–2309. [Google Scholar] [CrossRef]
- Tan, S.M.; Zhang, Y.; Connelly, K.A.; Gilbert, R.E.; Kelly, D.J. Targeted inhibition of activin receptor-like kinase 5 signaling attenuates cardiac dysfunction following myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H1415–H1425. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.R.; Abreu, R.D.S.; Vilar-Pereira, G.; Mello de Souza, E.; Ramos, I.P.; Bailly, S.; Feige, J.J.; Lannes-Vieira, J.; de Araújo-Jorge, T.C.; Waghabi, M.C. TGF-β inhibitor therapy decreases fibrosis and stimulates cardiac improvement in a pre-clinical study of chronic Chagas’ heart disease. PLoS Negl. Trop. Dis. 2019, 13, e0007602. [Google Scholar] [CrossRef] [PubMed]
- Ellmers, L.J.; Scott, N.J.A.; Medicherla, S.; Pilbrow, A.P.; Bridgman, P.G.; Yandle, T.G.; Richards, A.M.; Protter, A.A.; Cameron, V.A. Transforming Growth Factor-β Blockade Down-Regulates the Renin-Angiotensin System and Modifies Cardiac Remodeling after Myocardial Infarction. Endocrinology 2008, 149, 5828–5834. [Google Scholar] [CrossRef]
- Anderton, M.J.; Mellor, H.R.; Bell, A.; Sadler, C.; Pass, M.; Powell, S.; Steele, S.J.; Roberts, R.R.; Heier, A. Induction of heart valve lesions by small-molecule ALK5 inhibitors. Toxicol. Pathol. 2011, 39, 916–924. [Google Scholar] [CrossRef]
- Engebretsen, K.V.; Skårdal, K.; Bjørnstad, S.; Marstein, H.S.; Skrbic, B.; Sjaastad, I.; Christensen, G.; Bjørnstad, J.L.; Tønnessen, T. Attenuated development of cardiac fibrosis in left ventricular pressure overload by SM16, an orally active inhibitor of ALK5. J. Mol. Cell. Cardiol. 2014, 76, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Lucas, J.A.; Zhang, Y.; Li, P.; Gong, K.; Miller, A.P.; Hassan, E.; Hage, F.; Xing, D.; Wells, B.; Oparil, S.; et al. Inhibition of transforming growth factor-beta signaling induces left ventricular dilation and dysfunction in the pressure-overloaded heart. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H424–H432. [Google Scholar] [CrossRef] [PubMed]
- Hanna, A.; Humeres, C.; Frangogiannis, N.G. The role of Smad signaling cascades in cardiac fibrosis. Cell. Signal. 2021, 77, 109826. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Chen, B.; Humeres, C.; Alex, L.; Hanna, A.; Frangogiannis, N.G. The role of Smad2 and Smad3 in regulating homeostatic functions of fibroblasts in vitro and in adult mice. Biochim. Biophys. Acta Mol. Cell. Res. 2020, 1867, 118703. [Google Scholar] [CrossRef] [PubMed]
- Bujak, M.; Frangogiannis, N.G. The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc. Res. 2007, 74, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Bujak, M.; Ren, G.; Kweon, H.J.; Dobaczewski, M.; Reddy, A.; Taffet, G.; Wang, X.F.; Frangogiannis, N.G. Essential role of Smad3 in infarct healing and in the pathogenesis of cardiac remodeling. Circulation 2007, 116, 2127–2138. [Google Scholar] [CrossRef] [PubMed]
- Euler, G. Good and bad sides of TGFβ-signaling in myocardial infarction. Front. Physiol. 2015, 6, 66. [Google Scholar] [CrossRef]
- Biernacka, A.; Cavalera, M.; Wang, J.; Russo, I.; Shinde, A.; Kong, P.; Gonzalez-Quesada, C.; Rai, V.; Dobaczewski, M.; Lee, D.W.; et al. Smad3 Signaling Promotes Fibrosis While Preserving Cardiac and Aortic Geometry in Obese Diabetic Mice. Circ. Heart Fail. 2015, 8, 788–798. [Google Scholar] [CrossRef]
- Kong, P.; Shinde, A.V.; Su, Y.; Russo, I.; Chen, B.; Saxena, A.; Conway, S.J.; Graff, J.M.; Frangogiannis, N.G. Opposing Actions of Fibroblast and Cardiomyocyte Smad3 Signaling in the Infarcted Myocardium. Circulation 2018, 137, 707–724. [Google Scholar] [CrossRef]
- Umbarkar, P.; Singh, A.P.; Gupte, M.; Verma, V.K.; Galindo, C.L.; Guo, Y.; Zhang, Q.; McNamara, J.W.; Force, T.; Lal, H. Cardiomyocyte SMAD4-Dependent TGF-β Signaling is Essential to Maintain Adult Heart Homeostasis. JACC Basic Transl. Sci. 2019, 4, 41–53. [Google Scholar] [CrossRef]
- Yin, L.; Liu, M.-X.; Li, W.; Wang, F.-Y.; Tang, Y.-H.; Huang, C.-X. Over-Expression of Inhibitor of Differentiation 2 Attenuates Post-Infarct Cardiac Fibrosis Through Inhibition of TGF-β1/Smad3/HIF-1α/IL-11 Signaling Pathway. Front. Pharmacol. 2019, 10, 1349. [Google Scholar] [CrossRef]
- Becirovic-Agic, M.; Chalise, U.; Daseke, M.J., 2nd; Konfrst, S.; Salomon, J.D.; Mishra, P.K.; Lindsey, M.L. Infarct in the Heart: What’s MMP-9 Got to Do with It? Biomolecules 2021, 11, 491. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y. MT1-MMP: A key regulator of cell migration in tissue. IUBMB Life 2006, 58, 589–596. [Google Scholar] [CrossRef]
- Parks, W.C.; Wilson, C.L.; López-Boado, Y.S. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat. Rev. Immunol. 2004, 4, 617–629. [Google Scholar] [CrossRef]
- DeLeon-Pennell, K.Y.; Meschiari, C.A.; Jung, M.; Lindsey, M.L. Matrix Metalloproteinases in Myocardial Infarction and Heart Failure. Prog. Mol. Biol. Transl. Sci. 2017, 147, 75–100. [Google Scholar]
- Wang, M.; Zhao, D.; Spinetti, G.; Zhang, J.; Jiang, L.Q.; Pintus, G.; Monticone, R.; Lakatta, E.G. Matrix Metalloproteinase 2 Activation of Transforming Growth Factor-β1 (TGF-β1) and TGF-β1–Type II Receptor Signaling Within the Aged Arterial Wall. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1503–1509. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Stamenkovic, I. Localization of matrix metalloproteinase 9 to the cell surface provides a mechanism for CD44-mediated tumor invasion. Genes Dev. 1999, 13, 35–48. [Google Scholar] [CrossRef]
- Tatti, O.; Vehviläinen, P.; Lehti, K.; Keski-Oja, J. MT1-MMP releases latent TGF-β1 from endothelial cell extracellular matrix via proteolytic processing of LTBP-1. Exp. Cell. Res. 2008, 314, 2501–2514. [Google Scholar] [CrossRef]
- Karsdal, M.A.; Larsen, L.; Engsig, M.T.; Lou, H.; Ferreras, M.; Lochter, A.; Delaissé, J.M.; Foged, N.T. Matrix Metalloproteinase-dependent Activation of Latent Transforming Growth Factor-β Controls the Conversion of Osteoblasts into Osteocytes by Blocking Osteoblast Apoptosis. J. Biol. Chem. 2002, 277, 44061–44067. [Google Scholar] [CrossRef]
- Iyer, R.P.; de Castro Brás, L.E.; Patterson, N.L.; Bhowmick, M.; Flynn, E.R.; Asher, M.; Cannon, P.L.; Deleon-Pennell, K.Y.; Fields, G.B.; Lindsey, M.L. Early matrix metalloproteinase-9 inhibition post-myocardial infarction worsens cardiac dysfunction by delaying inflammation resolution. J. Mol. Cell. Cardiol. 2016, 100, 109–117. [Google Scholar] [CrossRef]
- Matsumura, S.; Iwanaga, S.; Mochizuki, S.; Okamoto, H.; Ogawa, S.; Okada, Y. Targeted deletion or pharmacological inhibition of MMP-2 prevents cardiac rupture after myocardial infarction in mice. J. Clin. Investig. 2005, 115, 599–609. [Google Scholar] [CrossRef]
- Chan, B.Y.H.; Roczkowsky, A.; Cho, W.J.; Poirier, M.; Sergi, C.; Keschrumrus, V.; Churko, J.M.; Granzier, H.; Schulz, R. MMP inhibitors attenuate doxorubicin cardiotoxicity by preventing intracellular and extracellular matrix remodelling. Cardiovasc. Res. 2021, 117, 188–200. [Google Scholar] [CrossRef]
- Zile, M.R.; Baicu, C.F.; Stroud, R.E.; Van Laer, A.O.; Jones, J.A.; Patel, R.; Mukherjee, R.; Spinale, F.G. Mechanistic relationship between membrane type-1 matrix metalloproteinase and the myocardial response to pressure overload. Circ. Heart Fail. 2014, 7, 340–350. [Google Scholar] [CrossRef]
- Ducharme, A.; Frantz, S.; Aikawa, M.; Rabkin, E.; Lindsey, M.; Rohde, L.E.; Schoen, F.J.; Kelly, R.A.; Werb, Z.; Libby, P.; et al. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J. Clin. Investig. 2000, 106, 55–62. [Google Scholar] [CrossRef]
- Yarbrough, W.M.; Mukherjee, R.; Escobar, G.P.; Mingoia, J.T.; Sample, J.A.; Hendrick, J.W.; Dowdy, K.B.; McLean, J.E.; Lowry, A.S.; O’Neill, T.P.; et al. Selective Targeting and Timing of Matrix Metalloproteinase Inhibition in Post-Myocardial Infarction Remodeling. Circulation 2003, 108, 1753–1759. [Google Scholar] [CrossRef]
- Hudson, M.P.; Armstrong, P.W.; Ruzyllo, W.; Brum, J.; Cusmano, L.; Krzeski, P.; Lyon, R.; Quinones, M.; Theroux, P.; Sydlowski, D. Effects of Selective Matrix Metalloproteinase Inhibitor (PG-116800) to Prevent Ventricular Remodeling After Myocardial Infarction: Results of the PREMIER (Prevention of Myocardial Infarction Early Remodeling) Trial. J. Am. Coll. Cardiol. 2006, 48, 15–20. [Google Scholar] [CrossRef]
- Fields, G.B. The Rebirth of Matrix Metalloproteinase Inhibitors: Moving Beyond the Dogma. Cells 2019, 8, 984. [Google Scholar] [CrossRef]
- Pang, X.; He, X.; Qiu, Z.; Zhang, H.; Xie, R.; Liu, Z.; Gu, Y.; Zhao, N.; Xiang, Q.; Cui, Y. Targeting integrin pathways: Mechanisms and advances in therapy. Signal Transduct Target. Ther. 2023, 8, 1. [Google Scholar] [CrossRef]
- Reed, N.I.; Jo, H.; Chen, C.; Tsujino, K.; Arnold, T.D.; DeGrado, W.F.; Sheppard, D. The αvβ1 integrin plays a critical in vivo role in tissue fibrosis. Sci. Transl. Med. 2015, 7, 288ra79. [Google Scholar] [CrossRef] [PubMed]
- Margadant, C.; Sonnenberg, A. Integrin-TGF-beta crosstalk in fibrosis, cancer and wound healing. EMBO Rep. 2010, 11, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Mu, D.; Cambier, S.; Fjellbirkeland, L.; Baron, J.L.; Munger, J.S.; Kawakatsu, H.; Sheppard, D.; Broaddus, V.C.; Nishimura, S.L. The integrin alpha(v)beta8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-beta1. J. Cell. Biol. 2002, 157, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Slack, R.J.; Macdonald, S.J.F.; Roper, J.A.; Jenkins, R.G.; Hatley, R.J.D. Emerging therapeutic opportunities for integrin inhibitors. Nat. Rev. Drug Discov. 2022, 21, 60–78. [Google Scholar] [CrossRef]
- Ong, C.H.; Tham, C.L.; Harith, H.H.; Firdaus, N.; Israf, D.A. TGF-β-induced fibrosis: A review on the underlying mechanism and potential therapeutic strategies. Eur. J. Pharmacol. 2021, 911, 174510. [Google Scholar] [CrossRef]
- Delacroix, C.; Achab-Ali, A.; Alayrac, P.; Gandon-Renard, M.; Dramé, F.; Sassoon, D.; Silvestre, J.S.; Hulot, J.S. Inhibition of itegrin alpha V (CD51) reduces inflammation and transition to heart failure following pressure overload. bioRxiv 2022, 2022, 2022-10. [Google Scholar]
- Bouvet, M.; Claude, O.; Roux, M.; Skelly, D.; Masurkar, N.; Mougenot, N.; Nadaud, S.; Blanc, C.; Delacroix, C.; Chardonnet, S.; et al. Anti-integrin α(v) therapy improves cardiac fibrosis after myocardial infarction by blunting cardiac PW1(+) stromal cells. Sci. Rep. 2020, 10, 11404. [Google Scholar] [CrossRef]
- Cilek, M.Z.; de Vega, S.; Shiozawa, J.; Yoshinaga, C.; Miyamae, Y.; Chijiiwa, M.; Mochizuki, S.; Ito, M.; Kaneko, H.; Kaneko, K.; et al. Synergistic upregulation of ADAMTS4 (aggrecanase-1) by cytokines and its suppression in knee osteoarthritic synovial fibroblasts. Lab. Investig. 2022, 102, 102–111. [Google Scholar] [CrossRef]
- Boyd, D.F.; Allen, E.K.; Randolph, A.G.; Guo, X.J.; Weng, Y.; Sanders, C.J.; Bajracharya, R.; Lee, N.K.; Guy, C.S.; Vogel, P.; et al. Exuberant fibroblast activity compromises lung function via ADAMTS4. Nature 2020, 587, 466–471. [Google Scholar] [CrossRef]
- Vojtusek, I.K.; Laganovic, M.; Burek Kamenaric, M.; Bulimbasic, S.; Hrkac, S.; Salai, G.; Ivkovic, V.; Coric, M.; Novak, R.; Grgurevic, L. First Characterization of ADAMTS-4 in Kidney Tissue and Plasma of Patients with Chronic Kidney Disease-A Potential Novel Diagnostic Indicator. Diagnostics 2022, 12, 648. [Google Scholar] [CrossRef]
- Al-Zahrani, A.A.; Gajewski, J.B. Long-term efficacy and tolerability of pentosan polysulphate sodium in the treatment of bladder pain syndrome. Can. Urol. Assoc. J. 2011, 5, 113. [Google Scholar] [CrossRef]
- Murphy-Ullrich, J.E.; Suto, M.J. Thrombospondin-1 regulation of latent TGF-β activation: A therapeutic target for fibrotic disease. Matrix Biol. 2018, 68–69, 28–43. [Google Scholar] [CrossRef] [PubMed]
- Belmadani, S.; Bernal, J.; Wei, C.-C.; Pallero, M.A.; Dell’italia, L.; Murphy-Ullrich, J.E.; Berecek, K.H. A thrombospondin-1 antagonist of transforming growth factor-beta activation blocks cardiomyopathy in rats with diabetes and elevated angiotensin II. Am. J. Pathol. 2007, 171, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Ruwanpura, S.M.; Thomas, B.J.; Bardin, P.G. Pirfenidone: Molecular Mechanisms and Potential Clinical Applications in Lung Disease. Am. J. Respir. Cell Mol. Biol. 2020, 62, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.T.; Ding, C.; Wilson, E.; Marcus, G.M.; Olgin, J.E. Pirfenidone mitigates left ventricular fibrosis and dysfunction after myocardial infarction and reduces arrhythmias. Heart Rhythm. 2010, 7, 1438–1445. [Google Scholar] [CrossRef]
- Lewis, G.A.; Dodd, S.; Clayton, D.; Bedson, E.; Eccleson, H.; Schelbert, E.B.; Naish, J.H.; Jimenez, B.D.; Williams, S.G.; Cunnington, C.; et al. Pirfenidone in heart failure with preserved ejection fraction: A randomized phase 2 trial. Nat. Med. 2021, 27, 1477–1482. [Google Scholar] [CrossRef]
- Jiang, C.; Huang, H.; Liu, J.; Wang, Y.; Lu, Z.; Xu, Z. Adverse events of pirfenidone for the treatment of pulmonary fibrosis: A meta-analysis of randomized controlled trials. PLoS ONE 2012, 7, e47024. [Google Scholar] [CrossRef]
- Darakhshan, S.; Pour, A.B. Tranilast: A review of its therapeutic applications. Pharmacol. Res. 2015, 91, 15–28. [Google Scholar] [CrossRef]
- Martin, J.; Kelly, D.J.; Mifsud, S.A.; Zhang, Y.; Cox, A.J.; See, F.; Krum, H.; Wilkinson-Berka, J.; Gilbert, R.E. Tranilast attenuates cardiac matrix deposition in experimental diabetes: Role of transforming growth factor-β. Cardiovasc. Res. 2005, 65, 694–701. [Google Scholar] [CrossRef]
- Zhu, J.; Ling, W.; Xue, C.; Zhou, Z.; Zhang, Y.; Yan, C.; Wu, M. Higenamine attenuates cardiac fibroblast abstract and fibrosis via inhibition of TGF-β1/Smad signaling. Eur. J. Pharmacol. 2021, 900, 174013. [Google Scholar] [CrossRef]
- Chen, G.; Xu, H.; Xu, T.; Ding, W.; Zhang, G.; Hua, Y.; Wu, Y.; Han, X.; Xie, L.; Liu, B.; et al. Calycosin reduces myocardial fibrosis and improves cardiac function in post-myocardial infarction mice by suppressing TGFBR1 signaling pathways. Phytomedicine 2022, 104, 154277. [Google Scholar] [CrossRef]
- Gao, H.; Bo, Z.; Wang, Q.; Luo, L.; Zhu, H.; Ren, Y. Salvanic acid B inhibits myocardial fibrosis through regulating TGF-β1/Smad signaling pathway. Biomed. Pharmacother. 2019, 110, 685–691. [Google Scholar] [CrossRef]
- Zeng, Z.; Wang, Q.; Yang, X.; Ren, Y.; Jiao, S.; Zhu, Q.; Guo, D.; Xia, K.; Wang, Y.; Li, C.; et al. Qishen granule attenuates cardiac fibrosis by regulating TGF-β /Smad3 and GSK-3β pathway. Phytomedicine 2019, 62, 152949. [Google Scholar] [CrossRef]
- Gao, L.; Wang, L.Y.; Liu, Z.Q.; Jiang, D.; Wu, S.Y.; Guo, Y.Q.; Tao, H.M.; Sun, M.; You, L.N.; Qin, S.; et al. TNAP inhibition attenuates cardiac fibrosis induced by myocardial infarction through deactivating TGF-β1/Smads and activating P53 signaling pathways. Cell Death Dis. 2020, 11, 44. [Google Scholar] [CrossRef]
- Song, S.; Liu, L.; Yu, Y.; Zhang, R.; Li, Y.; Cao, W.; Xiao, Y.; Fang, G.; Li, Z.; Wang, X.; et al. Inhibition of BRD4 attenuates transverse aortic constriction- and TGF-β-induced endothelial-mesenchymal transition and cardiac fibrosis. J. Mol. Cell. Cardiol. 2019, 127, 83–96. [Google Scholar] [CrossRef]
- See, F.; Thomas, W.; Way, K.; Tzanidis, A.; Kompa, A.; Lewis, D.; Itescu, S.; Krum, H. p38 mitogen-activated protein kinase inhibition improves cardiac function and attenuates left ventricular remodeling following myocardial infarction in the rat. J. Am. Coll. Cardiol. 2004, 44, 1679–1689. [Google Scholar] [CrossRef]
- Burke, R.M.; Dirkx, R.A.; Quijada, P.; Lighthouse, J.K.; Mohan, A.; O’Brien, M.; Wojciechowski, W.; Woeller, C.F.; Phipps, R.P.; Alexis, J.D.; et al. Prevention of Fibrosis and Pathological Cardiac Remodeling by Salinomycin. Circ. Res. 2021, 128, 1663–1678. [Google Scholar] [CrossRef]
- Morfino, P.; Aimo, A.; Castiglione, V.; Gálvez-Montón, C.; Emdin, M.; Bayes-Genis, A. Treatment of cardiac fibrosis: From neuro-hormonal inhibitors to CAR-T cell therapy. Heart Fail. Rev. 2023, 28, 555–569. [Google Scholar] [CrossRef]
- Murphy, A.M.; Wong, A.L.; Bezuhly, M. Modulation of angiotensin II signaling in the prevention of fibrosis. Fibrogenesis Tissue Repair 2015, 8, 7. [Google Scholar] [CrossRef]
- AlQudah, M.; Hale, T.M.; Czubryt, M.P. Targeting the renin-angiotensin-aldosterone system in fibrosis. Matrix Biol 2020, 91–92, 92–108. [Google Scholar] [CrossRef]
- Seeland, U.; Schäffer, A.; Selejan, S.; Hohl, M.; Reil, J.C.; Müller, P.; Rosenkranz, S.; Böhm, M. Effects of AT1- and beta-adrenergic receptor antagonists on TGF-beta1-induced fibrosis in transgenic mice. Eur. J. Clin. Investig. 2009, 39, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Shimada, Y.J.; Passeri, J.J.; Baggish, A.L.; O’Callaghan, C.; Lowry, P.A.; Yannekis, G.; Abbara, S.; Ghoshhajra, B.B.; Rothman, R.D.; Ho, C.Y.; et al. Effects of losartan on left ventricular hypertrophy and fibrosis in patients with nonobstructive hypertrophic cardiomyopathy. JACC Heart Fail. 2013, 1, 480–487. [Google Scholar] [CrossRef]
- Diez, J.; Querejeta, R.; Lopez, B.; Gonzalez, A.; Larman, M.; Martinez Ubago, J.L. Losartan-dependent regression of myocardial fibrosis is associated with reduction of left ventricular chamber stiffness in hypertensive patients. Circulation 2002, 105, 2512–2517. [Google Scholar] [CrossRef]
- Brilla, C.G.; Funck, R.C.; Rupp, H. Lisinopril-mediated regression of myocardial fibrosis in patients with hypertensive heart disease. Circulation 2000, 102, 1388–1393. [Google Scholar] [CrossRef] [PubMed]
- Blanter, J.B.; Frishman, W.H. The Preventive Role of Angiotensin Converting Enzyme Inhibitors/Angiotensin-II Receptor Blockers and β-Adrenergic Blockers in Anthracycline- and Trastuzumab-Induced Cardiotoxicity. Cardiol. Rev. 2019, 27, 256–259. [Google Scholar] [CrossRef] [PubMed]
- El-Said, N.T.; Mohamed, E.A.; Taha, R.A. Irbesartan suppresses cardiac toxicity induced by doxorubicin via regulating the p38-MAPK/NF-κB and TGF-β1 pathways. Naunyn-Schmiedeberg Arch. Pharmacol. 2019, 392, 647–658. [Google Scholar] [CrossRef]
- Lavall, D.; Jacobs, N.; Mahfoud, F.; Kolkhof, P.; Böhm, M.; Laufs, U. The non-steroidal mineralocorticoid receptor antagonist finerenone prevents cardiac fibrotic remodeling. Biochem. Pharmacol. 2019, 168, 173–183. [Google Scholar] [CrossRef]
- Franco, V.; Chen, Y.-F.; Feng, J.A.; Li, P.; Wang, D.; Hasan, E.; Oparil, S.; Perry, G.J. Eplerenone prevents adverse cardiac remodelling induced by pressure overload in atrial natriuretic peptide-null mice. Clin. Exp. Pharmacol. Physiol. 2006, 33, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Izawa, H.; Murohara, T.; Nagata, K.; Isobe, S.; Asano, H.; Amano, T.; Ichihara, S.; Kato, T.; Ohshima, S.; Murase, Y.; et al. Mineralocorticoid Receptor Antagonism Ameliorates Left Ventricular Diastolic Dysfunction and Myocardial Fibrosis in Mildly Symptomatic Patients With Idiopathic Dilated Cardiomyopathy. Circulation 2005, 112, 2940–2945. [Google Scholar] [CrossRef]
- McDiarmid, A.K.; Swoboda, P.P.; Erhayiem, B.; Bounford, K.A.; Bijsterveld, P.; Tyndall, K.; Fent, G.J.; Garg, P.; Dobson, L.E.; Musa, T.A.; et al. Myocardial Effects of Aldosterone Antagonism in Heart Failure With Preserved Ejection Fraction. J. Am. Heart Assoc. 2020, 9, e011521. [Google Scholar] [CrossRef]
- Cleland, J.G.; Tendera, M.; Adamus, J.; Freemantle, N.; Polonski, L.; Taylor, J. The perindopril in elderly people with chronic heart failure (PEP-CHF) study. Eur. Heart J. 2006, 27, 2338–2345. [Google Scholar] [CrossRef]
- Yusuf, S.; Pfeffer, M.A.; Swedberg, K.; Granger, C.B.; Held, P.; McMurray, J.J.; Michelson, E.L.; Olofsson, B.; Ostergren, J. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: The CHARM-Preserved Trial. Lancet 2003, 362, 777–781. [Google Scholar] [CrossRef]
- Pitt, B.; Pfeffer, M.A.; Assmann, S.F.; Boineau, R.; Anand, I.S.; Claggett, B.; Clausell, N.; Desai, A.S.; Diaz, R.; Fleg, J.L.; et al. Spironolactone for Heart Failure with Preserved Ejection Fraction. N. Engl. J. Med. 2014, 370, 1383–1392. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lin, X.; Chu, Y.; Boineau, R.; Anand, I.S.; Claggett, B.; Clausell, N.; Desai, A.S.; Diaz, R.; Fleg, J.L.; et al. Dapagliflozin: A sodium-glucose cotransporter 2 inhibitor, attenuates angiotensin II-induced cardiac fibrotic remodeling by regulating TGFβ1/Smad signaling. Cardiovasc. Diabetol. 2021, 20, 121. [Google Scholar] [CrossRef]
- Li, C.; Zhang, J.; Xue, M.; Li, X.; Han, F.; Liu, X.; Xu, L.; Lu, Y.; Cheng, Y.; Li, T.; et al. SGLT2 inhibition with empagliflozin attenuates myocardial oxidative stress and fibrosis in diabetic mice heart. Cardiovasc. Diabetol. 2019, 18, 15. [Google Scholar] [CrossRef]
- Palmiero, G.; Cesaro, A.; Vetrano, E.; Pafundi, P.C.; Galiero, R.; Caturano, A.; Moscarella, E.; Gragnano, F.; Salvatore, T.; Rinaldi, L.; et al. Impact of SGLT2 Inhibitors on Heart Failure: From Pathophysiology to Clinical Effects. Int. J. Mol. Sci. 2021, 22, 5863. [Google Scholar] [CrossRef] [PubMed]
- Shakour, N.; Karami, S.; Iranshahi, M.; Butler, A.E.; Sahebkar, A. Antifibrotic effects of sodium-glucose cotransporter-2 inhibitors: A comprehensive review. Diabetes Metab. Syndr. Clin. Res. Rev. 2024, 18, 102934. [Google Scholar] [CrossRef] [PubMed]
- Pfau, D.; Thorn, S.L.; Zhang, J.; Mikush, N.; Renaud, J.M.; Klein, R.; deKemp, R.A.; Wu, X.; Hu, X.; Sinusas, A.J.; et al. Angiotensin Receptor Neprilysin Inhibitor Attenuates Myocardial Remodeling and Improves Infarct Perfusion in Experimental Heart Failure. Sci. Rep. 2019, 9, 5791. [Google Scholar] [CrossRef] [PubMed]
- Burke, R.M.; Lighthouse, J.K.; Mickelsen, D.M.; Small, E.M. Sacubitril/Valsartan Decreases Cardiac Fibrosis in Left Ventricle Pressure Overload by Restoring PKG Signaling in Cardiac Fibroblasts. Circ. Heart Fail. 2019, 12, e005565. [Google Scholar] [CrossRef]
- Nordén, E.S.; Bendiksen, B.A.; Andresen, H.; Bergo, K.K.; Espe, E.K.; Hasic, A.; Hauge-Iversen, I.M.; Veras, I.; Hussain, R.I.; Sjaastad, I.; et al. Sacubitril/valsartan ameliorates cardiac hypertrophy and preserves diastolic function in cardiac pressure overload. ESC Heart Fail. 2021, 8, 918–927. [Google Scholar] [CrossRef]
- Arnold, S.V.; Silverman, D.N.; Gosch, K.; Nassif, M.E.; Infeld, M.; Litwin, S.; Meyer, M.; Fendler, T.J. Beta-Blocker Use and Heart Failure Outcomes in Mildly Reduced and Preserved Ejection Fraction. JACC Heart Fail. 2023, 11, 893–900. [Google Scholar] [CrossRef]
- López, B.; González, A.; Beaumont, J.; Querejeta, R.; Larman, M.; Díez, J. Identification of a Potential Cardiac Antifibrotic Mechanism of Torasemide in Patients With Chronic Heart Failure. J. Am. Coll. Cardiol. 2007, 50, 859–867. [Google Scholar] [CrossRef]
- Palazzuoli, A.; Caravita, S.; Paolillo, S.; Ghio, S.; Tocchetti, C.G.; Ruocco, G.; Correale, M.; Ambrosio, G.; Perrone Filardi, P.; Senni, M. Current gaps in HFpEF trials: Time to reconsider patients’ selection and to target phenotypes. Prog. Cardiovasc. Dis. 2021, 67, 89–97. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vistnes, M. Hitting the Target! Challenges and Opportunities for TGF-β Inhibition for the Treatment of Cardiac fibrosis. Pharmaceuticals 2024, 17, 267. https://doi.org/10.3390/ph17030267
Vistnes M. Hitting the Target! Challenges and Opportunities for TGF-β Inhibition for the Treatment of Cardiac fibrosis. Pharmaceuticals. 2024; 17(3):267. https://doi.org/10.3390/ph17030267
Chicago/Turabian StyleVistnes, Maria. 2024. "Hitting the Target! Challenges and Opportunities for TGF-β Inhibition for the Treatment of Cardiac fibrosis" Pharmaceuticals 17, no. 3: 267. https://doi.org/10.3390/ph17030267
APA StyleVistnes, M. (2024). Hitting the Target! Challenges and Opportunities for TGF-β Inhibition for the Treatment of Cardiac fibrosis. Pharmaceuticals, 17(3), 267. https://doi.org/10.3390/ph17030267