
Pharmacokinetics, Pharmacodynamics, and Side Effects of Midazolam: A Review and Case Example


{kind=link}
Abstract
:1. Introduction
2. TDM of Midazolam
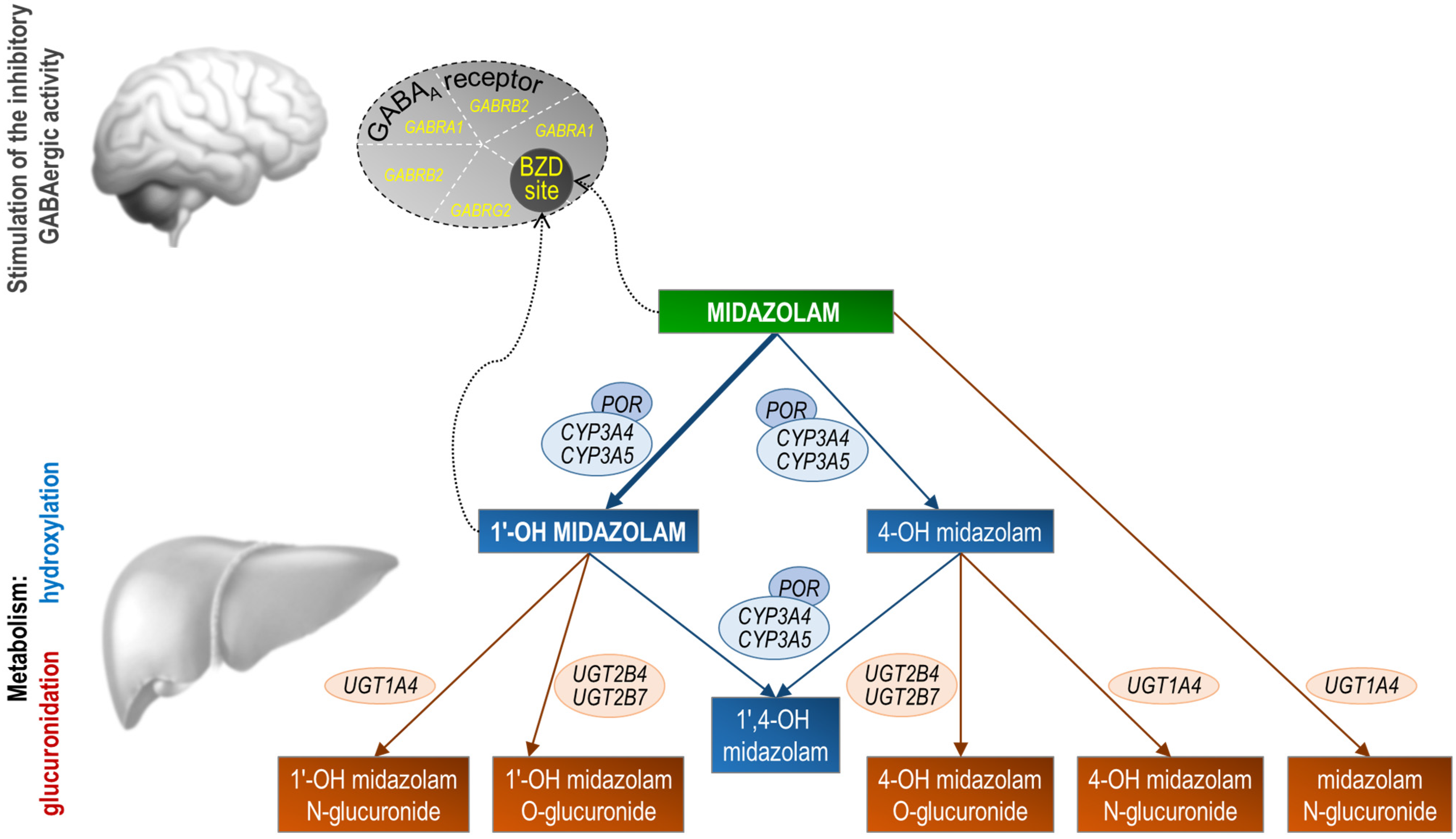
3. Transport and Metabolism of Midazolam
4. Pharmacokinetics and Pharmacodynamics of Midazolam Metabolites
5. Effect of Sex and Nutritional Status on the Pharmacokinetics of Midazolam
6. Impact of Inflammation on the Pharmacokinetics of Midazolam
7. Genetic Factors—CYP3A Polymorphisms
8. Transcriptional Regulation of CYP3A Genes
9. Regulation of CYP3A4 by Short Non-Coding Regulatory MicroRNAs
10. Genetic Factors—UGT1A Polymorphisms
11. Genetic Factors—Polymorphisms in GABAA Receptor Genes
12. Epigenetics
13. Case Example
14. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Altamimi, M.I.; Sammons, H.; Choonara, I. Inter-individual variation in midazolam clearance in children. Arch. Dis. Child. 2015, 100, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Lamba, J.K.; Lin, Y.S.; Schuetz, E.G.; Thummel, K.E. Genetic contribution to variable human CYP3A-mediated metabolism. Adv. Drug Deliv. Rev. 2002, 54, 1271–1294. [Google Scholar] [CrossRef]
- Backman, J.T.; Kivisto, K.T.; Olkkola, K.T.; Neuvonen, P.J. The area under the plasma concentration-time curve for oral midazolam is 400-fold larger during treatment with itraconazole than with rifampicin. Eur. J. Clin. Pharmacol. 1998, 54, 53–58. [Google Scholar] [CrossRef]
- Ozdemir, V.; Kalow, W.; Tang, B.K.; Paterson, A.D.; Walker, S.E.; Endrenyi, L.; Kashuba, A.D. Evaluation of the genetic component of variability in CYP3A4 activity: A repeated drug administration method. Pharmacogenetics 2000, 10, 373–388. [Google Scholar] [CrossRef] [PubMed]
- Matthaei, J.; Bonat, W.H.; Kerb, R.; Tzvetkov, M.V.; Strube, J.; Brunke, S.; Sachse-Seeboth, C.; Sehrt, D.; Hofmann, U.; von Bornemann Hjelmborg, J.; et al. Inherited and Acquired Determinants of Hepatic CYP3A Activity in Humans. Front. Genet. 2020, 11, 944. [Google Scholar] [CrossRef]
- Miao, J.; Jin, Y.; Marunde, R.L.; Gorski, C.J.; Kim, S.; Quinney, S.; Radovich, M.; Li, L.; Hall, S.D. Association of genotypes of the CYP3A cluster with midazolam disposition in vivo. Pharmacogenom. J. 2009, 9, 319–326. [Google Scholar] [CrossRef]
- He, P.; Court, M.H.; Greenblatt, D.J.; Von Moltke, L.L. Genotype-phenotype associations of cytochrome P450 3A4 and 3A5 polymorphism with midazolam clearance in vivo. Clin. Pharmacol. Ther. 2005, 77, 373–387. [Google Scholar] [CrossRef] [PubMed]
- Floyd, M.D.; Gervasini, G.; Masica, A.L.; Mayo, G.; George, A.L., Jr.; Bhat, K.; Kim, R.B.; Wilkinson, G.R. Genotype-phenotype associations for common CYP3A4 and CYP3A5 variants in the basal and induced metabolism of midazolam in European- and African-American men and women. Pharmacogenetics 2003, 13, 595–606. [Google Scholar] [CrossRef]
- Nies, R.J.; Muller, C.; Pfister, R.; Binder, P.S.; Nosseir, N.; Nettersheim, F.S.; Kuhr, K.; Wiesen, M.H.J.; Kochanek, M.; Michels, G. Monitoring of sedation depth in intensive care unit by therapeutic drug monitoring? A prospective observation study of medical intensive care patients. J. Intensive Care 2018, 6, 62. [Google Scholar] [CrossRef]
- Bremer, F.; Reulbach, U.; Schwilden, H.; Schuttler, J. Midazolam therapeutic drug monitoring in intensive care sedation: A 5-year survey. Ther. Drug Monit. 2004, 26, 643–649. [Google Scholar] [CrossRef]
- de Wildt, S.N.; de Hoog, M.; Vinks, A.A.; Joosten, K.F.; van Dijk, M.; van den Anker, J.N. Pharmacodynamics of midazolam in pediatric intensive care patients. Ther. Drug Monit. 2005, 27, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Oldenhof, H.; de Jong, M.; Steenhoek, A.; Janknegt, R. Clinical pharmacokinetics of midazolam in intensive care patients, a wide interpatient variability? Clin. Pharmacol. Ther. 1988, 43, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Takano, M.; Hasegawa, R.; Fukuda, T.; Yumoto, R.; Nagai, J.; Murakami, T. Interaction with P-glycoprotein and transport of erythromycin, midazolam and ketoconazole in Caco-2 cells. Eur. J. Pharmacol. 1998, 358, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Tolle-Sander, S.; Rautio, J.; Wring, S.; Polli, J.W.; Polli, J.E. Midazolam exhibits characteristics of a highly permeable P-glycoprotein substrate. Pharm. Res. 2003, 20, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Center for Drug Evaluation and Research. In Vitro Drug Interaction Studies—Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions Guidance for Industry. Available online: https://www.fda.gov/media/134582/download (accessed on 17 March 2024).
- Franke, R.M.; Baker, S.D.; Mathijssen, R.H.; Schuetz, E.G.; Sparreboom, A. Influence of solute carriers on the pharmacokinetics of CYP3A4 probes. Clin. Pharmacol. Ther. 2008, 84, 704–709. [Google Scholar] [CrossRef] [PubMed]
- Imanaga, J.; Kotegawa, T.; Imai, H.; Tsutsumi, K.; Yoshizato, T.; Ohyama, T.; Shirasaka, Y.; Tamai, I.; Tateishi, T.; Ohashi, K. The effects of the SLCO2B1 c.1457C > T polymorphism and apple juice on the pharmacokinetics of fexofenadine and midazolam in humans. Pharmacogenet. Genom. 2011, 21, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Jarvinen, E.; Deng, F.; Kiander, W.; Sinokki, A.; Kidron, H.; Sjostedt, N. The Role of Uptake and Efflux Transporters in the Disposition of Glucuronide and Sulfate Conjugates. Front. Pharmacol. 2021, 12, 802539. [Google Scholar] [CrossRef] [PubMed]
- Allonen, H.; Ziegler, G.; Klotz, U. Midazolam kinetics. Clin. Pharmacol. Ther. 1981, 30, 653–661. [Google Scholar] [CrossRef]
- Pentikainen, P.J.; Valisalmi, L.; Himberg, J.J.; Crevoisier, C. Pharmacokinetics of midazolam following intravenous and oral administration in patients with chronic liver disease and in healthy subjects. J. Clin. Pharmacol. 1989, 29, 272–277. [Google Scholar] [CrossRef]
- Lee, S.; Lee, Y.; Kim, A.H.; Yoon, S.; Lee, J.; Ji, S.C.; Yoon, S.H.; Lee, S.; Yu, K.S.; Jang, I.J.; et al. Urinary metabolic markers reflect on hepatic, not intestinal, CYP3A activity in healthy subjects. Drug Metab. Pharmacokinet. 2021, 36, 100374. [Google Scholar] [CrossRef]
- Stoch, S.A.; Friedman, E.; Maes, A.; Yee, K.; Xu, Y.; Larson, P.; Fitzgerald, M.; Chodakewitz, J.; Wagner, J.A. Effect of different durations of ketoconazole dosing on the single-dose pharmacokinetics of midazolam: Shortening the paradigm. J. Clin. Pharmacol. 2009, 49, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Tseng, E.; Walsky, R.L.; Luzietti, R.A., Jr.; Harris, J.J.; Kosa, R.E.; Goosen, T.C.; Zientek, M.A.; Obach, R.S. Relative contributions of cytochrome CYP3A4 versus CYP3A5 for CYP3A-cleared drugs assessed in vitro using a CYP3A4-selective inactivator (CYP3cide). Drug Metab. Dispos. 2014, 42, 1163–1173. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.S.; Cho, J.Y.; Jang, I.J.; Hong, K.S.; Chung, J.Y.; Kim, J.R.; Lim, H.S.; Oh, D.S.; Yi, S.Y.; Liu, K.H.; et al. Effect of the CYP3A5 genotype on the pharmacokinetics of intravenous midazolam during inhibited and induced metabolic states. Clin. Pharmacol. Ther. 2004, 76, 104–112. [Google Scholar] [CrossRef] [PubMed]
- von Moltke, L.L.; Greenblatt, D.J.; Schmider, J.; Duan, S.X.; Wright, C.E.; Harmatz, J.S.; Shader, R.I. Midazolam hydroxylation by human liver microsomes in vitro: Inhibition by fluoxetine, norfluoxetine, and by azole antifungal agents. J. Clin. Pharmacol. 1996, 36, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Seo, K.A.; Bae, S.K.; Choi, Y.K.; Choi, C.S.; Liu, K.H.; Shin, J.G. Metabolism of 1′- and 4-hydroxymidazolam by glucuronide conjugation is largely mediated by UDP-glucuronosyltransferases 1A4, 2B4, and 2B7. Drug Metab. Dispos. 2010, 38, 2007–2013. [Google Scholar] [CrossRef] [PubMed]
- Klieber, S.; Hugla, S.; Ngo, R.; Arabeyre-Fabre, C.; Meunier, V.; Sadoun, F.; Fedeli, O.; Rival, M.; Bourrie, M.; Guillou, F.; et al. Contribution of the N-glucuronidation pathway to the overall in vitro metabolic clearance of midazolam in humans. Drug Metab. Dispos. 2008, 36, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Hyland, R.; Osborne, T.; Payne, A.; Kempshall, S.; Logan, Y.R.; Ezzeddine, K.; Jones, B. In vitro and in vivo glucuronidation of midazolam in humans. Br. J. Clin. Pharmacol. 2009, 67, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, W.H.; Schalch, E.; Leishman, B.; Eckert, M. Comparison of the effects of intravenously administered midazolam, triazolam and their hydroxy metabolites. Br. J. Clin. Pharmacol. 1983, 16 (Suppl. S1), 63S–69S. [Google Scholar] [CrossRef] [PubMed]
- Mandema, J.W.; Tuk, B.; van Steveninck, A.L.; Breimer, D.D.; Cohen, A.F.; Danhof, M. Pharmacokinetic-pharmacodynamic modeling of the central nervous system effects of midazolam and its main metabolite alpha-hydroxymidazolam in healthy volunteers. Clin. Pharmacol. Ther. 1992, 51, 715–728. [Google Scholar] [CrossRef]
- Bauer, T.M.; Ritz, R.; Haberthur, C.; Ha, H.R.; Hunkeler, W.; Sleight, A.J.; Scollo-Lavizzari, G.; Haefeli, W.E. Prolonged sedation due to accumulation of conjugated metabolites of midazolam. Lancet 1995, 346, 145–147. [Google Scholar] [CrossRef]
- Stanski, D.; Hudson, R. Midazolam pharmacology and pharmacokinetics. Anesth. Rev. 1985, 12, 21–23. [Google Scholar]
- Kanto, J.H. Midazolam: The first water-soluble benzodiazepine. Pharmacology, pharmacokinetics and efficacy in insomnia and anesthesia. Pharmacotherapy 1985, 5, 138–155. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.N.; Rostami-Hodjegan, A.; Goddard, J.M.; Tanner, M.S.; Tucker, G.T. Contribution of midazolam and its 1-hydroxy metabolite to preoperative sedation in children: A pharmacokinetic-pharmacodynamic analysis. Br. J. Anaesth. 2002, 89, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Franken, L.G.; Masman, A.D.; de Winter, B.C.M.; Baar, F.P.M.; Tibboel, D.; van Gelder, T.; Koch, B.C.P.; Mathot, R.A.A. Hypoalbuminaemia and decreased midazolam clearance in terminally ill adult patients, an inflammatory effect? Br. J. Clin. Pharmacol. 2017, 83, 1701–1712. [Google Scholar] [CrossRef] [PubMed]
- Beierle, I.; Meibohm, B.; Derendorf, H. Gender differences in pharmacokinetics and pharmacodynamics. Int. J. Clin. Pharmacol. Ther. 1999, 37, 529–547. [Google Scholar] [PubMed]
- Gandhi, M.; Aweeka, F.; Greenblatt, R.M.; Blaschke, T.F. Sex differences in pharmacokinetics and pharmacodynamics. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 499–523. [Google Scholar] [CrossRef] [PubMed]
- Lamba, V.; Panetta, J.C.; Strom, S.; Schuetz, E.G. Genetic predictors of interindividual variability in hepatic CYP3A4 expression. J. Pharmacol. Exp. Ther. 2010, 332, 1088–1099. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.; Baumann, F.; Hanschmann, H.; Geissler, F.; Preiss, R. Gender difference in ifosfamide metabolism by human liver microsomes. Eur. J. Drug Metab. Pharmacokinet. 2001, 26, 193–200. [Google Scholar] [CrossRef]
- Wolbold, R.; Klein, K.; Burk, O.; Nussler, A.K.; Neuhaus, P.; Eichelbaum, M.; Schwab, M.; Zanger, U.M. Sex is a major determinant of CYP3A4 expression in human liver. Hepatology 2003, 38, 978–988. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, B.; Molony, C.; Chudin, E.; Hao, K.; Zhu, J.; Gaedigk, A.; Suver, C.; Zhong, H.; Leeder, J.S.; et al. Systematic genetic and genomic analysis of cytochrome P450 enzyme activities in human liver. Genome Res. 2010, 20, 1020–1036. [Google Scholar] [CrossRef]
- Gallagher, C.J.; Balliet, R.M.; Sun, D.; Chen, G.; Lazarus, P. Sex differences in UDP-glucuronosyltransferase 2B17 expression and activity. Drug Metab. Dispos. 2010, 38, 2204–2209. [Google Scholar] [CrossRef] [PubMed]
- Meibohm, B.; Beierle, I.; Derendorf, H. How important are gender differences in pharmacokinetics? Clin. Pharmacokinet. 2002, 41, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Cotreau, M.M.; von Moltke, L.L.; Greenblatt, D.J. The influence of age and sex on the clearance of cytochrome P450 3A substrates. Clin. Pharmacokinet. 2005, 44, 33–60. [Google Scholar] [CrossRef] [PubMed]
- Tsunoda, S.M.; Velez, R.L.; von Moltke, L.L.; Greenblatt, D.J. Differentiation of intestinal and hepatic cytochrome P450 3A activity with use of midazolam as an in vivo probe: Effect of ketoconazole. Clin. Pharmacol. Ther. 1999, 66, 461–471. [Google Scholar] [CrossRef]
- Thangavel, C.; Boopathi, E.; Shapiro, B.H. Intrinsic sexually dimorphic expression of the principal human CYP3A4 correlated with suboptimal activation of GH/glucocorticoid-dependent transcriptional pathways in men. Endocrinology 2011, 152, 4813–4824. [Google Scholar] [CrossRef] [PubMed]
- Dhir, R.N.; Dworakowski, W.; Thangavel, C.; Shapiro, B.H. Sexually dimorphic regulation of hepatic isoforms of human cytochrome p450 by growth hormone. J. Pharmacol. Exp. Ther. 2006, 316, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.Y.; Zhao, Y.S. Sex-dependent differences in cytochrome P450 3A activity as assessed by midazolam disposition in humans: A meta-analysis. Drug Metab. Dispos. 2010, 38, 817–823. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Ma, L.; Drusano, G.L.; Bertino, J.S., Jr.; Nafziger, A.N. Sex differences in CYP3A activity using intravenous and oral midazolam. Clin. Pharmacol. Ther. 2006, 80, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Zarezadeh, M.; Saedisomeolia, A.; Shekarabi, M.; Khorshidi, M.; Emami, M.R.; Muller, D.J. The effect of obesity, macronutrients, fasting and nutritional status on drug-metabolizing cytochrome P450s: A systematic review of current evidence on human studies. Eur. J. Nutr. 2021, 60, 2905–2921. [Google Scholar] [CrossRef]
- Lammers, L.A.; Achterbergh, R.; Romijn, J.A.; Mathot, R.A.A. Nutritional Status Differentially Alters Cytochrome P450 3A4 (CYP3A4) and Uridine 5′-Diphospho-Glucuronosyltransferase (UGT) Mediated Drug Metabolism: Effect of Short-Term Fasting and High Fat Diet on Midazolam Metabolism. Eur. J. Drug Metab. Pharmacokinet. 2018, 43, 751–767. [Google Scholar] [CrossRef]
- de Vries, E.M.; Lammers, L.A.; Achterbergh, R.; Klumpen, H.J.; Mathot, R.A.; Boelen, A.; Romijn, J.A. Fasting-Induced Changes in Hepatic P450 Mediated Drug Metabolism Are Largely Independent of the Constitutive Androstane Receptor CAR. PLoS ONE 2016, 11, e0159552. [Google Scholar] [CrossRef]
- Lammers, L.A.; Achterbergh, R.; de Vries, E.M.; van Nierop, F.S.; Klumpen, H.J.; Soeters, M.R.; Boelen, A.; Romijn, J.A.; Mathot, R.A. Short-term fasting alters cytochrome P450-mediated drug metabolism in humans. Drug Metab. Dispos. 2015, 43, 819–828. [Google Scholar] [CrossRef]
- Lammers, L.A.; Achterbergh, R.; van Schaik, R.H.N.; Romijn, J.A.; Mathot, R.A.A. Effect of Short-Term Fasting on Systemic Cytochrome P450-Mediated Drug Metabolism in Healthy Subjects: A Randomized, Controlled, Crossover Study Using a Cocktail Approach. Clin. Pharmacokinet. 2017, 56, 1231–1244. [Google Scholar] [CrossRef]
- Brill, M.J.; van Rongen, A.; Houwink, A.P.; Burggraaf, J.; van Ramshorst, B.; Wiezer, R.J.; van Dongen, E.P.; Knibbe, C.A. Midazolam pharmacokinetics in morbidly obese patients following semi-simultaneous oral and intravenous administration: A comparison with healthy volunteers. Clin. Pharmacokinet. 2014, 53, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Lenoir, C.; Rodieux, F.; Desmeules, J.A.; Rollason, V.; Samer, C.F. Impact of Inflammation on Cytochromes P450 Activity in Pediatrics: A Systematic Review. Clin. Pharmacokinet. 2021, 60, 1537–1555. [Google Scholar] [CrossRef]
- Aitken, A.E.; Morgan, E.T. Gene-specific effects of inflammatory cytokines on cytochrome P450 2C, 2B6 and 3A4 mRNA levels in human hepatocytes. Drug Metab. Dispos. 2007, 35, 1687–1693. [Google Scholar] [CrossRef]
- Aitken, A.E.; Richardson, T.A.; Morgan, E.T. Regulation of drug-metabolizing enzymes and transporters in inflammation. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 123–149. [Google Scholar] [CrossRef]
- Stanke-Labesque, F.; Gautier-Veyret, E.; Chhun, S.; Guilhaumou, R.; French Society of Pharmacology and Therapeutics. Inflammation is a major regulator of drug metabolizing enzymes and transporters: Consequences for the personalization of drug treatment. Pharmacol. Ther. 2020, 215, 107627. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.E.; Brown, K.C.; Werner, R.E.; Gotzkowsky, K.; Gaedigk, A.; Blake, M.; Hein, D.W.; van der Horst, C.; Kashuba, A.D. Variability in drug metabolizing enzyme activity in HIV-infected patients. Eur. J. Clin. Pharmacol. 2010, 66, 475–485. [Google Scholar] [CrossRef]
- Brussee, J.M.; Vet, N.J.; Krekels, E.H.J.; Valkenburg, A.J.; Jacqz-Aigrain, E.; van Gerven, J.M.A.; Swart, E.L.; van den Anker, J.N.; Tibboel, D.; de Hoog, M.; et al. Predicting CYP3A-mediated midazolam metabolism in critically ill neonates, infants, children and adults with inflammation and organ failure. Br. J. Clin. Pharmacol. 2018, 84, 358–368. [Google Scholar] [CrossRef]
- Le Carpentier, E.C.; Canet, E.; Masson, D.; Martin, M.; Deslandes, G.; Gaultier, A.; Dailly, E.; Bellouard, R.; Gregoire, M. Impact of Inflammation on Midazolam Metabolism in Severe COVID-19 Patients. Clin. Pharmacol. Ther. 2022, 112, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Jover, R.; Bort, R.; Gomez-Lechon, M.J.; Castell, J.V. Down-regulation of human CYP3A4 by the inflammatory signal interleukin-6: Molecular mechanism and transcription factors involved. FASEB J. 2002, 16, 1799–1801. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Hao, C.; Yang, D.; Shi, D.; Song, X.; Luan, X.; Hu, G.; Yan, B. Pregnane X receptor is required for interleukin-6-mediated down-regulation of cytochrome P450 3A4 in human hepatocytes. Toxicol. Lett. 2010, 197, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Ke, S.; Liu, D.; Sheng, T.; Thomas, P.E.; Rabson, A.B.; Gallo, M.A.; Xie, W.; Tian, Y. Role of NF-kappaB in regulation of PXR-mediated gene expression: A mechanism for the suppression of cytochrome P-450 3A4 by proinflammatory agents. J. Biol. Chem. 2006, 281, 17882–17889. [Google Scholar] [CrossRef]
- Keller, R.; Klein, M.; Thomas, M.; Drager, A.; Metzger, U.; Templin, M.F.; Joos, T.O.; Thasler, W.E.; Zell, A.; Zanger, U.M. Coordinating Role of RXRalpha in Downregulating Hepatic Detoxification during Inflammation Revealed by Fuzzy-Logic Modeling. PLoS Comput. Biol. 2016, 12, e1004431. [Google Scholar] [CrossRef] [PubMed]
- Patki, K.C.; Von Moltke, L.L.; Greenblatt, D.J. In vitro metabolism of midazolam, triazolam, nifedipine, and testosterone by human liver microsomes and recombinant cytochromes p450: Role of cyp3a4 and cyp3a5. Drug Metab. Dispos. 2003, 31, 938–944. [Google Scholar] [CrossRef]
- Pharmacogene Variation Consortium. CYP3A5. Available online: https://www.pharmvar.org/gene/CYP3A5 (accessed on 15 February 2024).
- Rodriguez-Antona, C.; Savieo, J.L.; Lauschke, V.M.; Sangkuhl, K.; Drogemoller, B.I.; Wang, D.; van Schaik, R.H.N.; Gilep, A.A.; Peter, A.P.; Boone, E.C.; et al. PharmVar GeneFocus: CYP3A5. Clin. Pharmacol. Ther. 2022, 112, 1159–1171. [Google Scholar] [CrossRef] [PubMed]
- Elens, L.; van Gelder, T.; Hesselink, D.A.; Haufroid, V.; van Schaik, R.H. CYP3A4*22: Promising newly identified CYP3A4 variant allele for personalizing pharmacotherapy. Pharmacogenomics 2013, 14, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Kim, B.J.; Lee, S.W.; Kang, H.; Kim, J.W.; Jang, I.J.; Kim, J.G. Influence of midazolam-related genetic polymorphism on conscious sedation during upper gastrointestinal endoscopy in a Korean population. Sci. Rep. 2019, 9, 16001. [Google Scholar] [CrossRef]
- Fromm, M.F.; Schwilden, H.; Bachmakov, I.; Konig, J.; Bremer, F.; Schuttler, J. Impact of the CYP3A5 genotype on midazolam pharmacokinetics and pharmacodynamics during intensive care sedation. Eur. J. Clin. Pharmacol. 2007, 63, 1129–1133. [Google Scholar] [CrossRef]
- MacPhee, I.A. Pharmacogenetic biomarkers: Cytochrome P450 3A5. Clin. Chim. Acta 2012, 413, 1312–1317. [Google Scholar] [CrossRef] [PubMed]
- Westlind-Johnsson, A.; Malmebo, S.; Johansson, A.; Otter, C.; Andersson, T.B.; Johansson, I.; Edwards, R.J.; Boobis, A.R.; Ingelman-Sundberg, M. Comparative analysis of CYP3A expression in human liver suggests only a minor role for CYP3A5 in drug metabolism. Drug Metab. Dispos. 2003, 31, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, G.R. Drug metabolism and variability among patients in drug response. N. Engl. J. Med. 2005, 352, 2211–2221. [Google Scholar] [CrossRef] [PubMed]
- Werk, A.N.; Cascorbi, I. Functional gene variants of CYP3A4. Clin. Pharmacol. Ther. 2014, 96, 340–348. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. Table of Pharmacogenomic Biomarkers in Drug Labeling. Available online: https://www.fda.gov/drugs/science-and-research-drugs/table-pharmacogenomic-biomarkers-drug-labeling (accessed on 15 February 2024).
- Pharmacogene Variation Consortium. CYP3A4. Available online: https://www.pharmvar.org/gene/CYP3A4 (accessed on 15 February 2024).
- PHARMGKB. Curation of DPWG Content into PharmGKB. Available online: https://www.pharmgkb.org/page/dpwgMapping#cyp3a4 (accessed on 15 February 2024).
- Garcia-Martin, E.; Martinez, C.; Pizarro, R.M.; Garcia-Gamito, F.J.; Gullsten, H.; Raunio, H.; Agundez, J.A. CYP3A4 variant alleles in white individuals with low CYP3A4 enzyme activity. Clin. Pharmacol. Ther. 2002, 71, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Alkattan, A.; Alsalameen, E. Polymorphisms of genes related to phase-I metabolic enzymes affecting the clinical efficacy and safety of clopidogrel treatment. Expert Opin. Drug Metab. Toxicol. 2021, 17, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Wojnowski, L.; Hustert, E.; Klein, K.; Goldammer, M.; Haberl, M.; Kirchheiner, J.; Koch, I.; Klattig, J.; Zanger, U.; Brockmoller, J. Re: Modification of clinical presentation of prostate tumors by a novel genetic variant in CYP3A4. J. Natl. Cancer Inst. 2002, 94, 630–631; author reply 631–632. [Google Scholar] [CrossRef]
- Lee, S.J.; Goldstein, J.A. Functionally defective or altered CYP3A4 and CYP3A5 single nucleotide polymorphisms and their detection with genotyping tests. Pharmacogenomics 2005, 6, 357–371. [Google Scholar] [CrossRef]
- Miyazaki, M.; Nakamura, K.; Fujita, Y.; Guengerich, F.P.; Horiuchi, R.; Yamamoto, K. Defective activity of recombinant cytochromes P450 3A4.2 and 3A4.16 in oxidation of midazolam, nifedipine, and testosterone. Drug Metab. Dispos. 2008, 36, 2287–2291. [Google Scholar] [CrossRef]
- Sata, F.; Sapone, A.; Elizondo, G.; Stocker, P.; Miller, V.P.; Zheng, W.; Raunio, H.; Crespi, C.L.; Gonzalez, F.J. CYP3A4 allelic variants with amino acid substitutions in exons 7 and 12: Evidence for an allelic variant with altered catalytic activity. Clin. Pharmacol. Ther. 2000, 67, 48–56. [Google Scholar] [CrossRef]
- Dai, D.; Tang, J.; Rose, R.; Hodgson, E.; Bienstock, R.J.; Mohrenweiser, H.W.; Goldstein, J.A. Identification of variants of CYP3A4 and characterization of their abilities to metabolize testosterone and chlorpyrifos. J. Pharmacol. Exp. Ther. 2001, 299, 825–831. [Google Scholar]
- Eiselt, R.; Domanski, T.L.; Zibat, A.; Mueller, R.; Presecan-Siedel, E.; Hustert, E.; Zanger, U.M.; Brockmoller, J.; Klenk, H.P.; Meyer, U.A.; et al. Identification and functional characterization of eight CYP3A4 protein variants. Pharmacogenetics 2001, 11, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, K.P.; Lin, Y.Y.; Cheng, C.L.; Lai, M.L.; Lin, M.S.; Siest, J.P.; Huang, J.D. Novel mutations of CYP3A4 in Chinese. Drug Metab. Dispos. 2001, 29, 268–273. [Google Scholar] [PubMed]
- Wang, A.; Yu, B.N.; Luo, C.H.; Tan, Z.R.; Zhou, G.; Wang, L.S.; Zhang, W.; Li, Z.; Liu, J.; Zhou, H.H. Ile118Val genetic polymorphism of CYP3A4 and its effects on lipid-lowering efficacy of simvastatin in Chinese hyperlipidemic patients. Eur. J. Clin. Pharmacol. 2005, 60, 843–848. [Google Scholar] [CrossRef]
- Al Maruf, A.; Ahmed, M.U.; Yasmin, H.; Ullah, M.A.; Azad, M.A.; Daly, A.K.; Hasnat, A. Genotypes and phenotypes of CYP3A in Bangladeshi population. Clin. Chim. Acta 2011, 412, 531–536. [Google Scholar] [CrossRef]
- Rais, N.; Chawla, Y.K.; Kohli, K.K. CYP3A phenotypes and genotypes in North Indians. Eur. J. Clin. Pharmacol. 2006, 62, 417–422. [Google Scholar] [CrossRef]
- Westlind-Johnsson, A.; Hermann, R.; Huennemeyer, A.; Hauns, B.; Lahu, G.; Nassr, N.; Zech, K.; Ingelman-Sundberg, M.; von Richter, O. Identification and characterization of CYP3A4*20, a novel rare CYP3A4 allele without functional activity. Clin. Pharmacol. Ther. 2006, 79, 339–349. [Google Scholar] [CrossRef]
- Zhou, Q.; Yu, X.; Shu, C.; Cai, Y.; Gong, W.; Wang, X.; Wang, D.M.; Hu, S. Analysis of CYP3A4 genetic polymorphisms in Han Chinese. J. Hum. Genet. 2011, 56, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Kumondai, M.; Gutierrez Rico, E.M.; Hishinuma, E.; Ueda, A.; Saito, S.; Saigusa, D.; Tadaka, S.; Kinoshita, K.; Nakayoshi, T.; Oda, A.; et al. Functional Characterization of 40 CYP3A4 Variants by Assessing Midazolam 1′-Hydroxylation and Testosterone 6beta-Hydroxylation. Drug Metab. Dispos. 2021, 49, 212–220. [Google Scholar] [CrossRef]
- Werk, A.N.; Lefeldt, S.; Bruckmueller, H.; Hemmrich-Stanisak, G.; Franke, A.; Roos, M.; Kuchle, C.; Steubl, D.; Schmaderer, C.; Brasen, J.H.; et al. Identification and characterization of a defective CYP3A4 genotype in a kidney transplant patient with severely diminished tacrolimus clearance. Clin. Pharmacol. Ther. 2014, 95, 416–422. [Google Scholar] [CrossRef]
- Apellaniz-Ruiz, M.; Inglada-Perez, L.; Naranjo, M.E.; Sanchez, L.; Mancikova, V.; Curras-Freixes, M.; de Cubas, A.A.; Comino-Mendez, I.; Triki, S.; Rebai, A.; et al. High frequency and founder effect of the CYP3A4*20 loss-of-function allele in the Spanish population classifies CYP3A4 as a polymorphic enzyme. Pharmacogenom. J. 2015, 15, 288–292. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Guo, Y.; Wrighton, S.A.; Cooke, G.E.; Sadee, W. Intronic polymorphism in CYP3A4 affects hepatic expression and response to statin drugs. Pharmacogenom. J. 2011, 11, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Ingelman-Sundberg, M.; Lauschke, V.M. Worldwide Distribution of Cytochrome P450 Alleles: A Meta-analysis of Population-scale Sequencing Projects. Clin. Pharmacol. Ther. 2017, 102, 688–700. [Google Scholar] [CrossRef] [PubMed]
- Pratt, V.M.; Cavallari, L.H.; Fulmer, M.L.; Gaedigk, A.; Hachad, H.; Ji, Y.; Kalman, L.V.; Ly, R.C.; Moyer, A.M.; Scott, S.A.; et al. CYP3A4 and CYP3A5 Genotyping Recommendations: A Joint Consensus Recommendation of the Association for Molecular Pathology, Clinical Pharmacogenetics Implementation Consortium, College of American Pathologists, Dutch Pharmacogenetics Working Group of the Royal Dutch Pharmacists Association, European Society for Pharmacogenomics and Personalized Therapy, and Pharmacogenomics Knowledgebase. J. Mol. Diagn. 2023, 25, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Eap, C.B.; Buclin, T.; Hustert, E.; Bleiber, G.; Golay, K.P.; Aubert, A.C.; Baumann, P.; Telenti, A.; Kerb, R. Pharmacokinetics of midazolam in CYP3A4- and CYP3A5-genotyped subjects. Eur. J. Clin. Pharmacol. 2004, 60, 231–236. [Google Scholar] [CrossRef]
- Schuetz, E.G. Lessons from the CYP3A4 promoter. Mol. Pharmacol. 2004, 65, 279–281. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Jimenez, C.P.; Jover, R.; Donato, M.T.; Castell, J.V.; Gomez-Lechon, M.J. Transcriptional regulation and expression of CYP3A4 in hepatocytes. Curr. Drug Metab. 2007, 8, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Antona, C.; Bort, R.; Jover, R.; Tindberg, N.; Ingelman-Sundberg, M.; Gomez-Lechon, M.J.; Castell, J.V. Transcriptional regulation of human CYP3A4 basal expression by CCAAT enhancer-binding protein alpha and hepatocyte nuclear factor-3 gamma. Mol. Pharmacol. 2003, 63, 1180–1189. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Jimenez, C.P.; Gomez-Lechon, M.J.; Castell, J.V.; Jover, R. Transcriptional regulation of the human hepatic CYP3A4: Identification of a new distal enhancer region responsive to CCAAT/enhancer-binding protein beta isoforms (liver activating protein and liver inhibitory protein). Mol. Pharmacol. 2005, 67, 2088–2101. [Google Scholar] [CrossRef]
- Tirona, R.G.; Lee, W.; Leake, B.F.; Lan, L.B.; Cline, C.B.; Lamba, V.; Parviz, F.; Duncan, S.A.; Inoue, Y.; Gonzalez, F.J.; et al. The orphan nuclear receptor HNF4alpha determines PXR- and CAR-mediated xenobiotic induction of CYP3A4. Nat. Med. 2003, 9, 220–224. [Google Scholar] [CrossRef]
- Tegude, H.; Schnabel, A.; Zanger, U.M.; Klein, K.; Eichelbaum, M.; Burk, O. Molecular mechanism of basal CYP3A4 regulation by hepatocyte nuclear factor 4alpha: Evidence for direct regulation in the intestine. Drug Metab. Dispos. 2007, 35, 946–954. [Google Scholar] [CrossRef]
- Jover, R.; Moya, M.; Gomez-Lechon, M.J. Transcriptional regulation of cytochrome p450 genes by the nuclear receptor hepatocyte nuclear factor 4-alpha. Curr. Drug Metab. 2009, 10, 508–519. [Google Scholar] [CrossRef]
- Biggs, J.S.; Wan, J.; Cutler, N.S.; Hakkola, J.; Uusimaki, P.; Raunio, H.; Yost, G.S. Transcription factor binding to a putative double E-box motif represses CYP3A4 expression in human lung cells. Mol. Pharmacol. 2007, 72, 514–525. [Google Scholar] [CrossRef]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef]
- Collins, J.M.; Wang, D. Co-expression of drug metabolizing cytochrome P450 enzymes and estrogen receptor alpha (ESR1) in human liver: Racial differences and the regulatory role of ESR1. Drug Metab. Pers. Ther. 2021, 36, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, B.; Hodgson, E.; D’Costa, D.J.; Robertson, G.R.; Liddle, C. Transcriptional regulation of the human CYP3A4 gene by the constitutive androstane receptor. Mol. Pharmacol. 2002, 62, 359–365. [Google Scholar] [CrossRef]
- Matsumura, K.; Saito, T.; Takahashi, Y.; Ozeki, T.; Kiyotani, K.; Fujieda, M.; Yamazaki, H.; Kunitoh, H.; Kamataki, T. Identification of a novel polymorphic enhancer of the human CYP3A4 gene. Mol. Pharmacol. 2004, 65, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Mathas, M.; Nestler, S.; Bengel, C.; Nem, D.; Godtel-Armbrust, U.; Lang, T.; Taudien, S.; Burk, O.; Wojnowski, L. The unique complexity of the CYP3A4 upstream region suggests a nongenetic explanation of its expression variability. Pharmacogenet. Genom. 2010, 20, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, J.M.; McKee, D.D.; Watson, M.A.; Willson, T.M.; Moore, J.T.; Kliewer, S.A. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J. Clin. Investig. 1998, 102, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, B.; Hodgson, E.; Liddle, C. The orphan human pregnane X receptor mediates the transcriptional activation of CYP3A4 by rifampicin through a distal enhancer module. Mol. Pharmacol. 1999, 56, 1329–1339. [Google Scholar] [CrossRef]
- Liu, F.J.; Song, X.; Yang, D.; Deng, R.; Yan, B. The far and distal enhancers in the CYP3A4 gene co-ordinate the proximal promoter in responding similarly to the pregnane X receptor but differentially to hepatocyte nuclear factor-4alpha. Biochem. J. 2008, 409, 243–250. [Google Scholar] [CrossRef]
- Lamba, J.; Lamba, V.; Strom, S.; Venkataramanan, R.; Schuetz, E. Novel single nucleotide polymorphisms in the promoter and intron 1 of human pregnane X receptor/NR1I2 and their association with CYP3A4 expression. Drug Metab. Dispos. 2008, 36, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.F.; Li, Z.H.; Liu, J.Y.; Liu, T.T.; Wang, P.; Fang, Y.; Zhou, J.; Cui, M.Z.; Gao, N.; Tian, X.; et al. Correlation of Cytochrome P450 Oxidoreductase Expression with the Expression of 10 Isoforms of Cytochrome P450 in Human Liver. Drug Metab. Dispos. 2016, 44, 1193–1200. [Google Scholar] [CrossRef]
- El-Sankary, W.; Plant, N.J.; Gibson, G.G.; Moore, D.J. Regulation of the CYP3A4 gene by hydrocortisone and xenobiotics: Role of the glucocorticoid and pregnane X receptors. Drug Metab. Dispos. 2000, 28, 493–496. [Google Scholar]
- Burk, O.; Koch, I.; Raucy, J.; Hustert, E.; Eichelbaum, M.; Brockmoller, J.; Zanger, U.M.; Wojnowski, L. The induction of cytochrome P450 3A5 (CYP3A5) in the human liver and intestine is mediated by the xenobiotic sensors pregnane X receptor (PXR) and constitutively activated receptor (CAR). J. Biol. Chem. 2004, 279, 38379–38385. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wong, T.; Hashizume, T.; Dickmann, L.Z.; Scian, M.; Koszewski, N.J.; Goff, J.P.; Horst, R.L.; Chaudhry, A.S.; Schuetz, E.G.; et al. Human UGT1A4 and UGT1A3 conjugate 25-hydroxyvitamin D3: Metabolite structure, kinetics, inducibility, and interindividual variability. Endocrinology 2014, 155, 2052–2063. [Google Scholar] [CrossRef]
- Xu, C.; Gao, J.; Zhang, H.F.; Gao, N.; Guo, Y.Y.; Fang, Y.; Wen, Q.; Qiao, H.L. Content and Activities of UGT2B7 in Human Liver In Vitro and Predicted In Vivo: A Bottom-Up Approach. Drug Metab. Dispos. 2018, 46, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Kliewer, S.A.; Goodwin, B.; Willson, T.M. The nuclear pregnane X receptor: A key regulator of xenobiotic metabolism. Endocr. Rev. 2002, 23, 687–702. [Google Scholar] [CrossRef]
- Vrzal, R.; Kubesova, K.; Pavek, P.; Dvorak, Z. Benzodiazepines medazepam and midazolam are activators of pregnane X receptor and weak inducers of CYP3A4: Investigation in primary cultures of human hepatocytes and hepatocarcinoma cell lines. Toxicol. Lett. 2010, 193, 183–188. [Google Scholar] [CrossRef]
- Gnerre, C.; Blattler, S.; Kaufmann, M.R.; Looser, R.; Meyer, U.A. Regulation of CYP3A4 by the bile acid receptor FXR: Evidence for functional binding sites in the CYP3A4 gene. Pharmacogenetics 2004, 14, 635–645. [Google Scholar] [CrossRef]
- Pascussi, J.M.; Gerbal-Chaloin, S.; Duret, C.; Daujat-Chavanieu, M.; Vilarem, M.J.; Maurel, P. The tangle of nuclear receptors that controls xenobiotic metabolism and transport: Crosstalk and consequences. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Duniec-Dmuchowski, Z.; Ellis, E.; Strom, S.C.; Kocarek, T.A. Regulation of CYP3A4 and CYP2B6 expression by liver X receptor agonists. Biochem. Pharmacol. 2007, 74, 1535–1540. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, T.; Yoshinari, K.; Aoyama, K.; Sugawara, M.; Sekiya, Y.; Nagata, K.; Yamazoe, Y. Role of vitamin D receptor in the lithocholic acid-mediated CYP3A induction in vitro and in vivo. Drug Metab. Dispos. 2008, 36, 2058–2063. [Google Scholar] [CrossRef] [PubMed]
- Klein, K.; Thomas, M.; Winter, S.; Nussler, A.K.; Niemi, M.; Schwab, M.; Zanger, U.M. PPARA: A novel genetic determinant of CYP3A4 in vitro and in vivo. Clin. Pharmacol. Ther. 2012, 91, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Schroder, A.; Wollnik, J.; Wrzodek, C.; Drager, A.; Bonin, M.; Burk, O.; Thomas, M.; Thasler, W.E.; Zanger, U.M.; Zell, A. Inferring statin-induced gene regulatory relationships in primary human hepatocytes. Bioinformatics 2011, 27, 2473–2477. [Google Scholar] [CrossRef] [PubMed]
- Rakhshandehroo, M.; Hooiveld, G.; Muller, M.; Kersten, S. Comparative analysis of gene regulation by the transcription factor PPARalpha between mouse and human. PLoS ONE 2009, 4, e6796. [Google Scholar] [CrossRef]
- Zhou, C.; Tabb, M.M.; Nelson, E.L.; Grun, F.; Verma, S.; Sadatrafiei, A.; Lin, M.; Mallick, S.; Forman, B.M.; Thummel, K.E.; et al. Mutual repression between steroid and xenobiotic receptor and NF-kappaB signaling pathways links xenobiotic metabolism and inflammation. J. Clin. Investig. 2006, 116, 2280–2289. [Google Scholar] [CrossRef] [PubMed]
- Slaviero, K.A.; Clarke, S.J.; Rivory, L.P. Inflammatory response: An unrecognised source of variability in the pharmacokinetics and pharmacodynamics of cancer chemotherapy. Lancet Oncol. 2003, 4, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.Z.; Gao, W.; Yu, A.M. MicroRNAs regulate CYP3A4 expression via direct and indirect targeting. Drug Metab. Dispos. 2009, 37, 2112–2117. [Google Scholar] [CrossRef]
- Ekstrom, L.; Skilving, I.; Ovesjo, M.L.; Aklillu, E.; Nylen, H.; Rane, A.; Diczfalusy, U.; Bjorkhem-Bergman, L. miRNA-27b levels are associated with CYP3A activity in vitro and in vivo. Pharmacol. Res. Perspect. 2015, 3, e00192. [Google Scholar] [CrossRef]
- Takagi, S.; Nakajima, M.; Mohri, T.; Yokoi, T. Post-transcriptional regulation of human pregnane X receptor by micro-RNA affects the expression of cytochrome P450 3A4. J. Biol. Chem. 2008, 283, 9674–9680. [Google Scholar] [CrossRef] [PubMed]
- Takagi, S.; Nakajima, M.; Kida, K.; Yamaura, Y.; Fukami, T.; Yokoi, T. MicroRNAs regulate human hepatocyte nuclear factor 4alpha, modulating the expression of metabolic enzymes and cell cycle. J. Biol. Chem. 2010, 285, 4415–4422. [Google Scholar] [CrossRef] [PubMed]
- Karbiener, M.; Fischer, C.; Nowitsch, S.; Opriessnig, P.; Papak, C.; Ailhaud, G.; Dani, C.; Amri, E.Z.; Scheideler, M. microRNA miR-27b impairs human adipocyte differentiation and targets PPARgamma. Biochem. Biophys. Res. Commun. 2009, 390, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Jennewein, C.; von Knethen, A.; Schmid, T.; Brune, B. MicroRNA-27b contributes to lipopolysaccharide-mediated peroxisome proliferator-activated receptor gamma (PPARgamma) mRNA destabilization. J. Biol. Chem. 2010, 285, 11846–11853. [Google Scholar] [CrossRef] [PubMed]
- Ou, Z.; Wada, T.; Gramignoli, R.; Li, S.; Strom, S.C.; Huang, M.; Xie, W. MicroRNA hsa-miR-613 targets the human LXRalpha gene and mediates a feedback loop of LXRalpha autoregulation. Mol. Endocrinol. 2011, 25, 584–596. [Google Scholar] [CrossRef] [PubMed]
- Schmeier, S.; Schaefer, U.; MacPherson, C.R.; Bajic, V.B. dPORE-miRNA: Polymorphic regulation of microRNA genes. PLoS ONE 2011, 6, e16657. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Yang, F.; Urban, T.J.; Li, L.; Chalasani, N.; Flockhart, D.A.; Liu, W. Impact of the Interaction between 3′-UTR SNPs and microRNA on the Expression of Human Xenobiotic Metabolism Enzyme and Transporter Genes. Front. Genet. 2012, 3, 248. [Google Scholar] [CrossRef]
- Kasteel, E.E.J.; Darney, K.; Kramer, N.I.; Dorne, J.; Lautz, L.S. Human variability in isoform-specific UDP-glucuronosyltransferases: Markers of acute and chronic exposure, polymorphisms and uncertainty factors. Arch. Toxicol. 2020, 94, 2637–2661. [Google Scholar] [CrossRef]
- Aueviriyavit, S.; Furihata, T.; Morimoto, K.; Kobayashi, K.; Chiba, K. Hepatocyte nuclear factor 1 alpha and 4 alpha are factors involved in interindividual variability in the expression of UGT1A6 and UGT1A9 but not UGT1A1, UGT1A3 and UGT1A4 mRNA in human livers. Drug Metab. Pharmacokinet. 2007, 22, 391–398. [Google Scholar] [CrossRef]
- Pharmacogenomics Laboratory at Université Laval. Available online: https://www.pharmacogenomics.pha.ulaval.ca/wp-content/uploads/2015/04/HAP-UGT1A4.htm (accessed on 15 February 2024).
- Ehmer, U.; Vogel, A.; Schutte, J.K.; Krone, B.; Manns, M.P.; Strassburg, C.P. Variation of hepatic glucuronidation: Novel functional polymorphisms of the UDP-glucuronosyltransferase UGT1A4. Hepatology 2004, 39, 970–977. [Google Scholar] [CrossRef]
- Erickson-Ridout, K.K.; Sun, D.; Lazarus, P. Glucuronidation of the second-generation antipsychotic clozapine and its active metabolite N-desmethylclozapine. Potential importance of the UGT1A1 A(TA)(7)TAA and UGT1A4 L48V polymorphisms. Pharmacogenetics Genom. 2012, 22, 561–576. [Google Scholar] [CrossRef] [PubMed]
- Erickson-Ridout, K.K.; Zhu, J.; Lazarus, P. Olanzapine metabolism and the significance of UGT1A448V and UGT2B1067Y variants. Pharmacogenetics Genom. 2011, 21, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Argikar, U.A.; Remmel, R.P. Functional analysis of UGT1A4(P24T) and UGT1A4(L48V) variant enzymes. Pharmacogenomics 2011, 12, 1671–1679. [Google Scholar] [CrossRef] [PubMed]
- Reimers, A.; Sjursen, W.; Helde, G.; Brodtkorb, E. Frequencies of UGT1A4*2 (P24T) and *3 (L48V) and their effects on serum concentrations of lamotrigine. Eur. J. Drug Metab. Pharmacokinet. 2016, 41, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Gulcebi, M.I.; Ozkaynakci, A.; Goren, M.Z.; Aker, R.G.; Ozkara, C.; Onat, F.Y. The relationship between UGT1A4 polymorphism and serum concentration of lamotrigine in patients with epilepsy. Epilepsy Res. 2011, 95, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Yang, L.Y.; Zhang, M.C.; Liu, S.Y. Correlation of the UGT1A4 gene polymorphism with serum concentration and therapeutic efficacy of lamotrigine in Han Chinese of Northern China. Eur. J. Clin. Pharmacol. 2014, 70, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Haslemo, T.; Loryan, I.; Ueda, N.; Mannheimer, B.; Bertilsson, L.; Ingelman-Sundberg, M.; Molden, E.; Eliasson, E. UGT1A4*3 encodes significantly increased glucuronidation of olanzapine in patients on maintenance treatment and in recombinant systems. Clin. Pharmacol. Ther. 2012, 92, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Lee, S.Y.; Yang, K.S.; Park, J.Y.; Yoon, S.Z.; Yoon, S.M. Polymorphism rs4263535 in GABRA1 intron 4 was related to deeper sedation by intravenous midazolam. J. Int. Med. Res. 2015, 43, 686–698. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y. Epigenetic regulation of pregnane X receptor activity. Drug Metab. Rev. 2013, 45, 166–172. [Google Scholar] [CrossRef]
- Meierhans, R.; Stover, J.F.; Bechir, M.; Keel, M.; Stocker, R. Reduced midazolam clearance must be considered in prolonged coma. Anaesth. Intensive Care 2008, 36, 915–916. [Google Scholar]
- Babu, T.A. Prolonged sedation following administration of oral midazolam. Indian Pediatr. 2013, 50, 342–343. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Ma, W.; Guo, Q.; Liu, J.; Li, W.; McLeod, H.L.; He, Y. The pharmacogenetics of medications used in general anesthesia. Pharmacogenomics 2018, 19, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.S.; Lockwood, G.F.; Graham, M.A.; Brian, W.R.; Loi, C.M.; Dobrinska, M.R.; Shen, D.D.; Watkins, P.B.; Wilkinson, G.R.; Kharasch, E.D.; et al. In-vivo phenotyping for CYP3A by a single-point determination of midazolam plasma concentration. Pharmacogenetics 2001, 11, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Troberg, J.; Finel, M. The Polymorphic Variant P24T of UDP-Glucuronosyltransferase 1A4 and Its Unusual Consequences. Drug Metab. Dispos. 2015, 43, 1769–1772. [Google Scholar] [CrossRef]
- Spina, S.P.; Ensom, M.H. Clinical pharmacokinetic monitoring of midazolam in critically ill patients. Pharmacotherapy 2007, 27, 389–398. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peter, J.-U.; Dieudonné, P.; Zolk, O. Pharmacokinetics, Pharmacodynamics, and Side Effects of Midazolam: A Review and Case Example. Pharmaceuticals 2024, 17, 473. https://doi.org/10.3390/ph17040473
Peter J-U, Dieudonné P, Zolk O. Pharmacokinetics, Pharmacodynamics, and Side Effects of Midazolam: A Review and Case Example. Pharmaceuticals. 2024; 17(4):473. https://doi.org/10.3390/ph17040473
Chicago/Turabian StylePeter, Jens-Uwe, Peter Dieudonné, and Oliver Zolk. 2024. "Pharmacokinetics, Pharmacodynamics, and Side Effects of Midazolam: A Review and Case Example" Pharmaceuticals 17, no. 4: 473. https://doi.org/10.3390/ph17040473
APA StylePeter, J. -U., Dieudonné, P., & Zolk, O. (2024). Pharmacokinetics, Pharmacodynamics, and Side Effects of Midazolam: A Review and Case Example. Pharmaceuticals, 17(4), 473. https://doi.org/10.3390/ph17040473