
Intestinal Fibrogenesis in Inflammatory Bowel Diseases: Exploring the Potential Role of Gut Microbiota Metabolites as Modulators
 , ,
, ,  ,
, 
 ,
, 
Abstract
:1. Overview of Inflammatory Bowel Diseases (IBDs)
1.1. Epidemiology and Global Prevalence
1.2. Clinical Manifestations and Disease Course
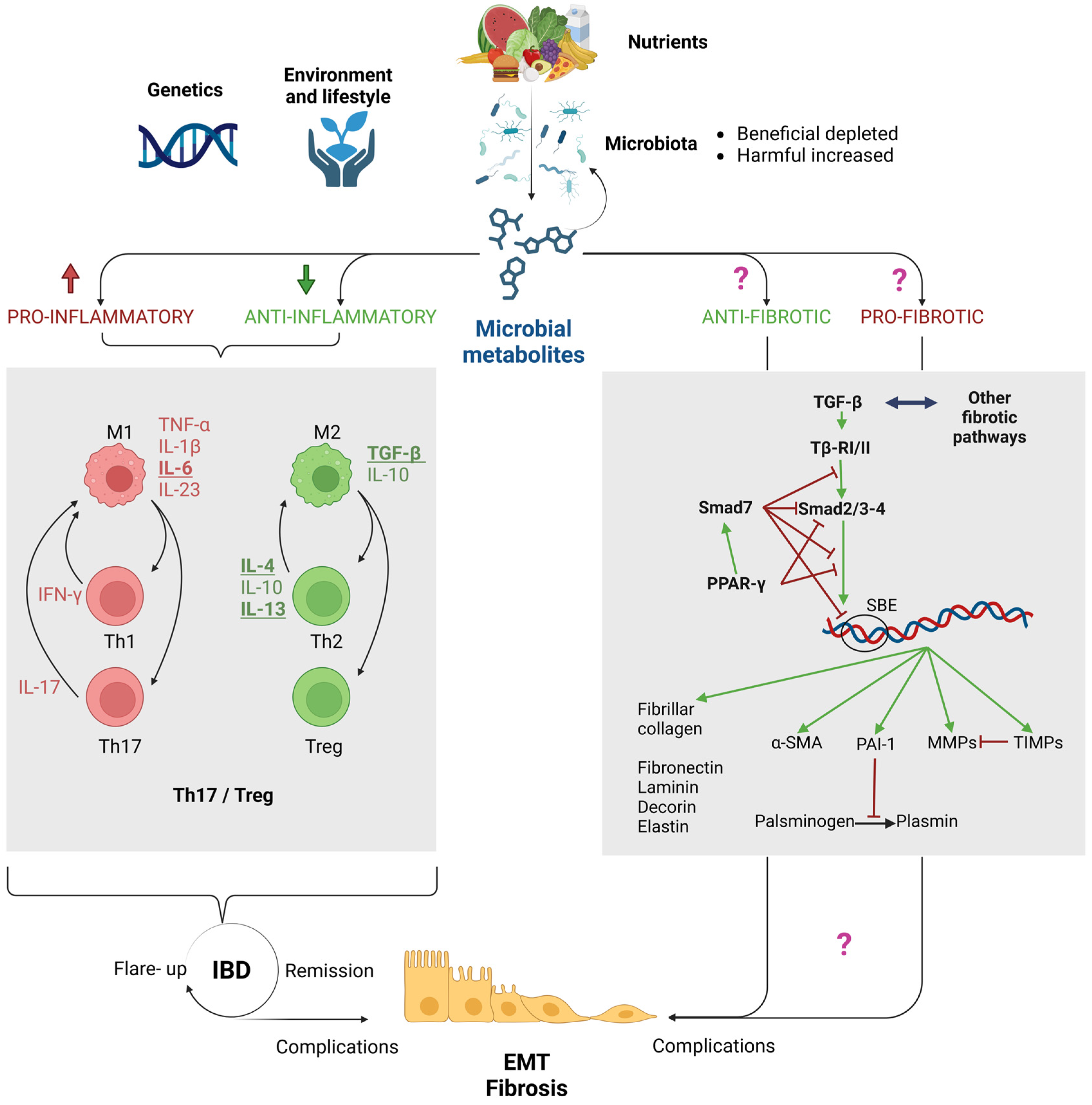
2. Purpose of this Review: Are the Microbiota and Its Metabolites Key Players in Fibrogenesis in IBDs?
3. The Intestinal Barrier in Health and in IBDs
3.1. Dysbiosis: Alterations in the Intestinal Microbiota
3.2. Impairment of the Mucus Layer
3.3. Epithelial Dysfunction and Increased Permeability: The “Leaky Gut”
3.4. Dysregulation of Mucosal Immunity
4. The Role of Immune Dysregulation in Driving Epithelial-to-Mesenchymal Transition (EMT) and Fibrosis in IBDs
5. Microbial Metabolites: Established Players in Intestinal Homeostasis and IBD Inflammation, with Potential Implications in Fibrosis
5.1. Short-Chain Fatty Acids (SCFAs)
5.2. Lactic Acid (LA)
5.3. Indoles

{kind=link}
| IAA | IPA | ILA | IS | IC | |
|---|---|---|---|---|---|
| AhR | Kidney: IPA suppresses the IS effect on the receptor [147]. | Gut: Supplementation with L. acidophilus, or its metabolite ILA, attenuates inflammation and restores IL-22 levels through AhR signaling in mice [142]. Similar results were observed in a mice model of DSS-induced colitis supplemented with two strains of ILA-producing B. bifidum [143]. | Liver: IS is an agonist of the AhR receptor [147]. | Gut: depletion of dietary IC is fatal in AhR IEC-deficient mice and worsens chronic colitis in C57BL/6 mice; in contrast, its administration reduces the Th17/Treg ratio in the same model [145,148]. | |
| TGF-β | Peritoneum: the novel IAA analogue MA-35 reduces TGF-β-positive cells in a murine model of peritoneal fibrosis [149]. | Kidney: IPA suppresses the IS effect on the receptor [147]. Liver: IPA aggravates CCl4-induced fibrosis by activating TGF-β1/Smads signaling in HSCs [150]. | Kidney: IS induces fibrosis through the stimulation of TGF-β1 [147]. | ||
| Smads | Kidney: the IAA novel analogue, MA-35, inhibits the phosphorylation of Smad3, thus reducing TGF-β1 signaling and related renal fibrosis [151]. | Liver: IPA aggravates CCl4-induced fibrosis by activating TGF-β1/Smads signaling in HSCs [150]. | |||
| PPAR-γ | Adipocytes: the administration of I3C restores the levels of PPAR-γ, which were deregulated in mice fed with a high-fat diet [146]. | ||||
| ECM | Peritoneum: the treatment with the novel IAA analogue MA-35 reduces α-SMA-positive myofibroblasts in a murine model of peritoneal fibrosis [149]. | Liver: IPA reduces α-SMA and collagen deposition and MMP expression while inducing TIMPs in TGF-β1-stimulated hepatic stellate cells [152]. Liver: IPA aggravates CCl4-induced fibrosis by activating TGF-β1/Smads signaling in HSCs [150]. | Kidney: IS enhances α-SMA expression [147]. | ||
| PXR | Gut: IPA reduces PXR-induced fibrosis in a mice model of colitis; IBD patients showed lower levels of PXR and fecal IPA [68]. |
| QUERY | (“IBD” OR “Gut”) AND (“TGF-Beta” OR “Smad” OR “PPAR-Gamma” OR “Fibrosis” OR “EMT” OR “Alpha-SMA” OR “MMP” OR “PAI-1” OR “TIMP”) | Title and Abstract Check | |
|---|---|---|---|
| AND | |||
| “butyrate” OR “butyric acid” | 83 | 16 | |
| “acetate” OR “acetic acid” | 58 | 4 | |
| “propionate” OR “propionic acid” | 41 | 5 | |
| “lactic acid” | 27 | 1 | |
| “indole-3-acetic acid” | 5 | 1 | |
| “indole-3-carbinol” | 2 | 0 | |
| “indole-3-lactic acid” | 0 | 0 | |
| “indole-3-propionic acid” | 5 | 2 | |
| “indoxyl sulfate” | 1 | 0 | |
| “urolithin” | 5 | 1 | |
| “hydrogen sulfide” | 1 | 0 | |
| “trimethylamine” OR “TMAO” OR “trimethylamine-N-oxide” | 52 | 8 | |
| Total | 280 | 38 | |
5.4. Urolithins (Uros)
5.5. Hydrogen Sulfide (H2S)
5.6. Trimethylamine (TMA) and Trimethylamine-N-Oxide (TMAO)
6. Discussion: Current Knowledge and Therapeutic Perspectives of Microbiota Metabolite Modulation in Intestinal Fibrogenesis
7. Methods
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Green, Z.; Beattie, R.M.; Ashton, J.J. Recent Developments in the Assessment and Management of Inflammatory Bowel Disease in Childhood: A Narrative Review. Transl. Pediatr. 2023, 12, 1853–1874. [Google Scholar] [CrossRef]
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Worldwide Incidence and Prevalence of Inflammatory Bowel Disease in the 21st Century: A Systematic Review of Population-Based Studies. Lancet 2017, 390, 2769–2778. [Google Scholar] [CrossRef]
- Nguyen, N.H.; Khera, R.; Dulai, P.S.; Boland, B.S.; Ohno-Machado, L.; Sandborn, W.J.; Singh, S. National Estimates of Financial Hardship From Medical Bills and Cost-Related Medication Nonadherence in Patients With Inflammatory Bowel Diseases in the United States. Inflamm. Bowel Dis. 2021, 27, 1068–1078. [Google Scholar] [CrossRef] [PubMed]
- Wehkamp, J.; Götz, M.; Herrlinger, K.; Steurer, W.; Stange, E.F. Inflammatory Bowel Disease: Crohn’s Disease and Ulcerative Colitis. Dtsch. Ärztebl. Int. 2016, 113, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Vavricka, S.R.; Schoepfer, A.; Scharl, M.; Lakatos, P.L.; Navarini, A.; Rogler, G. Extraintestinal Manifestations of Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2015, 21, 1982–1992. [Google Scholar] [CrossRef]
- Vancamelbeke, M.; Vermeire, S. The Intestinal Barrier: A Fundamental Role in Health and Disease. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 821–834. [Google Scholar] [CrossRef] [PubMed]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.; Gasbarrini, A.; Mele, M. What Is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms 2019, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Qiu, P.; Ishimoto, T.; Fu, L.; Zhang, J.; Zhang, Z.; Liu, Y. The Gut Microbiota in Inflammatory Bowel Disease. Front. Cell. Infect. Microbiol. 2022, 12, 733992. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.N.; St. Amand, A.L.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-Phylogenetic Characterization of Microbial Community Imbalances in Human Inflammatory Bowel Diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef] [PubMed]
- Machiels, K.; Joossens, M.; Sabino, J.; De Preter, V.; Arijs, I.; Eeckhaut, V.; Ballet, V.; Claes, K.; Van Immerseel, F.; Verbeke, K.; et al. A Decrease of the Butyrate-Producing Species Roseburia hominis and Faecalibacterium prausnitzii Defines Dysbiosis in Patients with Ulcerative Colitis. Gut 2014, 63, 1275–1283. [Google Scholar] [CrossRef]
- Vich Vila, A.; Imhann, F.; Collij, V.; Jankipersadsing, S.A.; Gurry, T.; Mujagic, Z.; Kurilshikov, A.; Bonder, M.J.; Jiang, X.; Tigchelaar, E.F.; et al. Gut Microbiota Composition and Functional Changes in Inflammatory Bowel Disease and Irritable Bowel Syndrome. Sci. Transl. Med. 2018, 10, eaap8914. [Google Scholar] [CrossRef]
- Kang, S.; Denman, S.E.; Morrison, M.; Yu, Z.; Dore, J.; Leclerc, M.; McSweeney, C.S. Dysbiosis of Fecal Microbiota in Crohn’s Disease Patients as Revealed by a Custom Phylogenetic Microarray. Inflamm. Bowel Dis. 2010, 16, 2034–2042. [Google Scholar] [CrossRef]
- Png, C.W.; Lindén, S.K.; Gilshenan, K.S.; Zoetendal, E.G.; McSweeney, C.S.; Sly, L.I.; McGuckin, M.A.; Florin, T.H.J. Mucolytic Bacteria With Increased Prevalence in IBD Mucosa Augment In Vitro Utilization of Mucin by Other Bacteria. Am. J. Gastroenterol. 2010, 105, 2420–2428. [Google Scholar] [CrossRef] [PubMed]
- Joossens, M.; Huys, G.; Cnockaert, M.; De Preter, V.; Verbeke, K.; Rutgeerts, P.; Vandamme, P.; Vermeire, S. Dysbiosis of the Faecal Microbiota in Patients with Crohn’s Disease and Their Unaffected Relatives. Gut 2011, 60, 631–637. [Google Scholar] [CrossRef]
- Sokol, H.; Seksik, P.; Furet, J.P.; Firmesse, O.; Nion-Larmurier, I.; Beaugerie, L.; Cosnes, J.; Corthier, G.; Marteau, P.; Doré, J. Low Counts of Faecalibacterium Prausnitzii in Colitis Microbiota. Inflamm. Bowel Dis. 2009, 15, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Sokol, H.; Seksik, P.; Rigottier-Gois, L.; Lay, C.; Lepage, P.; Podglajen, I.; Marteau, P.; Doré, J. Specificities of the Fecal Microbiota in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2006, 12, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Hansen, R.; Russell, R.K.; Reiff, C.; Louis, P.; McIntosh, F.; Berry, S.H.; Mukhopadhya, I.; Bisset, M.W.; Barclay, A.R.; Bishop, J.; et al. Microbiota of De-Novo Pediatric IBD: Increased Faecalibacterium Prausnitzii and Reduced Bacterial Diversity in Crohn’s But Not in Ulcerative Colitis. Am. J. Gastroenterol. 2012, 107, 1913–1922. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Chen, L.; Zhou, R.; Wang, X.; Song, L.; Huang, S.; Wang, G.; Xia, B. Increased Proportions of Bifidobacterium and the Lactobacillus Group and Loss of Butyrate-Producing Bacteria in Inflammatory Bowel Disease. J. Clin. Microbiol. 2014, 52, 398–406. [Google Scholar] [CrossRef]
- Fyderek, K.; Strus, M.; Kowalska-Duplaga, K.; Gosiewski, T.; Wędrychowicz, A.; Jedynak-Wąsowicz, U.; Sładek, M.; Pieczarkowski, S.; Adamski, P.; Kochan, P.; et al. Mucosal Bacterial Microflora and Mucus Layer Thickness Inadolescents with Inflammatory Bowel Disease. World J. Gastroenterol. 2009, 15, 5287. [Google Scholar] [CrossRef]
- Zhong, W.; Lu, X.; Shi, H.; Zhao, G.; Song, Y.; Wang, Y.; Zhang, J.; Jin, Y.; Wang, S. Distinct Microbial Populations Exist in the Mucosa-Associated Microbiota of Diarrhea Predominant Irritable Bowel Syndrome and Ulcerative Colitis. J. Clin. Gastroenterol. 2019, 53, 660–672. [Google Scholar] [CrossRef]
- Gophna, U.; Sommerfeld, K.; Gophna, S.; Doolittle, W.F.; Veldhuyzen Van Zanten, S.J.O. Differences between Tissue-Associated Intestinal Microfloras of Patients with Crohn’s Disease and Ulcerative Colitis. J. Clin. Microbiol. 2006, 44, 4136–4141. [Google Scholar] [CrossRef] [PubMed]
- Duranti, S.; Gaiani, F.; Mancabelli, L.; Milani, C.; Grandi, A.; Bolchi, A.; Santoni, A.; Lugli, G.A.; Ferrario, C.; Mangifesta, M.; et al. Elucidating the Gut Microbiome of Ulcerative Colitis: Bifidobacteria as Novel Microbial Biomarkers. FEMS Microbiol. Ecol. 2016, 92, fiw191. [Google Scholar] [CrossRef]
- Gibson, G.R.; Cummings, J.H.; Macfarlane, G.T. Growth and Activities of Sulphate-Reducing Bacteria in Gut Contents of Healthy Subjects and Patients with Ulcerative Colitis. FEMS Microbiol. Ecol. 1991, 9, 103–111. [Google Scholar] [CrossRef]
- Zheng, M.; Han, R.; Yuan, Y.; Xing, Y.; Zhang, W.; Sun, Z.; Liu, Y.; Li, J.; Mao, T. The Role of Akkermansia Muciniphila in Inflammatory Bowel Disease: Current Knowledge and Perspectives. Front. Immunol. 2023, 13, 1089600. [Google Scholar] [CrossRef] [PubMed]
- Oren, A.; Garrity, G.M. Valid Publication of the Names of Forty-Two Phyla of Prokaryotes. Int. J. Syst. Evol. Microbiol. 2021, 71, 005056. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Tomé, S.; Ortega Moreno, L.; Chaparro, M.; Gisbert, J.P. Gut Microbiota and Dietary Factors as Modulators of the Mucus Layer in Inflammatory Bowel Disease. Int. J. Mol. Sci. 2021, 22, 10224. [Google Scholar] [CrossRef]
- Bruewer, M.; Samarin, S.; Nusrat, A. Inflammatory Bowel Disease and the Apical Junctional Complex. Ann. N. Y. Acad. Sci. 2006, 1072, 242–252. [Google Scholar] [CrossRef]
- Soderholm, A.T.; Pedicord, V.A. Intestinal Epithelial Cells: At the Interface of the Microbiota and Mucosal Immunity. Immunology 2019, 158, 267–280. [Google Scholar] [CrossRef]
- Strober, W.; Asano, N.; Fuss, I.; Kitani, A.; Watanabe, T. Cellular and Molecular Mechanisms Underlying NOD 2 Risk-associated Polymorphisms in C Rohn’s Disease. Immunol. Rev. 2014, 260, 249–260. [Google Scholar] [CrossRef]
- Lu, Q.; Yang, M.; Liang, Y.; Xu, J.; Xu, H.; Nie, Y.; Wang, L.; Yao, J.; Li, D. Immunology of Inflammatory Bowel Disease: Molecular Mechanisms and Therapeutics. JIR 2022, 15, 1825–1844. [Google Scholar] [CrossRef]
- Tindemans, I.; Joosse, M.E.; Samsom, J.N. Dissecting the Heterogeneity in T-Cell Mediated Inflammation in IBD. Cells 2020, 9, 110. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Li, X.; Zhang, Q.; Yang, J.; Liu, G. Roles of Macrophages on Ulcerative Colitis and Colitis-Associated Colorectal Cancer. Front. Immunol. 2023, 14, 1103617. [Google Scholar] [CrossRef] [PubMed]
- Kałużna, A.; Olczyk, P.; Komosińska-Vassev, K. The Role of Innate and Adaptive Immune Cells in the Pathogenesis and Development of the Inflammatory Response in Ulcerative Colitis. J. Clin. Med. 2022, 11, 400. [Google Scholar] [CrossRef] [PubMed]
- Nakase, H.; Sato, N.; Mizuno, N.; Ikawa, Y. The Influence of Cytokines on the Complex Pathology of Ulcerative Colitis. Autoimmun. Rev. 2022, 21, 103017. [Google Scholar] [CrossRef]
- Heller, F.; Fromm, A.; Gitter, A.H.; Mankertz, J.; Schulzke, J.-D. Epithelial Apoptosis Is a Prominent Feature of the Epithelial Barrier Disturbance in Intestinal Inflammation: Effect of pro-Inflammatory Interleukin-13 on Epithelial Cell Function. Mucosal Immunol. 2008, 1, S58–S61. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; Sewell, G.W.; Levine, A.P.; Chew, T.S.; Dunne, J.; O’Shea, N.R.; Smith, P.J.; Harrison, P.J.; Macdonald, C.M.; Bloom, S.L.; et al. Disruption of Macrophage Pro-inflammatory Cytokine Release in C Rohn’s Disease Is Associated with Reduced Optineurin Expression in a Subset of Patients. Immunology 2015, 144, 45–55. [Google Scholar] [CrossRef]
- Petagna, L.; Antonelli, A.; Ganini, C.; Bellato, V.; Campanelli, M.; Divizia, A.; Efrati, C.; Franceschilli, M.; Guida, A.M.; Ingallinella, S.; et al. Pathophysiology of Crohn’s Disease Inflammation and Recurrence. Biol. Direct 2020, 15, 23. [Google Scholar] [CrossRef] [PubMed]
- Bunte, K.; Beikler, T. Th17 Cells and the IL-23/IL-17 Axis in the Pathogenesis of Periodontitis and Immune-Mediated Inflammatory Diseases. Int. J. Mol. Sci. 2019, 20, 3394. [Google Scholar] [CrossRef]
- Zhao, J.; Lu, Q.; Liu, Y.; Shi, Z.; Hu, L.; Zeng, Z.; Tu, Y.; Xiao, Z.; Xu, Q. Th17 Cells in Inflammatory Bowel Disease: Cytokines, Plasticity, and Therapies. J. Immunol. Res. 2021, 2021, 8816041. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Luo, M.; Chen, Z.; He, B. The Function and Role of the Th17/Treg Cell Balance in Inflammatory Bowel Disease. J. Immunol. Res. 2020, 2020, 8813558. [Google Scholar] [CrossRef]
- Giuffrida, P.; Cococcia, S.; Delliponti, M.; Lenti, M.V.; Di Sabatino, A. Controlling Gut Inflammation by Restoring Anti-Inflammatory Pathways in Inflammatory Bowel Disease. Cells 2019, 8, 397. [Google Scholar] [CrossRef] [PubMed]
- Goebel, E.J.; Hart, K.N.; McCoy, J.C.; Thompson, T.B. Structural Biology of the TGFβ Family. Exp. Biol. Med. 2019, 244, 1530–1546. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.-H.; Moustakas, A. Signaling Receptors for TGF-β Family Members. Cold Spring Harb. Perspect. Biol. 2016, 8, a022053. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Liu, Z.; Chen, Y. Regulation of TGF-β Signaling by Smad7. Acta Biochim. Biophys. Sin. 2009, 41, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-Smad Pathways in TGF-β Signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef]
- Coskun, M.; Olsen, J.; Seidelin, J.B.; Nielsen, O.H. MAP Kinases in Inflammatory Bowel Disease. Clin. Chim. Acta 2011, 412, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Jalil, A.T.; Hassan, N.F.; Abdulameer, S.J.; Farhan, Z.M.; Suleiman, A.A.; Al-Azzawi, A.K.; Zabibah, R.; Fadhil, A. Phosphatidylinositol 3-kinase Signaling Pathway and Inflammatory Bowel Disease: Current Status and Future Prospects. Fundam. Clin. Pharmacol. 2023, 37, 910–917. [Google Scholar] [CrossRef]
- Lijnen, P.; Petrov, V. Transforming Growth Factor-Beta 1-Induced Collagen Production Incultures of Cardiac Fibroblasts Is the Result of the Appearance Ofmyofibroblasts. Methods Find. Exp. Clin. Pharmacol. 2002, 24, 333. [Google Scholar] [CrossRef]
- Verrecchia, F.; Mauviel, A. Transforming Growth Factor-β Signaling Through the Smad Pathway: Role in Extracellular Matrix Gene Expression and Regulation. J. Investig. Dermatol. 2002, 118, 211–215. [Google Scholar] [CrossRef]
- Branton, M.H.; Kopp, J.B. TGF-β and Fibrosis. Microbes Infect. 1999, 1, 1349–1365. [Google Scholar] [CrossRef] [PubMed]
- Powell, D.W.; Pinchuk, I.V.; Saada, J.I.; Chen, X.; Mifflin, R.C. Mesenchymal Cells of the Intestinal Lamina Propria. Annu. Rev. Physiol. 2011, 73, 213–237. [Google Scholar] [CrossRef] [PubMed]
- Akhurst, R.J. From Shape-Shifting Embryonic Cells to Oncology: The Fascinating History of Epithelial Mesenchymal Transition. Semin. Cancer Biol. 2023, 96, 100–114. [Google Scholar] [CrossRef] [PubMed]
- Kardooni, A.; Bahrampour, A.; Golmohammadi, S.; Jalili, A.; Alishahi, M.M. The Role of Epithelial Mesenchymal Transition (EMT) in Pathogenesis of Cardiotoxicity: Diagnostic & Prognostic Approach. Mol. Biotechnol. 2023, 65, 1403–1413. [Google Scholar] [CrossRef] [PubMed]
- Hadpech, S.; Thongboonkerd, V. Epithelial–Mesenchymal Plasticity in Kidney Fibrosis. Genesis 2023, 62, e23529. [Google Scholar] [CrossRef]
- Mottais, A.; Riberi, L.; Falco, A.; Soccal, S.; Gohy, S.; De Rose, V. Epithelial–Mesenchymal Transition Mechanisms in Chronic Airway Diseases: A Common Process to Target? Int. J. Mol. Sci. 2023, 24, 12412. [Google Scholar] [CrossRef]
- Andoh, A.; Nishida, A. Molecular Basis of Intestinal Fibrosis in Inflammatory Bowel Disease. Inflamm. Intest. Dis. 2022, 7, 119–127. [Google Scholar] [CrossRef]
- Akhmetkaliyev, A.; Alibrahim, N.; Shafiee, D.; Tulchinsky, E. EMT/MET Plasticity in Cancer and Go-or-Grow Decisions in Quiescence: The Two Sides of the Same Coin? Mol. Cancer 2023, 22, 90. [Google Scholar] [CrossRef]
- Shen, Y.; Zhang, C.; Chen, Y. TGF-β in Inflammatory Bowel Diseases: A Tale of the Janus-Like Cytokine. Crit. Rev. Eukaryot. Gene Expr. 2015, 25, 335–347. [Google Scholar] [CrossRef]
- Di Gregorio, J.; Sferra, R.; Speca, S.; Vetuschi, A.; Dubuquoy, C.; Desreumaux, P.; Pompili, S.; Cristiano, L.; Gaudio, E.; Flati, V.; et al. Role of Glycogen Synthase Kinase-3β and PPAR-γ on Epithelial-to-Mesenchymal Transition in DSS-Induced Colorectal Fibrosis. PLoS ONE 2017, 12, e0171093. [Google Scholar] [CrossRef]
- Vallée, A.; Lecarpentier, Y.; Guillevin, R.; Vallée, J.-N. Interactions between TGF-Β1, Canonical WNT/β-Catenin Pathway and PPAR γ in Radiation-Induced Fibrosis. Oncotarget 2017, 8, 90579–90604. [Google Scholar] [CrossRef]
- Fang, J.; Wang, H.; Xue, Z.; Cheng, Y.; Zhang, X. PPARγ: The Central Mucus Barrier Coordinator in Ulcerative Colitis. Inflamm. Bowel Dis. 2021, 27, 732–741. [Google Scholar] [CrossRef]
- McCormack, N.; O’Dea, S. Regulation of Epithelial to Mesenchymal Transition by Bone Morphogenetic Proteins. Cell. Signal. 2013, 25, 2856–2862. [Google Scholar] [CrossRef]
- Hatamzade Esfahani, N.; Day, A.S. The Role of TGF-β, Activin and Follistatin in Inflammatory Bowel Disease. Gastrointest. Disord. 2023, 5, 167–186. [Google Scholar] [CrossRef]
- Cicchinelli, S.; Pignataro, G.; Gemma, S.; Piccioni, A.; Picozzi, D.; Ojetti, V.; Franceschi, F.; Candelli, M. PAMPs and DAMPs in Sepsis: A Review of Their Molecular Features and Potential Clinical Implications. Int. J. Mol. Sci. 2024, 25, 962. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef]
- Mukherjee, S.; Karmakar, S.; Babu, S.P.S. TLR2 and TLR4 Mediated Host Immune Responses in Major Infectious Diseases: A Review. Braz. J. Infect. Dis. 2016, 20, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Kelley, K.; Melichian, D.S.; Tamaki, Z.; Fang, F.; Su, Y.; Feng, G.; Pope, R.M.; Budinger, G.R.S.; Mutlu, G.M.; et al. Toll-Like Receptor 4 Signaling Augments Transforming Growth Factor-β Responses. Am. J. Pathol. 2013, 182, 192–205. [Google Scholar] [CrossRef]
- Flannigan, K.L.; Nieves, K.M.; Szczepanski, H.E.; Serra, A.; Lee, J.W.; Alston, L.A.; Ramay, H.; Mani, S.; Hirota, S.A. The Pregnane X Receptor and Indole-3-Propionic Acid Shape the Intestinal Mesenchyme to Restrain Inflammation and Fibrosis. Cell. Mol. Gastroenterol. Hepatol. 2023, 15, 765–795. [Google Scholar] [CrossRef]
- Salminen, S.; Collado, M.C.; Endo, A.; Hill, C.; Lebeer, S.; Quigley, E.M.M.; Sanders, M.E.; Shamir, R.; Swann, J.R.; Szajewska, H.; et al. The International Scientific Association of Probiotics and Prebiotics (ISAPP) Consensus Statement on the Definition and Scope of Postbiotics. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 649–667. [Google Scholar] [CrossRef]
- Akhtar, M.; Chen, Y.; Ma, Z.; Zhang, X.; Shi, D.; Khan, J.A.; Liu, H. Gut Microbiota-Derived Short Chain Fatty Acids Are Potential Mediators in Gut Inflammation. Anim. Nutr. 2022, 8, 350–360. [Google Scholar] [CrossRef]
- Comalada, M.; Bailón, E.; De Haro, O.; Lara-Villoslada, F.; Xaus, J.; Zarzuelo, A.; Gálvez, J. The Effects of Short-Chain Fatty Acids on Colon Epithelial Proliferation and Survival Depend on the Cellular Phenotype. J. Cancer Res. Clin. Oncol. 2006, 132, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Fusco, W.; Lorenzo, M.B.; Cintoni, M.; Porcari, S.; Rinninella, E.; Kaitsas, F.; Lener, E.; Mele, M.C.; Gasbarrini, A.; Collado, M.C.; et al. Short-Chain Fatty-Acid-Producing Bacteria: Key Components of the Human Gut Microbiota. Nutrients 2023, 15, 2211. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Liu, L.; Zhou, W.; Yang, C.; Mai, G.; Li, H.; Chen, Y. Gut Microbiota-Derived Butyrate Regulates Gut Mucus Barrier Repair by Activating the Macrophage/WNT/ERK Signaling Pathway. Clin. Sci. 2022, 136, 291–307. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, Y.; Zhang, B.; Lin, X.; Fu, X.; An, Y.; Zou, Y.; Wang, J.-X.; Wang, Z.; Yu, T. Lactate Metabolism in Human Health and Disease. Signal Transduct. Target. Ther. 2022, 7, 305. [Google Scholar] [CrossRef] [PubMed]
- Garrote, G.L.; Abraham, A.G.; Rumbo, M. Is Lactate an Undervalued Functional Component of Fermented Food Products? Front. Microbiol. 2015, 6, 629. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Pan, C.; Chen, X.; Jing, M.; Xie, J.; Gao, Y.; Huang, J.; Chen, X.; Gao, Y.; Xu, C.; et al. Probiotic Lactic Acid Bacteria Alleviate Pediatric IBD and Remodel Gut Microbiota by Modulating Macrophage Polarization and Suppressing Epithelial Apoptosis. Front. Microbiol. 2023, 14, 1168924. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.; Dokter-Fokkens, J.; Figueroa Lozano, S.; Zhang, Q.; De Haan, B.J.; Zhang, H.; Faas, M.M.; De Vos, P. Lactic Acid Bacteria May Impact Intestinal Barrier Function by Modulating Goblet Cells. Mol. Nutr. Food Res. 2018, 62, 1700572. [Google Scholar] [CrossRef] [PubMed]
- Tennoune, N.; Andriamihaja, M.; Blachier, F. Production of Indole and Indole-Related Compounds by the Intestinal Microbiota and Consequences for the Host: The Good, the Bad, and the Ugly. Microorganisms 2022, 10, 930. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, B.; Hu, Y.; Zhao, Y. New Insights Into Gut-Bacteria-Derived Indole and Its Derivatives in Intestinal and Liver Diseases. Front. Pharmacol. 2021, 12, 769501. [Google Scholar] [CrossRef]
- Lee, J.-H.; Wood, T.K.; Lee, J. Roles of Indole as an Interspecies and Interkingdom Signaling Molecule. Trends Microbiol. 2015, 23, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-G.; Lee, J.-H.; Cho, M.H.; Lee, J. Indole and 3-Indolylacetonitrile Inhibit Spore Maturation in Paenibacillus Alvei. BMC Microbiol. 2011, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Li, H.; Anjum, K.; Zhong, X.; Miao, S.; Zheng, G.; Liu, W.; Li, L. Dual Role of Indoles Derived From Intestinal Microbiota on Human Health. Front. Immunol. 2022, 13, 903526. [Google Scholar] [CrossRef]
- D’Amico, D.; Andreux, P.A.; Valdés, P.; Singh, A.; Rinsch, C.; Auwerx, J. Impact of the Natural Compound Urolithin A on Health, Disease, and Aging. Trends Mol. Med. 2021, 27, 687–699. [Google Scholar] [CrossRef] [PubMed]
- García-Villalba, R.; Giménez-Bastida, J.A.; Cortés-Martín, A.; Ávila-Gálvez, M.Á.; Tomás-Barberán, F.A.; Selma, M.V.; Espín, J.C.; González-Sarrías, A. Urolithins: A Comprehensive Update on Their Metabolism, Bioactivity, and Associated Gut Microbiota. Mol. Nutr. Food Res. 2022, 66, 2101019. [Google Scholar] [CrossRef] [PubMed]
- Abdelazeem, K.N.M.; Kalo, M.Z.; Beer-Hammer, S.; Lang, F. The Gut Microbiota Metabolite Urolithin A Inhibits NF-κB Activation in LPS Stimulated BMDMs. Sci. Rep. 2021, 11, 7117. [Google Scholar] [CrossRef] [PubMed]
- Blachier, F.; Andriamihaja, M.; Larraufie, P.; Ahn, E.; Lan, A.; Kim, E. Production of Hydrogen Sulfide by the Intestinal Microbiota and Epithelial Cells and Consequences for the Colonic and Rectal Mucosa. Am. J. Physiol.-Gastrointest. Liver Physiol. 2021, 320, G125–G135. [Google Scholar] [CrossRef] [PubMed]
- Rabus, R.; Venceslau, S.S.; Wöhlbrand, L.; Voordouw, G.; Wall, J.D.; Pereira, I.A.C. A Post-Genomic View of the Ecophysiology, Catabolism and Biotechnological Relevance of Sulphate-Reducing Prokaryotes. In Advances in Microbial Physiology; Elsevier: Amsterdam, The Netherlands, 2015; Volume 66, pp. 55–321. ISBN 978-0-12-803299-2. [Google Scholar]
- Singh, S.; Lin, H. Hydrogen Sulfide in Physiology and Diseases of the Digestive Tract. Microorganisms 2015, 3, 866–889. [Google Scholar] [CrossRef]
- Mironov, A.; Seregina, T.; Nagornykh, M.; Luhachack, L.G.; Korolkova, N.; Lopes, L.E.; Kotova, V.; Zavilgelsky, G.; Shakulov, R.; Shatalin, K.; et al. Mechanism of H 2 S-Mediated Protection against Oxidative Stress in Escherichia coli. Proc. Natl. Acad. Sci. USA 2017, 114, 6022–6027. [Google Scholar] [CrossRef]
- Ijssennagger, N.; Belzer, C.; Hooiveld, G.J.; Dekker, J.; Van Mil, S.W.C.; Müller, M.; Kleerebezem, M.; Van Der Meer, R. Gut Microbiota Facilitates Dietary Heme-Induced Epithelial Hyperproliferation by Opening the Mucus Barrier in Colon. Proc. Natl. Acad. Sci. USA 2015, 112, 10038–10043. [Google Scholar] [CrossRef] [PubMed]
- Roediger, W.E.W.; Moore, J.; Babidge, W. Colonic Sulfide in Pathogenesis and Treatment of Ulcerative Colitis. Dig. Dis. Sci. 1997, 42, 1571–1579. [Google Scholar] [CrossRef]
- Christl, S.U.; Eisner, H.-D.; Dusel, G.; Kasper, H.; Scheppach, W. Antagonistic Effects of Sulfide and Butyrate on Proliferation of Colonic Mucosa: A Potential Role for These Agents in the Pathogenesis of Ulcerative Colitis. Dig. Dis. Sci. 1996, 41, 2477–2481. [Google Scholar] [CrossRef]
- Hoyles, L.; Jiménez-Pranteda, M.L.; Chilloux, J.; Brial, F.; Myridakis, A.; Aranias, T.; Magnan, C.; Gibson, G.R.; Sanderson, J.D.; Nicholson, J.K.; et al. Metabolic Retroconversion of Trimethylamine N-Oxide and the Gut Microbiota. Microbiome 2018, 6, 73. [Google Scholar] [CrossRef] [PubMed]
- Rath, S.; Heidrich, B.; Pieper, D.H.; Vital, M. Uncovering the Trimethylamine-Producing Bacteria of the Human Gut Microbiota. Microbiome 2017, 5, 54. [Google Scholar] [CrossRef]
- Parada Venegas, D.; De La Fuente, M.K.; Landskron, G.; González, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short Chain Fatty Acids (SCFAs)-Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front. Immunol. 2019, 10, 277. [Google Scholar] [CrossRef]
- Xu, H.-M.; Zhao, H.-L.; Guo, G.-J.; Xu, J.; Zhou, Y.-L.; Huang, H.-L.; Nie, Y.-Q. Characterization of Short-Chain Fatty Acids in Patients with Ulcerative Colitis: A Meta-Analysis. BMC Gastroenterol. 2022, 22, 117. [Google Scholar] [CrossRef]
- Scheppach, W.; Sommer, H.; Kirchner, T.; Paganelli, G.-M.; Bartram, P.; Christl, S.; Richter, F.; Dusel, G.; Kasper, H. Effect of Butyrate Enemas on the Colonic Mucosa in Distal Ulcerative Colitis. Gastroenterology 1992, 103, 51–56. [Google Scholar] [CrossRef]
- Vieira, E.L.M.; Leonel, A.J.; Sad, A.P.; Beltrão, N.R.M.; Costa, T.F.; Ferreira, T.M.R.; Gomes-Santos, A.C.; Faria, A.M.C.; Peluzio, M.C.G.; Cara, D.C.; et al. Oral Administration of Sodium Butyrate Attenuates Inflammation and Mucosal Lesion in Experimental Acute Ulcerative Colitis. J. Nutr. Biochem. 2012, 23, 430–436. [Google Scholar] [CrossRef]
- Di Sabatino, A.; Cazzola, P.; Ciccocioppo, R.; Morera, R.; Biancheri, P.; Rovedatti, L.; Cantoro, L.; Vanoli, A.; Tinozzi, F.P.; Tinozzi, S.; et al. Efficacy of Butyrate in the Treatment of Mild to Moderate Crohn’s Disease. Dig. Liver Dis. Suppl. 2007, 1, 31–35. [Google Scholar] [CrossRef]
- Geirnaert, A.; Calatayud, M.; Grootaert, C.; Laukens, D.; Devriese, S.; Smagghe, G.; De Vos, M.; Boon, N.; Van De Wiele, T. Butyrate-Producing Bacteria Supplemented in Vitro to Crohn’s Disease Patient Microbiota Increased Butyrate Production and Enhanced Intestinal Epithelial Barrier Integrity. Sci. Rep. 2017, 7, 11450. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Vitetta, L. Butyrate in Inflammatory Bowel Disease Therapy. Gastroenterology 2020, 158, 1511. [Google Scholar] [CrossRef]
- Singh, V.; Lee, G.; Son, H.; Koh, H.; Kim, E.S.; Unno, T.; Shin, J.-H. Butyrate Producers, “The Sentinel of Gut”: Their Intestinal Significance with and beyond Butyrate, and Prospective Use as Microbial Therapeutics. Front. Microbiol. 2023, 13, 1103836. [Google Scholar] [CrossRef]
- Han, Y.; Zhao, Q.; Tang, C.; Li, Y.; Zhang, K.; Li, F.; Zhang, J. Butyrate Mitigates Weanling Piglets From Lipopolysaccharide-Induced Colitis by Regulating Microbiota and Energy Metabolism of the Gut–Liver Axis. Front. Microbiol. 2020, 11, 588666. [Google Scholar] [CrossRef] [PubMed]
- Souders, C.L.; Aristizabal-Henao, J.J.; Patuel, S.J.; Bowden, J.A.; Zubcevic, J.; Martyniuk, C.J. Interaction between Butyrate and Tumor Necrosis Factor α in Primary Rat Colonocytes. Biomolecules 2023, 13, 258. [Google Scholar] [CrossRef]
- Matsumoto, N.; Riley, S.; Fraser, D.; Al-Assaf, S.; Ishimura, E.; Wolever, T.; Phillips, G.O.; Phillips, A.O. Butyrate Modulates TGF-Β1 Generation and Function: Potential Renal Benefit for Acacia(Sen) SUPERGUMTM (Gum Arabic)? Kidney Int. 2006, 69, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, Y.-P.; Chen, H.-L.; Tsai, J.-N.; Lin, M.-Y.; Liao, J.-W.; Wei, M.-S.; Ko, J.-L.; Ou, C.-C. Administration of Lactobacillus Reuteri Combined with Clostridium Butyricum Attenuates Cisplatin-Induced Renal Damage by Gut Microbiota Reconstitution, Increasing Butyric Acid Production, and Suppressing Renal Inflammation. Nutrients 2021, 13, 2792. [Google Scholar] [CrossRef]
- Cheng, X.; Zhou, T.; He, Y.; Xie, Y.; Xu, Y.; Huang, W. The Role and Mechanism of Butyrate in the Prevention and Treatment of Diabetic Kidney Disease. Front. Microbiol. 2022, 13, 961536. [Google Scholar] [CrossRef]
- Li, X.; Li, R.; You, N.; Zhao, X.; Li, J.; Jiang, W. Butyric Acid Ameliorates Myocardial Fibrosis by Regulating M1/M2 Polarization of Macrophages and Promoting Recovery of Mitochondrial Function. Front. Nutr. 2022, 9, 875473. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, J.; Li, L.; Zhang, H.; Pang, D.; Ouyang, H.; Jin, X.; Tang, X. Clostridium Butyricum and Bifidobacterium Pseudolongum Attenuate the Development of Cardiac Fibrosis in Mice. Microbiol. Spectr. 2022, 10, e02524-22. [Google Scholar] [CrossRef]
- Alhusaini, A.M.; Alsoghayer, R.; Alhushan, L.; Alanazi, A.M.; Hasan, I.H. Acetyl-L-Carnitine and Liposomal Co-Enzyme Q10 Attenuate Hepatic Inflammation, Apoptosis, and Fibrosis Induced by Propionic Acid. Int. J. Mol. Sci. 2023, 24, 11519. [Google Scholar] [CrossRef] [PubMed]
- Gart, E.; Van Duyvenvoorde, W.; Toet, K.; Caspers, M.P.M.; Verschuren, L.; Nielsen, M.J.; Leeming, D.J.; Souto Lima, E.; Menke, A.; Hanemaaijer, R.; et al. Butyrate Protects against Diet-Induced NASH and Liver Fibrosis and Suppresses Specific Non-Canonical TGF-β Signaling Pathways in Human Hepatic Stellate Cells. Biomedicines 2021, 9, 1954. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Deng, M.; Gong, J.; Zhang, X.; Ge, S.; Zhao, L. Sodium Acetate Inhibit TGF-Β1-Induced Activation of Hepatic Stellate Cells by Restoring AMPK or c-Jun Signaling. Front. Nutr. 2021, 8, 729583. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; You, H.J.; Bajaj, J.S.; Joo, S.K.; Yu, J.; Park, S.; Kang, H.; Park, J.H.; Kim, J.H.; Lee, D.H.; et al. Distinct Signatures of Gut Microbiome and Metabolites Associated with Significant Fibrosis in Non-Obese NAFLD. Nat. Commun. 2020, 11, 4982. [Google Scholar] [CrossRef] [PubMed]
- Thing, M.; Werge, M.P.; Kimer, N.; Hetland, L.E.; Rashu, E.B.; Nabilou, P.; Junker, A.E.; Galsgaard, E.D.; Bendtsen, F.; Laupsa-Borge, J.; et al. Targeted Metabolomics Reveals Plasma Short-Chain Fatty Acids Are Associated with Metabolic Dysfunction-Associated Steatotic Liver Disease. BMC Gastroenterol. 2024, 24, 43. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Qiu, Y.; Gao, Z.; Wu, Y.-X.; Wan, B.; Liu, G.; Chen, J.; Zhou, Q.; Yu, R.; Pang, Q. Sodium Propionate Attenuates the Lipopolysaccharide-Induced Epithelial–Mesenchymal Transition via the PI3K/Akt/mTOR Signaling Pathway. J. Agric. Food Chem. 2020, 68, 6554–6563. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, D.; Kamada, N. Contribution of the Gut Microbiota to Intestinal Fibrosis in Crohn’s Disease. Front. Med. 2022, 9, 826240. [Google Scholar] [CrossRef]
- Zhou, T.; Xu, H.; Cheng, X.; He, Y.; Ren, Q.; Li, D.; Xie, Y.; Gao, C.; Zhang, Y.; Sun, X.; et al. Sodium Butyrate Attenuates Diabetic Kidney Disease Partially via Histone Butyrylation Modification. Mediat. Inflamm. 2022, 2022, 7643322. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; Guan, X.; Wang, X.; Qu, S.; Zhang, S.; Hui, W.; Men, L.; Chen, X. Dynamic Modulation of Gut Microbiota Improves Post-Myocardial Infarct Tissue Repair in Rats via Butyric Acid-Mediated Histone Deacetylase Inhibition. FASEB J. 2021, 35, e21385. [Google Scholar] [CrossRef] [PubMed]
- Gasaly, N.; Hermoso, M.A.; Gotteland, M. Butyrate and the Fine-Tuning of Colonic Homeostasis: Implication for Inflammatory Bowel Diseases. Int. J. Mol. Sci. 2021, 22, 3061. [Google Scholar] [CrossRef]
- Li, Y.J.; Ma, J.; Loh, Y.W.; Chadban, S.J.; Wu, H. Short-Chain Fatty Acids Directly Exert Anti-Inflammatory Responses in Podocytes and Tubular Epithelial Cells Exposed to High Glucose. Front. Cell Dev. Biol. 2023, 11, 1182570. [Google Scholar] [CrossRef]
- Lin, C.-J.; Cheng, Y.-C.; Chen, H.-C.; Chao, Y.-K.; Nicholson, M.W.; Yen, E.C.L.; Kamp, T.J.; Hsieh, P.C.H. Commensal Gut Microbiota-Derived Acetate and Propionate Enhance Heart Adaptation in Response to Cardiac Pressure Overload in Mice. Theranostics 2022, 12, 7319–7334. [Google Scholar] [CrossRef]
- Aoki, R.; Onuki, M.; Hattori, K.; Ito, M.; Yamada, T.; Kamikado, K.; Kim, Y.-G.; Nakamoto, N.; Kimura, I.; Clarke, J.M.; et al. Commensal Microbe-Derived Acetate Suppresses NAFLD/NASH Development via Hepatic FFAR2 Signalling in Mice. Microbiome 2021, 9, 188. [Google Scholar] [CrossRef]
- Martin-Gallausiaux, C.; Béguet-Crespel, F.; Marinelli, L.; Jamet, A.; Ledue, F.; Blottière, H.M.; Lapaque, N. Butyrate Produced by Gut Commensal Bacteria Activates TGF-Beta1 Expression through the Transcription Factor SP1 in Human Intestinal Epithelial Cells. Sci. Rep. 2018, 8, 9742. [Google Scholar] [CrossRef] [PubMed]
- Byndloss, M.X.; Olsan, E.E.; Rivera-Chávez, F.; Tiffany, C.R.; Cevallos, S.A.; Lokken, K.L.; Torres, T.P.; Byndloss, A.J.; Faber, F.; Gao, Y.; et al. Microbiota-Activated PPAR-γ Signaling Inhibits Dysbiotic Enterobacteriaceae Expansion. Science 2017, 357, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, Q.; Zhu, Y.; Zhang, Y.; Xia, Y.; Wei, Z.; Dai, Y. Rectal Administration of Butyrate Ameliorates Pulmonary Fibrosis in Mice through Induction of Hepatocyte Growth Factor in the Colon via the HDAC-PPARγ Pathway. Life Sci. 2022, 309, 120972. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.; He, L.; Zhong, Z.; Zhao, R.; Weng, S.; Mi, H.; Liu, F. Stigmasterol Restores the Balance of Treg/Th17 Cells by Activating the Butyrate-PPARγ Axis in Colitis. Front. Immunol. 2021, 12, 741934. [Google Scholar] [CrossRef] [PubMed]
- Simeoli, R.; Mattace Raso, G.; Pirozzi, C.; Lama, A.; Santoro, A.; Russo, R.; Montero-Melendez, T.; Berni Canani, R.; Calignano, A.; Perretti, M.; et al. An Orally Administered Butyrate-releasing Derivative Reduces Neutrophil Recruitment and Inflammation in Dextran Sulphate Sodium-induced Murine Colitis. Br. J. Pharmacol. 2017, 174, 1484–1496. [Google Scholar] [CrossRef] [PubMed]
- Ruan, G.; Chen, M.; Chen, L.; Xu, F.; Xiao, Z.; Yi, A.; Tian, Y.; Ping, Y.; Lv, L.; Cheng, Y.; et al. Roseburia Intestinalis and Its Metabolite Butyrate Inhibit Colitis and Upregulate TLR5 through the SP3 Signaling Pathway. Nutrients 2022, 14, 3041. [Google Scholar] [CrossRef]
- Cao, Y.; Gao, X.; Zhang, W.; Zhang, G.; Nguyen, A.K.; Liu, X.; Jimenez, F.; Cox, C.S.; Townsend, C.M.; Ko, T.C. Dietary Fiber Enhances TGF-β Signaling and Growth Inhibition in the Gut. Am. J. Physiol.-Gastrointest. Liver Physiol. 2011, 301, G156–G164. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Dong, Y.; Li, M.; Zhang, W.; Ding, Y.; Wang, X.; Chen, D.; Liu, T.; Wang, B.; Cao, H.; et al. Clostridium butyricum Inhibits Epithelial–Mesenchymal Transition of Intestinal Carcinogenesis through Downregulating METTL3. Cancer Sci. 2023, 114, 3114–3127. [Google Scholar] [CrossRef]
- Saez-Lara, M.J.; Gomez-Llorente, C.; Plaza-Diaz, J.; Gil, A. The Role of Probiotic Lactic Acid Bacteria and Bifidobacteria in the Prevention and Treatment of Inflammatory Bowel Disease and Other Related Diseases: A Systematic Review of Randomized Human Clinical Trials. BioMed Res. Int. 2015, 2015, 505878. [Google Scholar] [CrossRef]
- Sun, S.; Xu, X.; Liang, L.; Wang, X.; Bai, X.; Zhu, L.; He, Q.; Liang, H.; Xin, X.; Wang, L.; et al. Lactic Acid-Producing Probiotic Saccharomyces Cerevisiae Attenuates Ulcerative Colitis via Suppressing Macrophage Pyroptosis and Modulating Gut Microbiota. Front. Immunol. 2021, 12, 777665. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Jiang, L.; Han, D.; Si, D.; Sun, Z.; Wu, Z.; Dai, Z. Limosilactobacillus Mucosae and Lactobacillus Amylovorus Protect Against Experimental Colitis via Upregulation of Colonic 5-Hydroxytryptamine Receptor 4 and Transforming Growth Factor-Β2. J. Nutr. 2023, 153, 2512–2522. [Google Scholar] [CrossRef] [PubMed]
- Kottmann, R.M.; Kulkarni, A.A.; Smolnycki, K.A.; Lyda, E.; Dahanayake, T.; Salibi, R.; Honnons, S.; Jones, C.; Isern, N.G.; Hu, J.Z.; et al. Lactic Acid Is Elevated in Idiopathic Pulmonary Fibrosis and Induces Myofibroblast Differentiation via pH-Dependent Activation of Transforming Growth Factor-β. Am. J. Respir. Crit. Care Med. 2012, 186, 740–751. [Google Scholar] [CrossRef] [PubMed]
- Cervantes-García, D.; Jiménez, M.; Rivas-Santiago, C.E.; Gallegos-Alcalá, P.; Hernández-Mercado, A.; Santoyo-Payán, L.S.; Loera-Arias, M.D.J.; Saucedo-Cardenas, O.; Montes De Oca-Luna, R.; Salinas, E. Lactococcus lactis NZ9000 Prevents Asthmatic Airway Inflammation and Remodelling in Rats through the Improvement of Intestinal Barrier Function and Systemic TGF-β Production. Int. Arch. Allergy Immunol. 2021, 182, 277–291. [Google Scholar] [CrossRef]
- Jantararussamee, C.; Rodniem, S.; Taweechotipatr, M.; Showpittapornchai, U.; Pradidarcheep, W. Hepatoprotective Effect of Probiotic Lactic Acid Bacteria on Thioacetamide-Induced Liver Fibrosis in Rats. Probiotics Antimicrob. Proteins 2021, 13, 40–50. [Google Scholar] [CrossRef]
- Park, J.-Y.; Lee, J.Y.; Kim, Y.; Kang, C.-H. Lactic Acid Bacteria Improve the Photoprotective Effect via MAPK/AP-1/MMP Signaling Pathway on Skin Fibroblasts. Microorganisms 2022, 10, 2481. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Li, Y.; Xie, X.; Chen, M.; Xue, L.; Wang, J.; Ye, Q.; Wu, S.; Yang, R.; Zhao, H.; et al. Exploration of the Molecular Mechanisms Underlying the Anti-Photoaging Effect of Limosilactobacillus Fermentum XJC60. Front. Cell. Infect. Microbiol. 2022, 12, 838060. [Google Scholar] [CrossRef] [PubMed]
- Sakai, F.; Hosoya, T.; Ono-Ohmachi, A.; Ukibe, K.; Ogawa, A.; Moriya, T.; Kadooka, Y.; Shiozaki, T.; Nakagawa, H.; Nakayama, Y.; et al. Lactobacillus Gasseri SBT2055 Induces TGF-β Expression in Dendritic Cells and Activates TLR2 Signal to Produce IgA in the Small Intestine. PLoS ONE 2014, 9, e105370. [Google Scholar] [CrossRef]
- Madjirebaye, P.; Peng, F.; Mueed, A.; Huang, T.; Mahamat, B.; Pahane, M.M.; Xi, Q.; Chen, X.; Moussa, K.; Kadebe, Z.T.; et al. Exploring Impact of Probiotic-Fermented Soymilk on Dextran-Sulfate-Sodium-Induced Ulcerative Colitis via Modulating Inflammation and Gut Microbiota Profile. Mol. Nutr. Food Res. 2024, 68, 2300586. [Google Scholar] [CrossRef] [PubMed]
- Remund, B.; Yilmaz, B.; Sokollik, C. D-Lactate: Implications for Gastrointestinal Diseases. Children 2023, 10, 945. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Liu, C.; Li, R.; Zheng, M.; Feng, B.; Gao, J.; Long, X.; Li, L.; Li, S.; Zuo, X.; et al. Lactobacillus-Derived Indole-3-Lactic Acid Ameliorates Colitis in Cesarean-Born Offspring via Activation of Aryl Hydrocarbon Receptor. iScience 2023, 26, 108279. [Google Scholar] [CrossRef]
- Cui, Q.; Zhang, Z.; Tian, X.; Liang, X.; Lu, Y.; Shi, Y.; Kuerman, M.; Wang, R.; Zhou, Y.; Gong, P.; et al. Bifidobacterium bifidum Ameliorates DSS-Induced Colitis in Mice by Regulating AHR/NRF2/NLRP3 Inflammasome Pathways through Indole-3-Lactic Acid Production. J. Agric. Food Chem. 2023, 71, 1970–1981. [Google Scholar] [CrossRef]
- Wang, S.; Van Schooten, F.-J.; Jin, H.; Jonkers, D.; Godschalk, R. The Involvement of Intestinal Tryptophan Metabolism in Inflammatory Bowel Disease Identified by a Meta-Analysis of the Transcriptome and a Systematic Review of the Metabolome. Nutrients 2023, 15, 2886. [Google Scholar] [CrossRef] [PubMed]
- Qazi, A.; Comiskey, S.; Calzadilla, N.; Amin, F.; Sharma, A.; Khin, E.; Holton, N.; Weber, C.R.; Saksena, S.; Kumar, A.; et al. Potential Dietary and Therapeutic Strategies Involving Indole-3-Carbinole in Preclinical Models of Intestinal Inflammation. Nutrients 2023, 15, 4980. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Kim, Y.; Park, S.; Lee, K.W.; Park, T. Indole-3-Carbinol Prevents Diet-Induced Obesity through Modulation of Multiple Genes Related to Adipogenesis, Thermogenesis or Inflammation in the Visceral Adipose Tissue of Mice. J. Nutr. Biochem. 2012, 23, 1732–1739. [Google Scholar] [CrossRef] [PubMed]
- Yisireyili, M.; Takeshita, K.; Saito, S.; Murohara, T.; Niwa, T. Indole-3-Propionic Acid Suppresses Indoxyl Sulfate-Induced Expression of Fibrotic and Inflammatory Genes in Proximal Tubular Cells. Nagoya J. Med. Sci. 2017, 79, 477–486. [Google Scholar] [PubMed]
- Singh, N.P.; Singh, U.P.; Rouse, M.; Zhang, J.; Chatterjee, S.; Nagarkatti, P.S.; Nagarkatti, M. Dietary Indoles Suppress Delayed-Type Hypersensitivity by Inducing a Switch from Proinflammatory Th17 Cells to Anti-Inflammatory Regulatory T Cells through Regulation of MicroRNA. J. Immunol. 2016, 196, 1108–1122. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Torigoe, K.; Torigoe, M.; Muta, K.; Obata, Y.; Suzuki, T.; Suzuki, C.; Abe, T.; Koji, T.; Mukae, H.; et al. Mitochonic Acid-5 Ameliorates Chlorhexidine Gluconate-Induced Peritoneal Fibrosis in Mice. Med. Mol. Morphol. 2022, 55, 27–40. [Google Scholar] [CrossRef]
- Liu, F.; Sun, C.; Chen, Y.; Du, F.; Yang, Y.; Wu, G. Indole-3-Propionic Acid-Aggravated CCl4-Induced Liver Fibrosis via the TGF-Β1/Smads Signaling Pathway. J. Clin. Transl. Hepatol. 2021, 9, 917–930. [Google Scholar] [CrossRef]
- Shima, H.; Sasaki, K.; Suzuki, T.; Mukawa, C.; Obara, T.; Oba, Y.; Matsuo, A.; Kobayashi, T.; Mishima, E.; Watanabe, S.; et al. A Novel Indole Compound MA-35 Attenuates Renal Fibrosis by Inhibiting Both TNF-α and TGF-Β1 Pathways. Sci. Rep. 2017, 7, 1884. [Google Scholar] [CrossRef]
- Sehgal, R.; Ilha, M.; Vaittinen, M.; Kaminska, D.; Männistö, V.; Kärjä, V.; Tuomainen, M.; Hanhineva, K.; Romeo, S.; Pajukanta, P.; et al. Indole-3-Propionic Acid, a Gut-Derived Tryptophan Metabolite, Associates with Hepatic Fibrosis. Nutrients 2021, 13, 3509. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Pei, J.; Wang, X.; Tai, S.; Tang, L.; Hu, X. Gut Bacterial Metabolite Urolithin A Inhibits Myocardial Fibrosis through Activation of Nrf2 Pathway In Vitro and In Vivo. Mol. Med. 2022, 28, 19. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Tu, J.; Zhang, H.; Zhang, Y.; Zhou, B. Urolithin A Attenuates Renal Fibrosis by Inhibiting TGF-Β1/Smad and MAPK Signaling Pathways. J. Funct. Foods 2021, 83, 104547. [Google Scholar] [CrossRef]
- Duan, J.; Pan, J.; Sun, M.; Fang, Y. Comparative Multiomics Study of the Effects of Ellagic Acid on the Gut Environment in Young and Adult Mice. Food Res. Int. 2022, 161, 111819. [Google Scholar] [CrossRef]
- Cheng, F.; Dou, J.; Zhang, Y.; Wang, X.; Wei, H.; Zhang, Z.; Cao, Y.; Wu, Z. Urolithin A Inhibits Epithelial–Mesenchymal Transition in Lung Cancer Cells via P53-Mdm2-Snail Pathway. OncoTargets Ther. 2021, 14, 3199–3208. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Yan, R.; Zhou, X.; Ji, F.; Zhang, B. Hydrogen Sulfide Improves Colonic Barrier Integrity in DSS-Induced Inflammation in Caco-2 Cells and Mice. Int. Immunopharmacol. 2016, 39, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Mottawea, W.; Chiang, C.-K.; Mühlbauer, M.; Starr, A.E.; Butcher, J.; Abujamel, T.; Deeke, S.A.; Brandel, A.; Zhou, H.; Shokralla, S.; et al. Altered Intestinal Microbiota–Host Mitochondria Crosstalk in New Onset Crohn’s Disease. Nat. Commun. 2016, 7, 13419. [Google Scholar] [CrossRef] [PubMed]
- Chirindoth, S.S.; Cancarevic, I. Role of Hydrogen Sulfide in the Treatment of Fibrosis. Cureus 2021, 13, e18088. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Shi, X.; Wang, H.; Fan, J.; Feng, Y.; Lin, X.; Yang, J.; Cui, Q.; Tang, C.; Xu, G.; et al. Cystathionine γ Lyase–Hydrogen Sulfide Increases Peroxisome Proliferator-Activated Receptor γ Activity by Sulfhydration at C139 Site Thereby Promoting Glucose Uptake and Lipid Storage in Adipocytes. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2016, 1861, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Meng, J.; Wang, C.; Yang, C.; Wang, Y.; Li, Y.; Li, Y. Hydrogen Sulfide Alleviates Cigarette Smoke-Induced COPD through Inhibition of the TGF-β1/Smad Pathway. Exp. Biol. Med. 2020, 245, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Geng, J.; Zhao, J.; Ni, Q.; Zhao, C.; Zheng, Y.; Chen, X.; Wang, L. Trimethylamine N-Oxide Exacerbates Cardiac Fibrosis via Activating the NLRP3 Inflammasome. Front. Physiol. 2019, 10, 866. [Google Scholar] [CrossRef]
- Yang, W.; Zhang, S.; Zhu, J.; Jiang, H.; Jia, D.; Ou, T.; Qi, Z.; Zou, Y.; Qian, J.; Sun, A.; et al. Gut Microbe-Derived Metabolite Trimethylamine N-Oxide Accelerates Fibroblast-Myofibroblast Differentiation and Induces Cardiac Fibrosis. J. Mol. Cell. Cardiol. 2019, 134, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Kong, B.; Shuai, W.; Fu, H.; Jiang, X.; Huang, H. 3,3-Dimethyl-1-Butanol Attenuates Cardiac Remodeling in Pressure-Overload-Induced Heart Failure Mice. J. Nutr. Biochem. 2020, 78, 108341. [Google Scholar] [CrossRef]
- Moludi, J.; Saiedi, S.; Ebrahimi, B.; Alizadeh, M.; Khajebishak, Y.; Ghadimi, S.S. Probiotics Supplementation on Cardiac Remodeling Following Myocardial Infarction: A Single-Center Double-Blind Clinical Study. J. Cardiovasc. Trans. Res. 2021, 14, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, T.; Wu, H.; Shi, H.; Bai, J.; Zhao, W.; Jiang, D.; Jiang, X. Lactobacillus Rhamnosus GG Strain Mitigated the Development of Obstructive Sleep Apnea-Induced Hypertension in a High Salt Diet via Regulating TMAO Level and CD4+ T Cell Induced-Type I Inflammation. Biomed. Pharmacother. 2019, 112, 108580. [Google Scholar] [CrossRef]
- Kapetanaki, S.; Kumawat, A.K.; Persson, K.; Demirel, I. The Fibrotic Effects of TMAO on Human Renal Fibroblasts Is Mediated by NLRP3, Caspase-1 and the PERK/Akt/mTOR Pathway. Int. J. Mol. Sci. 2021, 22, 11864. [Google Scholar] [CrossRef] [PubMed]
- Fang, Q.; Zheng, B.; Liu, N.; Liu, J.; Liu, W.; Huang, X.; Zeng, X.; Chen, L.; Li, Z.; Ouyang, D. Trimethylamine N-Oxide Exacerbates Renal Inflammation and Fibrosis in Rats With Diabetic Kidney Disease. Front. Physiol. 2021, 12, 682482. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Miikeda, A.; Zuckerman, J.; Jia, X.; Charugundla, S.; Zhou, Z.; Kaczor-Urbanowicz, K.E.; Magyar, C.; Guo, F.; Wang, Z.; et al. Inhibition of Microbiota-Dependent TMAO Production Attenuates Chronic Kidney Disease in Mice. Sci. Rep. 2021, 11, 518. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-J.; Bale, S.; Verma, P.; Wan, Q.; Ma, F.; Gudjonsson, J.E.; Hazen, S.L.; Harms, P.W.; Tsou, P.-S.; Khanna, D.; et al. Gut Microbe-Derived Metabolite Trimethylamine N-Oxide Activates PERK to Drive Fibrogenic Mesenchymal Differentiation. iScience 2022, 25, 104669. [Google Scholar] [CrossRef]
- Wang, Q.; Sun, Y.; Zhou, T.; Jiang, C.; A, L.; Xu, W. Gut Microbiota-Dependent Trimethylamine n-Oxide Pathway Contributes to the Bidirectional Relationship between Intestinal Inflammation and Periodontitis. Front. Cell. Infect. Microbiol. 2023, 12, 1125463. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Buffa, J.A.; Roberts, A.B.; Sangwan, N.; Skye, S.M.; Li, L.; Ho, K.J.; Varga, J.; DiDonato, J.A.; Tang, W.H.W.; et al. Targeted Inhibition of Gut Microbial Trimethylamine N-Oxide Production Reduces Renal Tubulointerstitial Fibrosis and Functional Impairment in a Murine Model of Chronic Kidney Disease. Arter. Thromb. Vasc. Biol. 2020, 40, 1239–1255. [Google Scholar] [CrossRef]
- Organ, C.L.; Li, Z.; Sharp, T.E.; Polhemus, D.J.; Gupta, N.; Goodchild, T.T.; Tang, W.H.W.; Hazen, S.L.; Lefer, D.J. Nonlethal Inhibition of Gut Microbial Trimethylamine N-oxide Production Improves Cardiac Function and Remodeling in a Murine Model of Heart Failure. J. Am. Heart Assoc. 2020, 9, e016223. [Google Scholar] [CrossRef]
- Massey, W.; Mukherjee, P.; Nguyen, Q.T.; Mrdjen, M.; Wang, Z.; Brown, J.M.; Rieder, F. THE PATHOGENIC ROLE OF MICROBIAL TRIMETHYLAMINE IN IBD ASSOCIATED INTESTINAL FIBROSIS. Inflamm. Bowel Dis. 2024, 30, S64. [Google Scholar] [CrossRef]
- Jalandra, R.; Makharia, G.K.; Sharma, M.; Kumar, A. Inflammatory and Deleterious Role of Gut Microbiota-Derived Trimethylamine on Colon Cells. Front. Immunol. 2023, 13, 1101429. [Google Scholar] [CrossRef]
- Banno, Y.; Nomura, M.; Hara, R.; Asami, M.; Tanaka, K.; Mukai, Y.; Tomata, Y. Trimethylamine N-Oxide and Risk of Inflammatory Bowel Disease: A Mendelian Randomization Study. Medicine 2023, 102, e34758. [Google Scholar] [CrossRef]
- Wilson, A.; Teft, W.A.; Morse, B.L.; Choi, Y.-H.; Woolsey, S.; DeGorter, M.K.; Hegele, R.A.; Tirona, R.G.; Kim, R.B. Trimethylamine-N-Oxide: A Novel Biomarker for the Identification of Inflammatory Bowel Disease. Dig. Dis. Sci. 2015, 60, 3620–3630. [Google Scholar] [CrossRef] [PubMed]
- Bunt, D.; Minnaard, A.; El Aidy, S. Potential Modulatory Microbiome Therapies for Prevention or Treatment of Inflammatory Bowel Diseases. Pharmaceuticals 2021, 14, 506. [Google Scholar] [CrossRef]
- Zhang, Q.; Yu, N.; Lee, C. Mysteries of TGF-Î2 Paradox in Benign and Malignant Cells. Front. Oncol. 2014, 4, 94. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, G.P.; Lee, S.M.; Mazmanian, S.K. Gut Biogeography of the Bacterial Microbiota. Nat. Rev. Microbiol. 2016, 14, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Stenke, E.; Bourke, B.; Knaus, U. Crohn’s Strictures—Moving Away from the Knife. Front. Pediatr. 2017, 5, 141. [Google Scholar] [CrossRef] [PubMed]
- Golob, J.; Rao, K.; Berinstein, J.; Chey, W.; Owyang, C.; Kamada, N.; Higgins, P.; Young, V.; Bishu, S.; Lee, A. The Fecal Microbiome in Quiescent Crohn’s Disease with Persistent Gastrointestinal Symptoms Show Enrichment of Oral Microbes But Depletion of Butyrate and Indole Producers. medRxiv 2023. [Google Scholar] [CrossRef]
- Shashni, B.; Tajika, Y.; Ikeda, Y.; Nishikawa, Y.; Nagasaki, Y. Self-Assembling Polymer-Based Short Chain Fatty Acid Prodrugs Ameliorate Non-Alcoholic Steatohepatitis and Liver Fibrosis. Biomaterials 2023, 295, 122047. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Hao, W.; Wang, X.; Zhou, R.; Lin, Q. Probiotics for the Treatment of Ulcerative Colitis: A Review of Experimental Research from 2018 to 2022. Front. Microbiol. 2023, 14, 1211271. [Google Scholar] [CrossRef] [PubMed]

| Phylum | Class | Order | Family | Genus | Species | CD | Ref. | UC | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Firmicutes | Clostridia | ↓ | [9] | ↓ | [9] | ||||
| Clostridiales | Lachnospiraceae | Roseburia | R. hominis | ↓ | [10] | ||||
| R. intestinalis | ↓ | [11] | ↓ | [11] | |||||
| Ruminococcus | R. albus | ↓ | [12] | ||||||
| R. callidus | ↓ | [12] | |||||||
| R. bromii | ↓ | [12] | |||||||
| R. gnavus | ↑ | [13] | ↑ | [13] | |||||
| R. torques | ↑ | [13] | ↑ | [13] | |||||
| Acidaminococcaceae | Dialister | D. invisus | ↓ | [14] | |||||
| Eubacteriaceae | Eubacterium | E. rectale | ↓ | [12] | |||||
| Clostridiaceae | Clostridium | C. difficile | ↑ | [12] | |||||
| C. coccoides | ↓ | [15] | ↓ | [15,16] | |||||
| C. leptum | ↓ | [12,15,16] | ↓ | [15] | |||||
| Faecalibacterium | F. prausnitzii | ↑ | [17] | ↓ | [10,11,15] | ||||
| ↓ | [11,12,14,15] | ||||||||
| Bacilli | ↑ | [9] | ↑ | [9] | |||||
| Bacillales | Listeriaceae | Listeria | ↑ | [12] | |||||
| Lactobacillales | Enterococcaceae | Enterococcus | ↑ | [12] | |||||
| Lactobacillaceae | Lactobacillus | ↑ ↓ | [12,18] [19] | ↓ | [20] | ||||
| Bacteroidetes | Bacteroidetes | ↓ | [9] | ↓ | [9] | ||||
| Bacteroidales | Bacteroidaceae | Bacteroides | B. fragilis | ↓ | [11,12] | ↓ | [11] | ||
| ↑ | [21] | ||||||||
| B. vulgatus | ↓ | [11,12] | ↓ | [11] | |||||
| ↑ | [21] | ||||||||
| Actinobacteria | Actinobacteria | ↑ | [9] | ↓ | [22] | ||||
| Bifidobacteriales | Bifidobacteriaceae | Bifidobacterium | B. longum | ↑ | [11] | ||||
| B. bifidum | ↓ | [22] | |||||||
| Proteobacteria | ↑ | [9] | ↑ | [9] | |||||
| δ | Desulfovibrionales | Desulfovibrionaceae | Desulfovibrio | ↑ | [23] | ||||
| γ | Enterobacteriales | Enterobacteriaceae | Escherichia | ↑ | [11,21] | ||||
| Shigella | ↑ | [11] | |||||||
| S. flexneri | ↑ | [12] | |||||||
| Pseudomonadales | Moraxellaceae | Acinetobacter | A. junii | ↑ | [21] | ||||
| Verrucomicrobia | Verrucomicrobiae | ↓ | [13,24] | ↓ | [13,24] | ||||
| Verrucomicrobiales | Verrucomicrobiaceae | Akkermansia | A. muciniphila | ↓ | [13,24] | ↓ | [13,24] |
| Main Effects on the Parts of the Gut Barrier | |||||||
|---|---|---|---|---|---|---|---|
| Microbial Metabolites | Precursor | Species Involved in the Metabolism | Microbiota | Mucus | Epithelium | IIS | Ref. |
Short-chain fatty acids (SCFAs)
| Non-digestible dietary fibers, amino acids, and lactate. |
| SCFAs interact with other bacteria such as Lactobacilli and Bifidobacteria, enhancing their growth. | SCFAs stimulate goblet cells and induce the MUC2 gene. | SCFAs are the principal energetic source for colonocytes and contribute to the integrity of the APC. | SCFAs regulate TLR and FFAR activation, the differentiation of Tregs, and IL-10 secretion. | [70,71,72,73] |
| Lactic acid (LA) | Fermented foods: carbohydrate fermentation. | “LAB”, Gram-positive catalase-negative bacteria resistant to low pH, mainly belonging to the Lactobacillus genus. | LAB produce bacteriocins, peptides involved in the mucosal defense. | Various strains of LAB differently affect goblet cell functions and the expression of mucus-related genes, MUC2 included. | LA promotes the TCA for energy production, maintains the cellular redox state, stimulates the ACC for fatty acid synthesis, and contributes to normal epithelial proliferation. | LAB administration promotes macrophage M2 polarization and a reduction in pro-inflammatory cytokines (e.g., IL-1β and IL-6) | [74,75,76,77] |
| Indoles | Tryptophan, the essential amino acid found in meat, fish, dairy, eggs, nuts, seeds, legumes, and whole grains. | Tryptophanase-expressing bacteria, such as Clostridium, Bacteroides, Lactobacillus, and Bifidobacterium spp. | Indoles influence bacterial communication, limiting virulence gene expression and bacterial invasiveness, in a dose-dependent manner. | Indoles boost MUC2 and MUC4 expression and goblet cell activity. | Indoles reduce the epithelial permeability by enhancing tight junctions. | [78,79,80,81,82] | |
| Urolithin A (UA) | Polyphenolic compounds (ellagitannins) in fruits, nuts, and tea. | In the small intestine, ellagitannins are hydrolyzed to ellagic and gallic acid intermediates, and further metabolized by Gordonibacter urolithinfaciens and Ellagibactrer into UA. Only about 40% of elderly humans possess a suitable gut microbiota able to produce UA. | UA and its synthetic analogue, UAS03, have been reported to upregulate tight junction proteins. | UA reduces the production of ROS and suppresses the TLR4, MAPK, and PI3K pathways, with decrease in the expression of pro-inflammatory mi-RNA and cytokines (IL-1β, IL-6, and TNF-α). | [83,84,85] | ||
| Hydrogen sulfide (H2S) | Sulfate (SO42−) derived from amino acids (mainly cysteine and methionine), additives, preservatives, and IEC production (CBS activity). | Sulfate-reducing bacteria (SRB), like colonic Desulfovibrio, Desulfotomaculum, and Bilophila, utilize SO42− as a terminal electron acceptor in their metabolic pathways, reducing it to H2S. | Exogenous H2S confers to the bacteria’s high resistance to oxidative stress. | High concentrations of H2S destabilize the disulfide bonds of the mucin-2 network, resulting in increased contact between bacteria and the epithelium. | H2S is the primary mineral energy substrate for colonocytes, but in high concentrations, it inhibits the mitochondrial respiratory chain. Also, it negatively interferes with butyrate metabolism. | [86,87,88,89,90,91,92] | |
| Trimethylamine (TMA) | Choline, carnitine, and betaine, contained in red meat, eggs, fish, and dairy. | Several bacterial species (e.g., E. coli, Enterococcus, Clostridium, Proteus, Shigella, Klebsiella, and Providentia spp.) transform the precursors in TMA, which is further oxidized in the liver to form TMAO. | TMA and TMAO modulate the composition of the microbiota. | [93,94] | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cicchinelli, S.; Gemma, S.; Pignataro, G.; Piccioni, A.; Ojetti, V.; Gasbarrini, A.; Franceschi, F.; Candelli, M. Intestinal Fibrogenesis in Inflammatory Bowel Diseases: Exploring the Potential Role of Gut Microbiota Metabolites as Modulators. Pharmaceuticals 2024, 17, 490. https://doi.org/10.3390/ph17040490
Cicchinelli S, Gemma S, Pignataro G, Piccioni A, Ojetti V, Gasbarrini A, Franceschi F, Candelli M. Intestinal Fibrogenesis in Inflammatory Bowel Diseases: Exploring the Potential Role of Gut Microbiota Metabolites as Modulators. Pharmaceuticals. 2024; 17(4):490. https://doi.org/10.3390/ph17040490
Chicago/Turabian StyleCicchinelli, Sara, Stefania Gemma, Giulia Pignataro, Andrea Piccioni, Veronica Ojetti, Antonio Gasbarrini, Francesco Franceschi, and Marcello Candelli. 2024. "Intestinal Fibrogenesis in Inflammatory Bowel Diseases: Exploring the Potential Role of Gut Microbiota Metabolites as Modulators" Pharmaceuticals 17, no. 4: 490. https://doi.org/10.3390/ph17040490
APA StyleCicchinelli, S., Gemma, S., Pignataro, G., Piccioni, A., Ojetti, V., Gasbarrini, A., Franceschi, F., & Candelli, M. (2024). Intestinal Fibrogenesis in Inflammatory Bowel Diseases: Exploring the Potential Role of Gut Microbiota Metabolites as Modulators. Pharmaceuticals, 17(4), 490. https://doi.org/10.3390/ph17040490