
Development and Validation of a Capillary Zone Electrophoresis–Tandem Mass Spectrometry Method for Simultaneous Quantification of Eight β-Lactam Antibiotics and Two β-Lactamase Inhibitors in Plasma Samples



Abstract
:1. Introduction
2. Results and Discussion
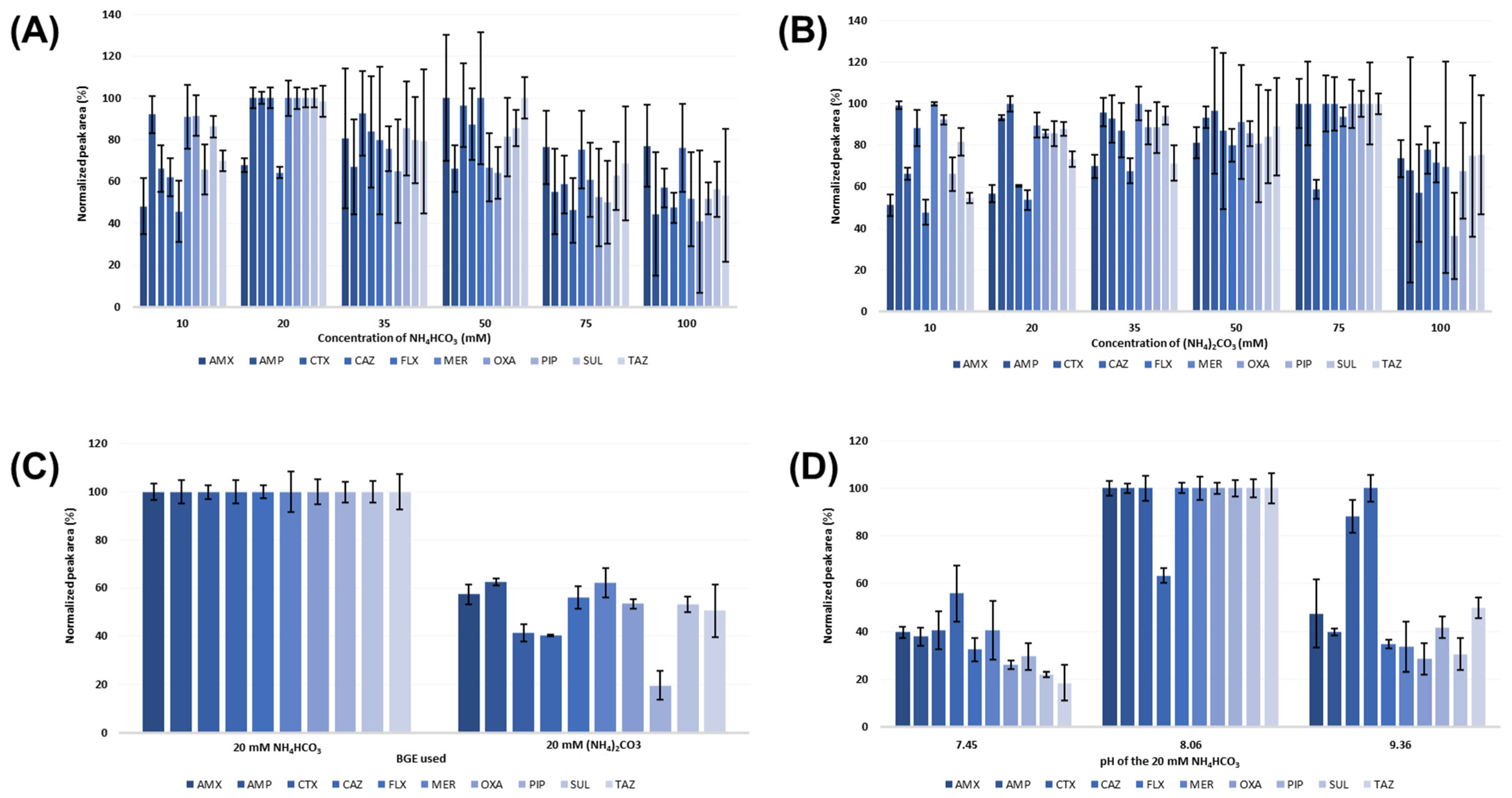
2.1. Optimization of the CZE Separation Conditions
2.2. Optimization of MS Detection Conditions
2.2.1. ESI Optimization
2.2.2. Triple Quadrupole (QqQ) MS Optimization
2.3. Optimization of Plasma Sample Preparation
2.4. Method Validation
2.5. Method Practicality and Greenness Evaluation
2.6. Limitations and Future Perspectives of the Study
3. Materials and Methods
3.1. Chemicals and Samples
3.2. Instrumentation
3.3. Capillary Treatment
3.4. Preparation of Standard Solutions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Carlier, M.; Stove, V.; De Waele, J.J.; Verstraete, A.G. Ultrafast Quantification of β-Lactam Antibiotics in Human Plasma Using UPLC–MS/MS. J. Chromatogr. B 2015, 978–979, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Dhaese, S.; Van Vooren, S.; Boelens, J.; De Waele, J. Therapeutic Drug Monitoring of β-Lactam Antibiotics in the ICU. Expert. Rev. Anti Infect. Ther. 2020, 18, 1155–1164. [Google Scholar] [CrossRef] [PubMed]
- Cusumano, J.A.; Klinker, K.P.; Huttner, A.; Luther, M.K.; Roberts, J.A.; LaPlante, K.L. Towards Precision Medicine: Therapeutic Drug Monitoring-Guided Dosing of Vancomycin and β-Lactam Antibiotics to Maximize Effectiveness and Minimize Toxicity. Am. J. Health Syst. Pharm. 2020, 77, 1104–1112. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.A.; Paul, S.K.; Akova, M.; Bassetti, M.; De Waele, J.J.; Dimopoulos, G.; Kaukonen, K.-M.; Koulenti, D.; Martin, C.; Montravers, P.; et al. DALI: Defining Antibiotic Levels in Intensive Care Unit Patients: Are Current β-Lactam Antibiotic Doses Sufficient for Critically Ill Patients? Clin. Infect. Dis. 2014, 58, 1072–1083. [Google Scholar] [CrossRef] [PubMed]
- Stašek, J.; Keller, F.; Kočí, V.; Klučka, J.; Klabusayová, E.; Wiewiorka, O.; Strašilová, Z.; Beňovská, M.; Škardová, M.; Maláska, J. Update on Therapeutic Drug Monitoring of Beta-Lactam Antibiotics in Critically Ill Patients—A Narrative Review. Antibiotics 2023, 12, 568. [Google Scholar] [CrossRef] [PubMed]
- Dilworth, T.J.; Schulz, L.T.; Micek, S.T.; Kollef, M.H.; Rose, W.E. β-Lactam Therapeutic Drug Monitoring in Critically Ill Patients: Weighing the Challenges and Opportunities to Assess Clinical Value. Crit. Care Explor. 2022, 4, e0726. [Google Scholar] [CrossRef]
- Gatti, M.; Cojutti, P.G.; Pascale, R.; Tonetti, T.; Laici, C.; Dell’Olio, A.; Siniscalchi, A.; Giannella, M.; Viale, P.; Pea, F. Assessment of a PK/PD Target of Continuous Infusion Beta-Lactams Useful for Preventing Microbiological Failure and/or Resistance Development in Critically Ill Patients Affected by Documented Gram-Negative Infections. Antibiotics 2021, 10, 1311. [Google Scholar] [CrossRef] [PubMed]
- Maguigan, K.L.; Al-Shaer, M.H.; Peloquin, C.A. Beta-Lactams Dosing in Critically Ill Patients with Gram-Negative Bacterial Infections: A PK/PD Approach. Antibiotics 2021, 10, 1154. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.N.; Papich, M.G.; Drusano, G.L. Dosing Regimen Matters: The Importance of Early Intervention and Rapid Attainment of the Pharmacokinetic/Pharmacodynamic Target. Antimicrob. Agents Chemother. 2012, 56, 2795–2805. [Google Scholar] [CrossRef]
- Owens, R.C.; Shorr, A.F. Rational Dosing of Antimicrobial Agents: Pharmacokinetic and Pharmacodynamic Strategies. Am. J. Health-Syst. Pharm. 2009, 66, S23–S30. [Google Scholar] [CrossRef]
- Legrand, T.; Vodovar, D.; Tournier, N.; Khoudour, N.; Hulin, A. Simultaneous Determination of Eight β-Lactam Antibiotics, Amoxicillin, Cefazolin, Cefepime, Cefotaxime, Ceftazidime, Cloxacillin, Oxacillin, and Piperacillin, in Human Plasma by Using Ultra-High-Performance Liquid Chromatography with Ultraviolet Detection. Antimicrob. Agents Chemother. 2016, 60, 4734–4742. [Google Scholar] [CrossRef]
- Carlier, M.; Noe, M.; De Waele, J.J.; Stove, V.; Verstraete, A.G.; Lipman, J.; Roberts, J.A. Population Pharmacokinetics and Dosing Simulations of Amoxicillin/Clavulanic Acid in Critically Ill Patients. J. Antimicrob. Chemother. 2013, 68, 2600–2608. [Google Scholar] [CrossRef]
- Huttner, A.; Harbarth, S.; Hope, W.W.; Lipman, J.; Roberts, J.A. Therapeutic Drug Monitoring of the β-Lactam Antibiotics: What Is the Evidence and Which Patients Should We Be Using It for? J. Antimicrob. Chemother. 2015, 70, dkv201. [Google Scholar] [CrossRef]
- Gonçalves-Pereira, J.; Póvoa, P. Antibiotics in Critically Ill Patients: A Systematic Review of the Pharmacokinetics of β-Lactams. Crit. Care 2011, 15, R206. [Google Scholar] [CrossRef]
- Abdulla, A.; Ewoldt, T.M.J.; Hunfeld, N.G.M.; Muller, A.E.; Rietdijk, W.J.R.; Polinder, S.; van Gelder, T.; Endeman, H.; Koch, B.C.P. The Effect of Therapeutic Drug Monitoring of Beta-Lactam and Fluoroquinolones on Clinical Outcome in Critically Ill Patients: The DOLPHIN Trial Protocol of a Multi-Centre Randomised Controlled Trial. BMC Infect. Dis. 2020, 20, 57. [Google Scholar] [CrossRef]
- Blondiaux, N.; Wallet, F.; Favory, R.; Onimus, T.; Nseir, S.; Courcol, R.J.; Durocher, A.; Roussel-Delvallez, M. Daily Serum Piperacillin Monitoring Is Advisable in Critically Ill Patients. Int. J. Antimicrob. Agents 2010, 35, 500–503. [Google Scholar] [CrossRef]
- Kurihara, Y.; Kizu, J.; Hori, S. Simple and Rapid Determination of Serum Carbapenem Concentrations by High-Performance Liquid Chromatography. J. Infect. Chemother. 2008, 14, 30–34. [Google Scholar] [CrossRef]
- Denooz, R.; Charlier, C. Simultaneous Determination of Five β-Lactam Antibiotics (Cefepim, Ceftazidim, Cefuroxim, Meropenem and Piperacillin) in Human Plasma by High-Performance Liquid Chromatography with Ultraviolet Detection. J. Chromatogr. B 2008, 864, 161–167. [Google Scholar] [CrossRef]
- Pinder, N.; Brenner, T.; Swoboda, S.; Weigand, M.A.; Hoppe-Tichy, T. Therapeutic Drug Monitoring of Beta-Lactam Antibiotics—Influence of Sample Stability on the Analysis of Piperacillin, Meropenem, Ceftazidime and Flucloxacillin by HPLC-UV. J. Pharm. Biomed. Anal. 2017, 143, 86–93. [Google Scholar] [CrossRef]
- Verhoven, S.M.; Groszek, J.J.; Fissell, W.H.; Seegmiller, A.; Colby, J.; Patel, P.; Verstraete, A.; Shotwell, M. Therapeutic Drug Monitoring of Piperacillin and Tazobactam by RP-HPLC of Residual Blood Specimens. Clin. Chim. Acta 2018, 482, 60–64. [Google Scholar] [CrossRef]
- Gaikwad, A.; Gavali, S.; Narendiran; Katale, D.; Bonde, S.; Bhadane, R.P. An LC–MS–MS Method for the Simultaneous Quantification of Amoxicillin and Clavulanic Acid in Human Plasma and Its Pharmacokinetic Application. J. Pharm. Res. 2013, 6, 804–812. [Google Scholar] [CrossRef]
- Piestansky, J.; Cizmarova, I.; Mikus, P.; Parrak, V.; Babiak, P.; Secnik, P.; Secnik, P.; Kovac, A. An Ultra-High-Performance Liquid Chromatography-Tandem Mass Spectrometry Method for Simultaneous Determination of 4 β-Lactam Antibiotics, Tazobactam, and Linezolid in Human Plasma Samples. Ther. Drug Monit. 2022, 44, 784–790. [Google Scholar] [CrossRef]
- Colin, P.; De Bock, L.; T’jollyn, H.; Boussery, K.; Van Bocxlaer, J. Development and Validation of a Fast and Uniform Approach to Quantify β-Lactam Antibiotics in Human Plasma by Solid Phase Extraction-Liquid Chromatography–Electrospray-Tandem Mass Spectrometry. Talanta 2013, 103, 285–293. [Google Scholar] [CrossRef]
- Paal, M.; Zoller, M.; Schuster, C.; Vogeser, M.; Schütze, G. Simultaneous Quantification of Cefepime, Meropenem, Ciprofloxacin, Moxifloxacin, Linezolid and Piperacillin in Human Serum Using an Isotope-Dilution HPLC–MS/MS Method. J. Pharm. Biomed. Anal. 2018, 152, 102–110. [Google Scholar] [CrossRef]
- Farid, N.F.; Abdelwahab, N.S. New ecological method for determination of different β-lactams: Application to real human plasma samples. RSC Adv. 2019, 9, 19539–19548. [Google Scholar] [CrossRef]
- Nishi, H.; Fukuyama, T.; Matsuo, M. Separation and Determination of Aspoxicillin in Human Plasma by Micellar Electrokinetic Chromatography with Direct Sample Injection. J. Chromatogr. A 1990, 515, 245–255. [Google Scholar] [CrossRef]
- Mrestani, Y.; Neubert, R.; Schiewe, J.; Härtl, A. Application of Capillary Zone Electrophoresis in Cephalosporin Analysis. J. Chromatogr. B Biomed. Sci. Appl. 1997, 690, 321–326. [Google Scholar] [CrossRef]
- Castaneda Penalvo, G.; Kelly, M.; Maillols, H.; Fabre, H. Evaluation of Capillary Zone Electrophoresis and Micellar Electrokinetic Capillary Chromatography with Direct Injection of Plasma for the Determination of Cefotaxime and Its Metabolite. Anal. Chem. 1997, 69, 1364–1369. [Google Scholar] [CrossRef]
- Mrestani, Y.; Neubert, R.; Nagel, F. Capillary Zone Electrophoresis Determination of Meropenem in Biological Media Using a High Sensitivity Cell. J. Pharm. Biomed. Anal. 1999, 20, 899–903. [Google Scholar] [CrossRef]
- Mayer, B.X.; Hollenstein, U.; Brunner, M.; Eichler, H.-G.; Müller, M. Micellar Electrokinetic Chromatography for the Analysis of Cefpirome in Microdialysis and Plasma Samples Obtainedin Vivo from Human Volunteers. Electrophoresis 2000, 21, 1558–1564. [Google Scholar] [CrossRef]
- Gáspár, A.; Kardos, S.; Andrási, M.; Klekner, Á. Capillary Electrophoresis for the Direct Determination of Cephalosporins in Clinical Samples. Chromatographia 2002, 56, S109–S114. [Google Scholar] [CrossRef]
- Mayer, B.X.; Petsch, M.; Tschernko, E.M.; Müller, M. Strategies for the Determination of Cefazolin in Plasma and Microdialysis Samples by Short-End Capillary Zone Electrophoresis. Electrophoresis 2003, 24, 1215–1220. [Google Scholar] [CrossRef]
- Kitahashi, T.; Furuta, I. Determination of Meropenem by Capillary Electrophoresis Using Direct Injection of Serum. J. Chromatogr. Sci. 2005, 43, 430–433. [Google Scholar] [CrossRef]
- Chou, Y.-W.; Yang, Y.-H.; Chen, J.-H.; Kuo, C.-C.; Chen, S.-H. Quantification of Meropenem in Plasma and Cerebrospinal Fluid by Micellar Electrokinetic Capillary Chromatography and Application in Bacterial Meningitis Patients. J. Chromatogr. B 2007, 856, 294–301. [Google Scholar] [CrossRef]
- Andrási, M.; Gáspár, A.; Klekner, Á. Analysis of Cephalosporins in Bronchial Secretions by Capillary Electrophoresis after Simple Pretreatment. J. Chromatogr. B 2007, 846, 355–358. [Google Scholar] [CrossRef]
- Solangi, A.; Mallah, A.; Memon, S. Determination of Ceftriaxone, Ceftizoxime, Paracetamol, and Diclofenac Sodium by Capillary Zone Electrophoresis in Pharmaceutical Formulations and in Human Blood Serum. Turk. J. Chem. 2010, 34, 921–933. [Google Scholar] [CrossRef]
- Tůma, P.; Jaček, M.; Fejfarová, V.; Polák, J. Electrophoretic Stacking for Sensitive Determination of Antibiotic Ceftazidime in Human Blood and Microdialysates from Diabetic Foot. Anal. Chim. Acta 2016, 942, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Šlampová, A.; Kubáň, P. Micro-Electromembrane Extraction through Volatile Free Liquid Membrane for the Determination of β-Lactam Antibiotics in Biological and Environmental Samples. Talanta 2023, 252, 123831. [Google Scholar] [CrossRef]
- Liang, H.-H.; Lin, Y.-C.; Hung, C.-C.; Hou, Y.-C.; Lin, Y.-H. Method Development for Determination of Doripenem in Human Plasma via Capillary Electrophoresis Coupled with Field-Enhanced Sample Stacking and Sweeping. Int. J. Mol. Sci. 2023, 24, 13751. [Google Scholar] [CrossRef]
- Al-Attas, A.; Nasr, J.J.; El-Enany, N.; Belal, F. A green capillary zone electrophoresis method for the simultaneous determination of piperacillin, tazobactam and cefepime in pharmaceutical formulations and human plasma. Biomed. Chromatogr. 2015, 29, 1811–1818. [Google Scholar] [CrossRef]
- Hancu, G.; Neacsu, A.; Papp, L.A.; Ciurba, A. Simultaneous determination of amoxicillin and clavulanic acid in pharmaceutical preparations by capillary zone electrophoresis. Bras. J. Pharm. Sci. 2016, 52, 281–286. [Google Scholar] [CrossRef]
- Pham, T.N.M.; Le, T.B.; Le, D.D.; Ha, T.H.; Nguyen, N.S.; Pham, T.D.; Hauser, P.C.; Nguyen, T.A.H.; Mai, T.D. Determination of Carbapenem Antibiotics Using a Purpose-Made Capillary Electrophoresis Instrument with Contactless Conductivity Detection. J. Pharm. Biomed. Anal. 2020, 178, 112906. [Google Scholar] [CrossRef] [PubMed]
- Tůma, P.; Jaček, M.; Sommerová, B.; Dlouhý, P.; Jarošíková, R.; Husáková, J.; Wosková, V.; Fejfarová, V. Monitoring of Amoxicilline and Ceftazidime in the Microdialysate of Diabetic Foot and Serum by Capillary Electrophoresis with Contactless Conductivity Detection. Electrophoresis 2022, 43, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.A.H.; Pham, T.N.M.; Le, T.B.; Le, D.C.; Tran, T.T.P.; Nguyen, T.Q.H.; Nguyen, T.K.T.; Hauser, P.C.; Mai, T.D. Cost-effective capillary electrophoresis with contactless conductivity detection for quality control of beta-lactam antibiotics. J. Chromatogr. A 2019, 1605, 360356. [Google Scholar] [CrossRef]
- Capillary Electrophoresis: Trends and Developments in Pharmaceutical Research; Kanchi, S.; Sagrado, S.; Sabela, M.; Bisetty, K. (Eds.) Routledge: Abingdon, UK, 2017; ISBN 978-1-315-22538-8. [Google Scholar]
- Lefeuvre, S.; Bois-Maublanc, J.; Hocqueloux, L.; Bret, L.; Francia, T.; Eleout-Da Violante, C.; Billaud, E.M.; Barbier, F.; Got, L. A Simple Ultra-High-Performance Liquid Chromatography-High Resolution Mass Spectrometry Assay for the Simultaneous Quantification of 15 Antibiotics in Plasma. J. Chromatogr. B 2017, 1065–1066, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Radovanovic, M.; Day, R.O.; Jones, G.D.R.; Galettis, P.; Norris, R.L.G. LC–MS/MS Method for Simultaneous Quantification of Ten Antibiotics in Human Plasma for Routine Therapeutic Drug Monitoring. J. Mass. Spectrom. Adv. Clin. Lab. 2022, 26, 48–59. [Google Scholar] [CrossRef]
- Straub, R.F.; Voyksner, R.D. Determination of Penicillin G, Ampicillin, Amoxicillin, Cloxacillin and Cephapirin by High-Performance Liquid Chromatography—Electrospray Mass Spectrometry. J. Chromatogr. A 1993, 647, 167–181. [Google Scholar] [CrossRef]
- Zaikin, V.G.; Borisov, R.S. Options of the Main Derivatization Approaches for Analytical ESI and MALDI Mass Spectrometry. Crit. Rev. Anal. Chem. 2022, 52, 1287–1342. [Google Scholar] [CrossRef]
- Klampfl, C.W.; Himmelsbach, M. Sheath Liquids in CE-MS: Role, Parameters, and Optimization. In Capillary Electrophoresis-Mass Spectrometry (CE-MS): Principles and Applications; de Jong, G., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2016; pp. 41–65. ISBN 978-3-527-33924-2. [Google Scholar]
- Carlier, M.; Stove, V.; Roberts, J.A.; Van De Velde, E.; De Waele, J.J.; Verstraete, A.G. Quantification of Seven β-Lactam Antibiotics and Two β-Lactamase Inhibitors in Human Plasma Using a Validated UPLC-MS/MS Method. Int. J. Antimicrob. Agents 2012, 40, 416–422. [Google Scholar] [CrossRef]
- Mandell, G.L.; Bennett, J.E.; Dolin, R. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases, 7th ed.; Churchill Livingstone, Elsevier: Philadelphia, PA, USA, 2010; ISBN 978-0-443-06839-3. [Google Scholar]
- Enna, S.J.; Bylund, D.B. xPharm: The Comprehensive Pharmacology Reference; Elsevier: Amsterdam, The Netherlands, 2008; ISBN 978-0-08-055232-3. [Google Scholar]
- Handbook of Drug Monitoring Methods: Therapeutics and Drugs of Abuse; Dasgupta, A. (Ed.) Humana Press: Totowa, NJ, USA, 2008; ISBN 978-1-58829-780-8. [Google Scholar]
- Sillén, H.; Mitchell, R.; Sleigh, R.; Mainwaring, G.; Catton, K.; Houghton, R.; Glendining, K. Determination of Avibactam and Ceftazidime in Human Plasma Samples by LC–MS. Bioanalysis 2015, 7, 1423–1434. [Google Scholar] [CrossRef]
- Popowicz, N.D.; O’Halloran, S.J.; Fitzgerald, D.; Lee, Y.C.G.; Joyce, D.A. A Rapid, LC-MS/MS Assay for Quantification of Piperacillin and Tazobactam in Human Plasma and Pleural Fluid; Application to a Clinical Pharmacokinetic Study. J. Chromatogr. B 2018, 1081–1082, 58–66. [Google Scholar] [CrossRef] [PubMed]
- US Department of Health and Human Services Food and Drug Administration, Center for Drug; Evaluation and Research (CDER) Center for Veterinary Medicine (CVM). Bioanalytical Method Validation, Guidance for Industry. 2018. Available online: https://www.fda.gov/files/drugs/published/Bioanalytical-Method-Validation-Guidance-for-Industry.pdf (accessed on 28 February 2024).
- Schulz, M.; Schmoldt, A.; Andresen-Streichert, H.; Iwersen-Bergmann, S. Revisited: Therapeutic and Toxic Blood Concentrations of More than 1100 Drugs and Other Xenobiotics. Crit Care 2020, 24, 195. [Google Scholar] [CrossRef] [PubMed]
- Manousi, N.; Wojnowski, W.; Płotka-Wasylka, J.; Samanidou, V. Blue Applicability Grade Index (BAGI) and Software: A New Tool for the Evaluation of Method Practicality. Green Chem. 2023, 25, 7598–7604. [Google Scholar] [CrossRef]
- Pena-Pereira, F.; Wojnowski, W.; Tobiszewski, M. AGREE—Analytical GREEnness Metric Approach and Software. Anal. Chem. 2020, 92, 10076–10082. [Google Scholar] [CrossRef]
- Plotka-Wasylka, J. A new tool for the evaluation of the analytical procedure: Green Analytical Procedure Index. Talanta 2018, 181, 204–209. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Matrix | Sample Preparation | Separation Conditions | LOD (μg·mL−1) | Analytes | Reference |
|---|---|---|---|---|---|---|
| MEKC-UV (λ = 210 nm) | plasma | - | uncoated fused silica capillary; ID 50 µm; Leff = 50 cm; BGE = 20 mM phosphate–borate buffer + 50 mM SDS, pH = 8.5 | 1.3 | aspoxicillin | [26] |
| CZE-UV (λ = 210 nm) | plasma | dilution | uncoated fused silica capillary; ID 50 µm; Leff = 40 cm; BGE = 20 mM phosphate buffer, pH = 6 | 4 | cefodizime | [27] |
| 2 | cefuroxime | |||||
| 6 | cefpirome | |||||
| 2 | cefotaxime | |||||
| CZE-UV (λ = 254 nm) | plasma | precipitation | uncoated fused silica capillary; ID 75 µm; Leff = 50 cm; BGE = 40 mM borate buffer, pH = 9.2 | 2 | cefotaxime | [28] |
| MEKC-UV (λ = 254 nm) | plasma | dilution | uncoated fused silica capillary; ID 75 µm; Leff = 50 cm; BGE = 30 mM phosphate buffer + 165 mM SDS, pH = 8 | 1 | deacetylcefotaxime | |
| CZE-DAD (λ = 200 nm, 303 nm) | plasma | dilution/ precipitation | uncoated fused silica capillary; ID 75 µm; Leff = 72 cm; BGE = 10 mM phosphate buffer, pH = 7.2 | 4 | meropenem | [29] |
| MEKC-UV (λ = 270 nm) | plasma | ultracentrifugation | uncoated fused silica capillary; ID 50 µm; Leff = 56 cm; BGE = 20 mM citrate buffer + 50 mM SDS, pH = 2.8 | 0.2 | cefpirome | [30] |
| MEKC-UV (λ = 270 nm) | serum | - | uncoated fused silica capillary; ID 50 µm; Leff = 48.5 cm; BGE = 25 mM borate buffer + 100 mM SDS, pH = 9.2 | - | cefuroxime cefotaxime ceftriaxone ceftazidime | [31] |
| CZE-UV (λ = 270 nm) | plasma | precipitation | uncoated fused silica capillary; ID 50 µm; Leff = 8.5 cm; BGE = 20 mM sodium hydrogen phosphate, pH = 6.4 | 1 | cefazolin | [32] |
| microdialysates | dilution | 2 | ||||
| MEKC-UV (λ = 197 nm) | serum | - | uncoated fused silica capillary; ID 75 µm; Leff = 50 cm; BGE = 25 mM borate buffer + 90 mM SDS, pH = 10 | 2 | meropenem | [33] |
| MEKC-UV (λ = 300 nm) | plasma | SPE | uncoated fused silica capillary; ID 50 µm; Leff = 30 cm; BGE = 40 mM TRIS buffer + SDS, pH = 8 | 0.2 | meropenem | [34] |
| MEKC-UV (λ = 270 nm) | serum | - | Polyimide-coated silica capillary; ID 50 µm; Leff = 40 cm; BGE = 25 mM borate buffer + 50 mM SDS, pH = 9.1 | - | cefazolin cefepime cefamandole cefuroxime ceftazidime ceftriaxone | [35] |
| CZE-UV (λ = 214 nm) | serum | precipitation | uncoated fused silica capillary; ID 75 µm; Leff = 50 cm BGE = 50 mM tetraborate, pH = 9 | 5 | ceftriaxone | [36] |
| 1 | ceftizoxime | |||||
| CZE-UV (λ = 260 nm, 200 nm) | plasma microdialysates | precipitation | INST-coated fused silica capillary; ID 25 µm; Leff = 31.5 cm; BGE = 50 mM chloroacetic acid + 20% MeOH + 0.5% INST, pH = 2.32 | 0.42 | ceftazidime | [37] |
| CZE-C4D | plasma | SPE | uncoated fused silica capillary; ID 50 µm; Leff = 50 cm; BGE = 10 mM TRIS, pH = 8 | 0.45 | doripenem meropenem imipenem ertapenem | [42] |
| CZE-C4D | serum microdialysates | precipitation | INST-coated silica capillary; ID 25 µm; Leff = 18 cm; BGE = 0.5 M acetic acid | 0.043 | amoxicillin | [43] |
| INST-coated silica capillary; ID 25 µm; Leff = 18 cm; BGE = 3.2 M acetic acid + 13% MeOH (for ceftazidime) | 0.096 | ceftazidime | ||||
| MEKC-UV (λ = 200 nm) | serum | μ-EME | uncoated fused silica capillary; ID 75 µm; Leff = 39 cm; BGE = 25 mM phosphate buffer + 50 mM SDS, pH = 8.13 | 0.17 | penicillin | [38] |
| 0.2 | phenoxypenicillin | |||||
| 0.13 | ampicillin | |||||
| 0.12 | amoxicillin | |||||
| MEKC-DAD (λ = 300 nm) | plasma | FESS, sweeping, SPE | uncoated fused silica capillary; ID 50 µm; Leff = 50 cm; high-conductivity buffer = 150 mM phosphate (pH = 2.5) + 20% MeOH; low-conductivity buffer = 50 mM phosphate buffer (pH = 2.5) + 100 mM SDS | 0.4 | doripenem | [39] |
| CZE-UV (λ = 215 nm) | plasma | protein precipitation | uncoated fused silica capillary; ID 50 µm; Leff = 40.2 cm; BGE = 15 mM sodium borate buffer (pH 9.3) | 0.56 0.95 2.09 | Piperacillin Tazobactam cefepime | [40] |
| Analyte | Parent Ion m/z [M+H]+ | Quantifier m/z [M+H]+ | Qualifier m/z [M+H]+ | Fragmentor Voltage (V) | Collision Energy (eV) | Internal Standard |
|---|---|---|---|---|---|---|
| Sulbactam | 234 | 123.8 | 141.5 | 80 | 15 | [13C2, 15N3]-tazobactam |
| Tazobactam | 301.1 | 168 | 207.1 | 120 | 15 | [13C2, 15N3]-tazobactam |
| [13C2, 15N3]-tazobactam | 306 | 210 | 120 | 15 | ||
| Ampicillin | 350 | 106.1 | 191.9 | 100 | 15 | [2H5]-piperacillin |
| Amoxicillin | 366.1 | 113.7 | 349.1 | 80 | 10 | [2H5]-piperacillin |
| Meropenem | 384 | 113.7 | 274.8 | 100 | 15 | [2H6]-meropenem |
| [2H6]-meropenem | 390.2 | 147.2 | 100 | 15 | ||
| Oxacillin | 402.1 | 160 | 242.7 | 100 | 10 | [2H5]-piperacillin |
| Flucloxacillin | 454.1 | 295 | 237.5 | 140 | 10 | [2H5]-piperacillin |
| Cefotaxime | 456 | 166.7 | 211.1 | 120 | 15 | [13C, 2H3]-cefotaxime |
| [13C, 2H3]-cefotaxime | 460.1 | 166.9 | 120 | 15 | ||
| Piperacillin | 518 | 143 | 159.9 | 140 | 10 | [2H5]-piperacillin |
| [2H5]-piperacillin | 532.2 | 148.1 | 140 | 10 | ||
| Ceftazidime | 547.1 | 395.8 | 467.6 | 120 | 15 | [13C, 2H3]-cefotaxime |
| Calibration Range (µg·mL−1) | tm (min), n = 6 | RSDtm (%), n = 6 | RSDarea (%), n = 6 | a (Counts) | SDa | b (Counts·µg−1·mL) | SDb | r2 | LOD (µg·mL−1) | LLOQ (µg·mL−1) | N | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Plasma matrix | AMX | 1–40 | 17.06 | 0.6 | 4.8 | 0.130 | 0.003 | −0.020 | 0.063 | 0.992 | 0.100 | 1.000 | 5762 |
| AMP | 0.5–40 | 16.76 | 0.3 | 1.5 | 0.932 | 0.031 | −0.586 | 0.514 | 0.988 | 0.010 | 0.500 | 32,125 | |
| CTX | 0.48–38.15 | 16.02 | 0.1 | 9.4 | 0.312 | 0.005 | 0.158 | 0.114 | 0.995 | 0.010 | 0.477 | 53,143 | |
| CAZ | 1–40 | 15.57 | 0.3 | 9.5 | 0.050 | 0.001 | 0.000 | 0.023 | 0.993 | 0.500 | 1.000 | 5860 | |
| FLX | 0.48–38.15 | 16.3 | 0.2 | 11.4 | 0.139 | 0.004 | −0.069 | 0.063 | 0.991 | 0.010 | 0.477 | 8772 | |
| MER | 0.88–35.06 | 16.58 | 0.5 | 13.5 | 0.097 | 0.002 | 0.078 | 0.034 | 0.995 | 0.438 | 0.876 | 2925 | |
| OXA | 0.45–36.38 | 16.44 | 0.2 | 2.4 | 0.730 | 0.025 | −0.023 | 0.437 | 0.985 | 0.009 | 0.455 | 20,774 | |
| PIP | 0.96–38.30 | 15.63 | 0.1 | 1.5 | 0.186 | 0.005 | 0.089 | 0.098 | 0.991 | 0.010 | 0.957 | 7191 | |
| SUL | 0.91–36.55 | 19.74 | 0.5 | 7.1 | 0.581 | 0.013 | −0.218 | 0.264 | 0.992 | 0.091 | 0.914 | 51,854 | |
| TAZ | 1–40 | 18.68 | 0.3 | 5.8 | 0.493 | 0.009 | 0.199 | 0.195 | 0.995 | 0.050 | 1.000 | 40,860 | |
| Water matrix | AMX | 1–40 | 21.43 | 1.1 | 8.1 | 0.129 | 0.005 | 0.216 | 0.099 | 0.987 | 0.050 | 1.000 | 5586 |
| AMP | 0.5–40 | 21.50 | 0.6 | 6.4 | 0.782 | 0.020 | 0.846 | 0.353 | 0.993 | 0.001 | 0.500 | 8279 | |
| CTX | 0.48–38.15 | 19.50 | 0.1 | 5.1 | 0.329 | 0.007 | −0.021 | 0.147 | 0.993 | 0.001 | 0.477 | 282,724 | |
| CAZ | 1–40 | 18.82 | 0.2 | 3.0 | 0.064 | 0.002 | −0.066 | 0.030 | 0.992 | 0.010 | 1.000 | 21,266 | |
| FLX | 0.48–38.15 | 19.96 | 0.4 | 3.1 | 0.125 | 0.004 | 0.094 | 0.067 | 0.990 | 0.010 | 0.477 | 49,069 | |
| MER | 0.88–35.06 | 20.28 | 0.1 | 1.3 | 0.088 | 0.003 | 0.160 | 0.054 | 0.991 | 0.438 | 0.876 | 50,658 | |
| OXA | 0.45–36.38 | 20.14 | 0.4 | 7.5 | 0.704 | 0.021 | 0.133 | 0.340 | 0.992 | 0.001 | 0.455 | 379,984 | |
| PIP | 0.96–38.30 | 18.88 | 0.1 | 5.6 | 0.184 | 0.005 | 0.038 | 0.098 | 0.991 | 0.010 | 0.957 | 342,736 | |
| SUL | 0.91–36.55 | 25.13 | 0.1 | 1.5 | 0.760 | 0.021 | −0.784 | 0.383 | 0.993 | 0.009 | 0.914 | 401,423 | |
| TAZ | 1–40 | 23.53 | 0.1 | 4.6 | 0.470 | 0.011 | 0.156 | 0.202 | 0.994 | 0.010 | 1.000 | 435,107 |
| QC Low | QC Medium | QC High | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nominal (µg·mL−1) | Found (µg·mL−1) | RSD (%) | RE (%) | Nominal (µg·mL−1) | Found (µg·mL−1) | RSD (%) | RE (%) | Nominal (µg·mL−1) | Found (µg·mL−1) | RSD (%) | RE (%) | ||
| Intra-day, n = 3 | AMX | 2.50 | 2.18 | 6.8 | −14.5 | 15.00 | 17.17 | 10.4 | 12.6 | 35.00 | 31.7 | 12.5 | −10.5 |
| AMP | 2.50 | 2.67 | 1.6 | 6.3 | 15.00 | 16.04 | 10.3 | 6.5 | 35.00 | 36.2 | 14.3 | 3.4 | |
| CTX | 2.38 | 2.65 | 8.2 | 10.2 | 14.31 | 15.56 | 7.0 | 8.1 | 33.39 | 32.8 | 5.4 | −1.8 | |
| CAZ | 2.50 | 3.08 | 10.7 | 18.9 | 15.00 | 15.28 | 11.7 | 1.8 | 35.00 | 30.6 | 7.7 | −14.3 | |
| FLX | 2.38 | 2.19 | 3.4 | −8.9 | 14.31 | 16.12 | 5.1 | 11.3 | 33.38 | 31.5 | 12.0 | −5.9 | |
| MER | 2.19 | 2.39 | 12.3 | 8.2 | 13.15 | 14.19 | 3.3 | 7.4 | 30.68 | 28.5 | 5.1 | −7.6 | |
| OXA | 2.27 | 2.14 | 2.4 | −6.1 | 13.64 | 15.38 | 13.5 | 11.3 | 31.83 | 30.8 | 12.1 | −3.3 | |
| PIP | 2.39 | 2.84 | 1.1 | 15.6 | 14.36 | 13.74 | 7.2 | −4.5 | 33.51 | 31.4 | 6.4 | −6.8 | |
| SUL | 2.28 | 2.44 | 8.4 | 6.2 | 13.71 | 13.53 | 14.6 | −1.4 | 31.98 | 34.9 | 11.9 | 8.3 | |
| TAZ | 2.50 | 2.50 | 9.9 | 0.1 | 15.00 | 15.78 | 10.3 | 4.9 | 35.00 | 36.5 | 4.0 | 4.1 | |
| Inter-day, n = 16 | AMX | 2.50 | 2.58 | 14.0 | 3.1 | 15.00 | 14.73 | 12.4 | −1.8 | 35.00 | 35.6 | 10.5 | 1.7 |
| AMP | 2.50 | 2.64 | 14.3 | 5.3 | 15.00 | 14.55 | 10.7 | −3.1 | 35.00 | 34.8 | 11.8 | −0.7 | |
| CTX | 2.38 | 2.39 | 13.2 | 1.9 | 14.31 | 15.29 | 10.6 | 6.5 | 33.39 | 32.5 | 7.8 | −2.6 | |
| CAZ | 2.50 | 2.49 | 14.9 | −1.0 | 15.00 | 14.58 | 9.8 | −2.9 | 35.00 | 34.3 | 12.0 | −1.9 | |
| FLX | 2.38 | 2.51 | 14.6 | 6.4 | 14.31 | 14.18 | 13.2 | −0.9 | 33.38 | 34.2 | 10.6 | 2.3 | |
| MER | 2.19 | 2.40 | 13.6 | 8.7 | 13.15 | 13.78 | 6.9 | 4.6 | 30.68 | 31.4 | 9.0 | 2.3 | |
| OXA | 2.27 | 2.41 | 14.7 | 5.6 | 13.64 | 14.40 | 11.7 | 5.3 | 31.83 | 30.6 | 11.8 | −3.9 | |
| PIP | 2.39 | 2.48 | 14.5 | 3.4 | 14.36 | 13.88 | 8.0 | −3.5 | 33.51 | 33.6 | 8.7 | 0.4 | |
| SUL | 2.28 | 2.46 | 13.6 | 7.1 | 13.71 | 13.60 | 11.7 | −0.8 | 31.98 | 32.8 | 13.9 | 2.6 | |
| TAZ | 2.50 | 2.45 | 14.0 | −2.2 | 15.00 | 15.35 | 7.9 | 2.3 | 35.00 | 35.3 | 10.0 | 0.9 | |
| QC Low | QC Medium | QC High | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nominal (µg·mL−1) | Found (µg·mL−1) | RSD (%) | RE (%) | Nominal (µg·mL−1) | Found (µg·mL−1) | RSD (%) | RE (%) | Nominal (µg·mL−1) | Found (µg·mL−1) | RSD (%) | RE (%) | ||
| Autosampler stability, n = 5 | AMX | 2.50 | 2.14 | 3.3 | −1.9 | 15.00 | 19.84 | 5.8 | 13.5 | 35.00 | 36.31 | 3.3 | 12.8 |
| AMP | 2.50 | 2.21 | 4.6 | −4.1 | 15.00 | 18.38 | 11.2 | 12.7 | 35.00 | 41.02 | 3.4 | 11.7 | |
| CTX | 2.38 | 2.72 | 2.5 | 2.4 | 14.31 | 15.93 | 2.2 | 2.3 | 33.39 | 31.50 | 3.2 | −4.1 | |
| CAZ | 2.50 | 3.29 | 6.2 | 6.4 | 15.00 | 13.45 | 3.8 | −13.6 | 35.00 | 29.09 | 4.3 | −5.3 | |
| FLX | 2.38 | 2.44 | 11.9 | 10.4 | 14.31 | 16.77 | 13.5 | 3.9 | 33.38 | 37.00 | 2.9 | 14.8 | |
| MER | 2.19 | 2.17 | 8.7 | −10.2 | 13.15 | 12.48 | 9.7 | −13.7 | 30.68 | 28.30 | 8.4 | −0.7 | |
| OXA | 2.27 | 2.12 | 12.2 | −1.0 | 13.64 | 17.83 | 2.9 | 13.8 | 31.83 | 34.74 | 4.8 | 11.2 | |
| PIP | 2.39 | 2.40 | 3.2 | 6.7 | 14.36 | 13.07 | 3.9 | −5.2 | 33.51 | 30.24 | 5.8 | −3.7 | |
| SUL | 2.28 | 2.55 | 13.0 | 4.6 | 13.71 | 15.25 | 3.9 | 11.3 | 31.98 | 35.79 | 11.1 | 2.5 | |
| TAZ | 2.50 | 2.50 | 5.6 | 0.1 | 15.00 | 16.76 | 6.3 | 5.8 | 35.00 | 35.56 | 12.9 | −2.7 | |
| Freeze-to-thaw stability, n = 5 | AMX | 2.50 | 2.01 | 9.8 | −9.4 | 15.00 | 14.12 | 12.7 | −1.4 | 35.00 | 34.41 | 13.6 | −6.0 |
| AMP | 2.50 | 2.14 | 11.5 | 0.4 | 15.00 | 16.55 | 7.6 | 12.9 | 35.00 | 35.09 | 9.3 | 2.2 | |
| CTX | 2.38 | 1.81 | 8.9 | −14.8 | 14.31 | 18.85 | 6.7 | 11.6 | 33.39 | 27.35 | 9.2 | −12.8 | |
| CAZ | 2.50 | 2.45 | 13.9 | 13.6 | 15.00 | 17.52 | 8.7 | 9.4 | 35.00 | 29.80 | 12.4 | −6.3 | |
| FLX | 2.38 | 2.42 | 12.7 | 14.7 | 14.31 | 14.85 | 10.2 | 14.2 | 33.38 | 32.38 | 7.0 | −14.0 | |
| MER | 2.19 | 4.25 | 5.6 | −2.3 | 13.15 | 16.33 | 11.4 | 9.3 | 30.68 | 31.56 | 8.7 | 3.1 | |
| OXA | 2.27 | 5.32 | 8.7 | −3.7 | 13.64 | 16.58 | 11.4 | 14.3 | 31.83 | 30.80 | 14.4 | 8.1 | |
| PIP | 2.39 | 2.09 | 5.0 | 2.3 | 14.36 | 14.88 | 3.7 | 12.9 | 33.51 | 27.17 | 9.3 | −13.6 | |
| SUL | 2.28 | 2.31 | 8.3 | 14.8 | 13.71 | 16.73 | 4.9 | 8.7 | 31.98 | 25.35 | 7.7 | −12.1 | |
| TAZ | 2.50 | 2.55 | 2.7 | 8.9 | 15.00 | 17.03 | 8.1 | 5.5 | 35.00 | 31.59 | 6.0 | −1.2 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cizmarova, I.; Mikus, P.; Svidrnoch, M.; Piestansky, J. Development and Validation of a Capillary Zone Electrophoresis–Tandem Mass Spectrometry Method for Simultaneous Quantification of Eight β-Lactam Antibiotics and Two β-Lactamase Inhibitors in Plasma Samples. Pharmaceuticals 2024, 17, 526. https://doi.org/10.3390/ph17040526
Cizmarova I, Mikus P, Svidrnoch M, Piestansky J. Development and Validation of a Capillary Zone Electrophoresis–Tandem Mass Spectrometry Method for Simultaneous Quantification of Eight β-Lactam Antibiotics and Two β-Lactamase Inhibitors in Plasma Samples. Pharmaceuticals. 2024; 17(4):526. https://doi.org/10.3390/ph17040526
Chicago/Turabian StyleCizmarova, Ivana, Peter Mikus, Martin Svidrnoch, and Juraj Piestansky. 2024. "Development and Validation of a Capillary Zone Electrophoresis–Tandem Mass Spectrometry Method for Simultaneous Quantification of Eight β-Lactam Antibiotics and Two β-Lactamase Inhibitors in Plasma Samples" Pharmaceuticals 17, no. 4: 526. https://doi.org/10.3390/ph17040526
APA StyleCizmarova, I., Mikus, P., Svidrnoch, M., & Piestansky, J. (2024). Development and Validation of a Capillary Zone Electrophoresis–Tandem Mass Spectrometry Method for Simultaneous Quantification of Eight β-Lactam Antibiotics and Two β-Lactamase Inhibitors in Plasma Samples. Pharmaceuticals, 17(4), 526. https://doi.org/10.3390/ph17040526