
Synergistic Biomedical Potential and Molecular Docking Analyses of Coumarin–Triazole Hybrids as Tyrosinase Inhibitors: Design, Synthesis, In Vitro Profiling, and In Silico Studies
 ,
,
Abstract
:1. Introduction
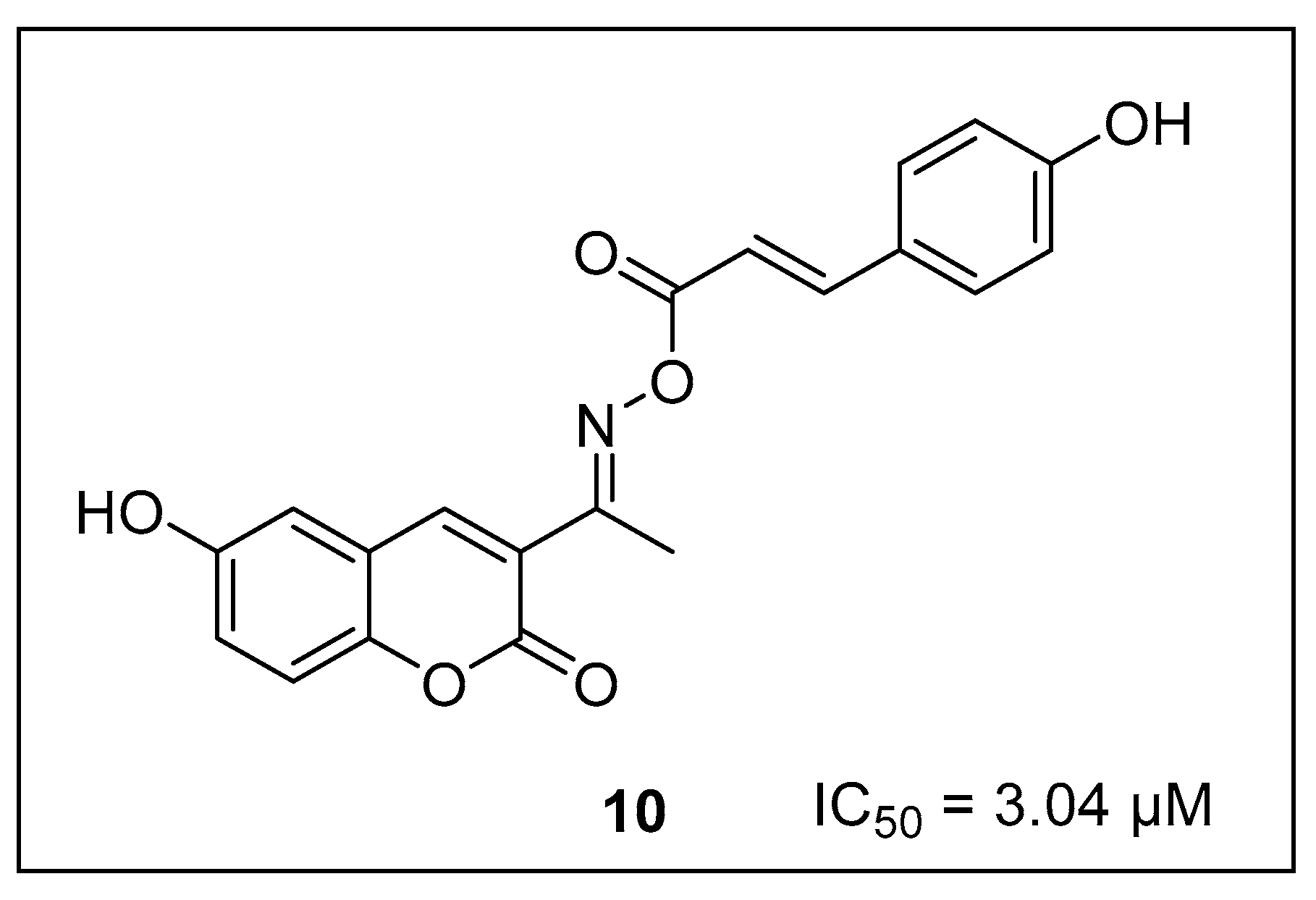
2. Result and Discussion
2.1. Synthesis
2.2. Spectral Description of the Most Potent Compound (17e)
2.3. Anti-Tyrosinase Activity of Target Compounds
2.4. Structure–Activity Relationship (SAR) of Potent Anti-Tyrosinase Derivatives
2.5. In Silico Modeling of Representative Potent Tyrosinase Inhibitor (17e)
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. General Synthetic Protocol for Synthesis of Substituted 4-Methyl-2-oxo-2H-chromen-7-yl-2-((5-(benzofuran-2-yl)-4H-1,2,4-triazol-3-yl)thio)acetates (17a–h)
3.2.1. 4-Methyl-2-oxo-2H-chromen-7-yl-2-((5-(benzofuran-2-yl)-4-phenyl-4H-1,2,4-triazol-3-yl)thio)acetate (17a)
- Coarse off-white solid; yield: 65%; mp 152 °C; 1H NMR (CDCl3, 400 MHz) δ (ppm): 2.40 (s, 3H); 4.32 (s, 2H); 6.25 (s, 1H); 6.55 (s, 1H); 7.13 (s, 1H); 7.18 (d, J = 8 Hz, 1H); 7.27 (s, 1H); 7.35 (s, 1H); 7.37–7.42 (m, 3H); 7.54–7.68 (m, 6H). 13C NMR (CDCl3, 100 MHz): δ (ppm) 18.7, 34.5, 107.5, 110.2, 113.2, 114.6, 116.7, 118.0, 118.1, 124.4, 125.5, 125.6, 127.3, 127.3, 129.1, 129.28, 130.3, 130.3, 131.1, 132.7, 143.2, 147.5, 151.8, 152.8, 153.5, 154.0, 160.3, 166.3. MS m/z: 509.9 [M]+. Anal. Elem. Calc. for C28H19N3O5 S: C, 66.00: H, 3.76: N, 8.25: Found: C, 66.02: H, 3.78: N, 8.24.
3.2.2. 4-Methyl-2-oxo-2H-chromen-7-yl-2-((5-(7-methoxybenzofuran-2-yl)-4-phenyl-4H-1,2,4-triazol-3-yl)thio)acetate (17b)
- Amorphous white solid; yield: 75%; mp 140 °C; 1H NMR (CDCl3, 400 MHz) δ (ppm): 2.40 (s, 3H); 3.81 (s, 3H); 4.34 (s, 2H); 6.25 (s, 1H); 6.77 (t, J = 20 Hz 2H); 7.05–7.12 (m, 3H); 7.17 (d, J = 12 Hz 1H); 7.41 (d, J = 8 Hz 2H); 7.57–7.62 (m, 4H). 13C NMR (CDCl3, 100 MHz): δ (ppm) 18.7, 34.5, 56.4, 108.6, 109.3, 109.4, 110.2, 114.6, 118.0, 118.1, 124.3, 125.5, 127.4, 127.5, 129.0, 130.0, 130.1, 130.6, 130.7, 133.1, 142.2, 144.5, 145.5, 148.0, 151.9, 152.8, 154.0, 160.4, 166.4. MS m/z: calcd. 539.9 [M]+. Anal. Elem. Calc. for C29H21N3O6S: C, 64.56: H, 3.92: N, 7.79: Found: C, 64.53: H, 3.93: N, 7.80.
3.2.3. 4-Methyl-2-oxo-2H-chromen-7-yl-2-((5-(7-methoxybenzofuran-2-yl)-4-(4-methoxyphenyl)-4H-1,2,4-triazol-3-yl)thio)acetate (17c)
- Coarse off-white powder; yield, 64%; mp 137 °C; 1H NMR (CDCl3, 400 MHz) δ (ppm): 2.36 (s, 3H); 3.82 (s, 3H); 3.85 (s, 3H); 4.27 (s, 2H); 6.2 (s, 1H); 6.57 (s, 1H); 6.74 (d, J = 8 Hz, 1H); 7.0–7.08 (m, 5H); 7.13 (d, J = 8 Hz, 1H); 7.26 (d, J = 8 Hz, 2H); 7.54 (d, J = 8 Hz, 1H). 13C NMR (CDCl3, 100 MHz): δ (ppm) 18.7, 34.4, 55.7, 56.4, 108.2, 109.0, 110.2, 113.8, 114.6, 114.6, 115.2, 115.3, 118.0, 124.2, 125.5, 125.5, 128.6, 128.6, 129.1, 142.5, 144.5, 145.5, 148.3, 151.9, 152.1, 152.8, 154.0, 160.4, 161.1, 166.5. MS m/z: 569.9 [M]+. Anal. Elem. Calc. for C30H23N3O7S: C, 63.26: H, 4.07: N, 7.38: Found: C, 63.25: H, 4.09: N, 7.39.
3.2.4. 4-Methyl-2-oxo-2H-chromen-7-yl-2-((5-(5-bromobenzofuran-2-yl)-4-phenyl-4H-1,2,4-triazol-3-yl)thio)acetate (17d)
- Coarse white powder; yield, 70%; mp 232 °C; 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 2.49 (s, 3H); 4.45 (s, 2H); 6.41 (s, 1H); 6.50 (s, 1H); 7.23 (d, J = 28 Hz 2H); 7.51–7.69 (m, 7 H); 7.87 (s, 2H). 13C NMR (DMSO-d6, 100 MHz): δ (ppm) 18.6, 34.5, 107.0, 110.2, 113.9, 114.3, 118.5, 118.5, 125.0, 125.0, 125.0, 127.1, 128.0, 128.1, 128.1, 129.3, 130.7, 133.3, 133.3, 144.3, 144.3, 144.5, 147.5, 153.0, 153.3, 153.3, 160.0, 167.4. MS m/z: 587.6 [M]+. Anal. Elem. Calc. for C28H18BrN3O5S: C, 57.15: H, 3.08: N, 7.14: Found: C, 57.17: H, 3.09: N, 7.17.
3.2.5. 4-Methyl-2-oxo-2H-chromen-7-yl-2-((5-(5-bromobenzofuran-2-yl)-4-(4-methoxyphenyl)-4H-1,2,4-triazol-3-yl)thio)acetate (17e)
- Coarse gray solid; yield, 62%; mp 220 °C 1H NMR (CDCl3, 400 MHz) δ (ppm): 2.40 (s, 3H); 3.91 (s, 3H); 4.34 (s, 2H); 6.25 (s, 1H); 6.42 (s, 1H); 7.08 (d, J = 8 Hz, 2H); 7.13 (s, 1H); 7.17 (d, J = 8 Hz, 1H); 7.31 (d, J = 8 Hz, 3H); 7.39 (d, J = 8 Hz, 1H); 7.59 (d, J = 8 Hz, 2H). 13C NMR (CDCl3, 100 MHz): δ (ppm) 18.7, 34.6, 55.7, 106.8, 110.2, 113.2, 113.2, 114.6, 115.4, 116.6, 118.0, 118.1, 124.2, 125.0, 125.5, 128.6, 129.1, 129.2, 143.6, 148.0, 148.0, 151.8, 152.7, 152.8, 153.5, 154.0, 160.3, 161.3, 166.4. MS m/z: 617.6 [M]+. Anal. Elem. Calc. for C29H20BrN3O6S: C, 56.62: H, 3.26: N, 6.79: Found: C, 56.65: H, 3.27: N, 6.80.
3.2.6. 4-Methyl-2-oxo-2H-chromen-7-yl-2-((5-(5-chlorobenzofuran-2-yl)-4-phenyl-4H-1,2,4-triazol-3-yl)thio)acetate (17f)
- Amorphous white solid; yield, 72%; mp 226 °C; 1H NMR (DMSO-d6, 400 MHz) δ (ppm): 2.49 (s, 3H); 4.45 (s, 2H); 6.41 (s, 1H); 6.50 (s, 1H); 7.23 (t, J = 28 Hz, 1H); 7.38 (d, J = 12 Hz, 2H); 7.57–7.85 (m, 7H); 7.88 (d, J = 12 Hz, 1H). 13C NMR (DMSO-d6, 100 MHz): δ (ppm) 18.6, 34.5, 107.1, 110.2, 113.4, 114.3, 114.3, 118.5, 122.0, 126.6, 126.6, 126.6, 127.1, 128.0, 128.5, 129.1, 129.1, 130.7, 131.4, 133.32, 144.5, 144.5, 147.5, 152.6, 152.6, 153.9, 160.0, 167.4. MS m/z: 543.9 [M]+. Anal. Elem. Calc. for C28H18ClN3O5S: C, 61.82: H, 3.34: N, 7.72: Found: C, 61.84: H, 3.35: N, 7.70.
3.2.7. 4-Methyl-2-oxo-2H-chromen-7-yl2-((5-(5-chlorobenzofuran-2-yl)-4-(4-methoxyphenyl)-4H-1,2,4-triazol-3-yl)thio)acetate (17g)
- Coarse off-white solid; yield, 67%; mp 200 °C; 1H NMR (CDCl3, 400 MHz) δ (ppm): 2.32 (s, 3H); 3.68–4.12 (m, 3H); 4.12 (s, 2H); 6.01 (s, 1H); 6.74 (t, J = 16 Hz, 2H); 6.83–6.92 (m, 1H); 7.07 (t, J = 16 Hz, 2H); 7.15–7.30 (m, 5H); 7.35 (t, J = 24 Hz 1H). 13C NMR (CDCl3, 100 MHz): δ (ppm) 18.6, 21.0, 22.6, 31.5, 55.4, 60.4, 103.3, 110.1, 110.7, 112.7, 113.5, 114.3, 115.4, 115.6, 118.0, 121.2, 125.6, 125.6, 126.5, 128.6, 128.9, 128.9, 152.8, 152.8, 153.4, 155.0, 160.9, 162.2, 171.3. MS m/z: 571.9 [M]+. Anal. Elem. Calc. for C29H20ClN3O6S: C, 60.68: H, 3.51: N, 7.32: Found: C, 60.71: H, 3.52: N, 7.34.
3.2.8. 4-Methyl-2-oxo-2H-chromen-7-yl-2-((5-(naphtho[2,1-b]furan-2-yl)-4-phenyl-4H-1,2,4-triazol-3-yl)thio)acetate (17h)
- Yellow solid; yield, 63%; mp 218 °C; 1H NMR (CDCl3, 400 MHz) δ (ppm): 2.35 (s, 3H); 4.32 (s, 2H); 6.19 (s, 1H); 7.08 (s, 1H); 7.14–7.21 (m, 3H); 7.42 (d, J = 8 Hz, 3H); 7.48 (t, J = 16 Hz 1H); 7.54–7.63 (m, 5H); 7.66–7.87 (m, 2H). 13C NMR (CDCl3, 100 MHz): δ (ppm) 18.7, 34.5, 110.2, 110.2, 112.2, 112.2, 114.6, 118.0, 122.8, 123.2, 125.7, 125.6, 126.9, 126.9, 127.8, 127.8, 127.8, 128.8, 128.8, 128.8, 128.8, 130.3, 130.4, 131.1, 132.9, 140.98, 151.8, 152.8, 152.8, 154.0, 160.4, 166.3. MS m/z: 559.9 [M]+. Anal. Elem. Calc. for C32H21ClN3O5S: C, 68.68: H, 3.78: N, 7.51: Found: C, 68.70: H, 3.76: N, 7.52.
3.3. Tyrosine Assay of Inhibitory Activity of Target Compounds
3.4. In Silico Modeling Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, H.-Y.; Yeh, Y.-C. Detection of tyrosine and monitoring tyrosinase activity using an enzyme cascade-triggered colorimetric reaction. RSC. Adv. 2020, 10, 29745–29750. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Wen, H.; Li, J.; Lu, L.; Zhao, W.; Jiang, X.; Bai, R. Small-molecule agents for treating skin diseases. Eur. J. Med. Chem. 2024, 268, 116269. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.D.; Choi, H.; Abekura, F.; Park, J.Y.; Yang, W.S.; Yang, S.H.; Kim, C.H. Naturally-occurring tyrosinase inhibitors classified by enzyme kinetics and copper chelation. Int. J. Mol. Sci. 2023, 24, 8226. [Google Scholar] [CrossRef] [PubMed]
- Irfan, A.; Zahoor, A.F.; Kamal, S.; Hassan, M.; Kloczkowski, A. Ultrasonic-Assisted Synthesis of Benzofuran Appended Oxadiazole Molecules as Tyrosinase Inhibitors: Mechanistic Approach through Enzyme Inhibition, Molecular Docking, Chemoinformatics, ADMET and Drug-Likeness Studies. Int. J. Mol. Sci. 2022, 23, 10979. [Google Scholar] [CrossRef] [PubMed]
- Nagatsu, T.; Nakashima, A.; Watanabe, H.; Ito, S.; Wakamatsu, K. Neuromelanin in Parkinson’s Disease: Tyrosine Hydroxylase and Tyrosinase. Int. J. Mol. Sci. 2022, 23, 4176. [Google Scholar] [CrossRef]
- Fang, X.; Dai, L.; Ding, T.M.; Zhu, Y.; Zan, J.F.; Chen, L.L.; Ding, X.P.; Liu, J.F. Screening and identification of tyrosinase inhibitors in edible plant materials by on-line UPLC-enzyme reactor coupled with UHPLC-FTMS. Food Chem. 2023, 403, 134331. [Google Scholar] [CrossRef] [PubMed]
- Xue, S.; Li, Z.; Ze, X.; Wu, X.; He, C.; Shuai, W.; Marlow, M.; Chen, J.; Scurr, D.; Zhu, Z.; et al. Design, synthesis, and biological evaluation of novel hybrids containing dihydrochalcone as tyrosinase inhibitors to treat skin hyperpigmentation. J. Med. Chem. 2023, 66, 5099–5117. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.; Shahzadi, S.; Kloczkowski, A. Tyrosinase inhibitors naturally present in plants and synthetic modifications of these natural products as anti-melanogenic agents: A review. Molecules 2023, 28, 378. [Google Scholar] [CrossRef] [PubMed]
- Nagatsu, T.; Nakashima, A.; Watanabe, H.; Ito, S.; Wakamatsu, K.; Zucca, F.A.; Zecca, L.; Youdim, M.; Wulf, M.; Riederer, P.; et al. The role of tyrosine hydroxylase as a key player in neuromelanin synthesis and the association of neuromelanin with Parkinson’s disease. J. Neural Transm. 2023, 130, 611–625. [Google Scholar] [CrossRef]
- Marieshwari, B.N.; Bhuvaragavan, S.; Sruthi, K.; Mullainadhan, P.; Janarthanan, S. Insect phenoloxidase and its diverse roles: Melanogenesis and beyond. J. Comp. Physiol. B 2023, 193, 1–23. [Google Scholar] [CrossRef]
- Carradori, S.; Melfi, F.; Rešetar, J.; Şimşek, R. Tyrosinase enzyme and its inhibitors: An update of the literature. Metalloenzymes 2024, 533–546. [Google Scholar] [CrossRef]
- Vaezi, M. Structure and inhibition mechanism of some synthetic compounds and phenolic derivatives as tyrosinase inhibitors: Review and new insight. J. Biomol. Struct. Dyn. 2023, 41, 4798–4810. [Google Scholar] [CrossRef] [PubMed]
- Baber, M.A.; Crist, C.M.; Devolve, N.L.; Patrone, J.D. Tyrosinase Inhibitors: A Perspective. Molecules 2023, 28, 5762. [Google Scholar] [CrossRef] [PubMed]
- Loizzo, R.; Tundis, R.; Menichini, F. Natural and Synthetic Tyrosinase Inhibitors as Antibrowning Agents: An Update. Compr. Rev. Food Sci. Food Saf. 2012, 4, 378–398. [Google Scholar] [CrossRef]
- Chandarajoti, K.; Kara, J.; Suwanhom, P.; Nualnoi, T.; Puripattanavong, J.; Lee, V.S.; Tipmanee, V.; Lomlim, L. Synthesis and evaluation of coumarin derivatives on antioxidative, tyrosinase inhibitory activities, melanogenesis, and in silico investigations. Sci. Rep. 2024, 14, 5535. [Google Scholar] [CrossRef] [PubMed]
- Bhosale, R.S.; Sarda, S.R.; Ardhapure, S.S.; Jadhav, W.N.; Bhusare, S.R.; Pawar, R.P. An efficient protocol for the synthesis of quinoxaline derivatives at room temperature using molecular iodine as the catalyst. Tetrahedron Lett. 2005, 46, 7183–7186. [Google Scholar] [CrossRef]
- Irfan, A.; Faisal, S.; Zahoor, A.F.; Noreen, R.; Al-Hussain, S.A.; Tuzun, B.; Javaid, R.; Elhenawy, A.A.; Zaki, M.E.A.; Ahmad, S.; et al. In Silico Development of Novel Benzofuran-1,3,4-Oxadiazoles as Lead Inhibitors of M. tuberculosis Polyketide Synthase 13. Pharmaceuticals 2023, 16, 829. [Google Scholar] [CrossRef]
- Banik, B.K.; Sahoo, B.M.; Kumar, B.V.; Panda, K.C.; Jena, J.; Mahaputra, M.K.; Borah, P. Green Synthetic Approach: An Efficient eco-friendly tool for synthesis of biologically active oxadiazole derivatives. Molecules 2021, 26, 1163. [Google Scholar] [CrossRef]
- Ahmad, S.; Khan, M.; Alam, A.; Ajmal, A.; Wadood, A.; Khan, A.; AlAsmari, A.F.; Alharbi, M.; Alshammari, A.; Shakoor, A. Novel flurbiprofen clubbed oxadiazole derivatives as potential urease inhibitors and their molecular docking study. RSC Adv. 2023, 13, 25717–25728. [Google Scholar] [CrossRef]
- Shahzadi, I.; Zahoor, A.F.; Rasul, A.; Rasool, N.; Raza, Z.; Faisal, S.; Parveen, B.; Kamal, S.; Zia-ur-Rehman, M.; Zahid, F.M. Synthesis, anticancer, and computational studies of 1, 3, 4-oxadiazole-purine derivatives. J. Heterocycl. Chem. 2020, 57, 2782–2794. [Google Scholar] [CrossRef]
- Shahzadi, I.; Zahoor, A.F.; Rasul, A.; Mansha, A.; Ahmad, S.; Raza, Z. Synthesis, hemolytic studies, and in silico modeling of novel acefylline–1, 2, 4-triazole hybrids as potential anti-cancer agents against MCF-7 and A549. ACS Omega 2021, 6, 11943–11953. [Google Scholar] [CrossRef] [PubMed]
- Korlyukov, A.A.; Stash, A.I.; Romanenko, A.R.; Trzybiński, D.; Woźniak, K.; Vologzhanina, A.V. Ligand-Receptor Interactions of Lamivudine: A View from Charge Density Study and QM/MM Calculations. Biomedicines 2023, 11, 743. [Google Scholar] [CrossRef] [PubMed]
- Hatami, M.; Basri, Z.; Sakhvidi, B.K.; Mortazavi, M. Thiadiazole—A promising structure in design and development of anti-Alzheimer agents. Int. Immunopharmacol. 2023, 118, 110027. [Google Scholar] [CrossRef]
- Rasgania, J.; Gavadia, R.; Nimesh, S.; Loveleen, L.; Mor, S.; Singh, D.; Jakhar, K. Synthesis of isatin-tagged thiadiazoles as anti-breast cancer leads: In-vitro and in-silico investigations. J. Mol. Struct. 2023, 1294, 136464. [Google Scholar] [CrossRef]
- Taiwo, F.O.; Abioye, O.E.; Obafemi, C.A.; Akinpelu, D.A. Synthesis and Antibacterial Potency of Some 3-methyl-2-oxo-1,2-dihydroquinoxaline-6-sulfonohydrazone Derivatives. J. Adv. Microb. 2023, 23, 56–70. [Google Scholar] [CrossRef]
- Akhtar, R.; Zahoor, A.F.; Rasul, A.; Khan, S.G.; Ali, K.G. In-vitro cytotoxic evaluation of newly designed ciprofloxacinoxadiazole hybrids against human liver tumor cell line (Huh7). Pak. J. Pharm. Sci. 2021, 34, 1143–1148. [Google Scholar]
- Li, Q.; Yang, Y.; Li, Y.; Mi, Y.; Ma, X.; Jiang, A.; Guo, Z. Enhanced biological activities of coumarin-functionalized polysaccharide derivatives: Chemical modification and activity assessment. Int. J. Biol. Macromol. 2023, 253, 126691. [Google Scholar] [CrossRef]
- Sadgir, N.V.; Adole, V.A.; Dhonnar, S.L.; Jagdale, B.S. Synthesis and biological evaluation of coumarin appended thiazole hybrid heterocycles: Antibacterial and antifungal study. J. Moe. Struct. 2023, 1293, 136229. [Google Scholar] [CrossRef]
- Yang, X.C.; Zeng, C.M.; Avula, S.R.; Peng, X.M.; Geng, R.X.; Zhou, C.H. Novel coumarin aminophosphonates as potential multitargeting antibacterial agents against Staphylococcus aureus. Eur. J. Med. Chem. 2023, 245, 114891. [Google Scholar] [CrossRef]
- Patel, K.B.; Mukherjee, S.; Bhatt, H.; Rajani, D.; Ahmad, I.; Patel, H.; Kumari, P. Synthesis, docking, and biological investigations of new coumarin-piperazine hybrids as potential antibacterial and anticancer agents. J. Mol. Struct. 2023, 1276, 134755. [Google Scholar] [CrossRef]
- Rai, U.S.; Isloor, A.M.; Shetty, P.; Vijesh, A.M.; Prabhu, N.; Isloor, S. Novel Chromeno [2,3-b]-pyrimidine Derivatives as Potential Anti-Microbial Agents. Eur. J. Med. Chem. 2010, 45, 2695–2699. [Google Scholar]
- Kostova, I. Coumarins as Inhibitors of HIV Reverse Transcriptase. Curr. HIV Res. 2006, 4, 347–363. [Google Scholar] [CrossRef] [PubMed]
- Ramsis, T.M.; Ebrahim, M.A.; Fayed, E.A. Synthetic Coumarin Derivatives with Anticoagulation and Antiplatelet Aggregation Inhibitory Effects. Med. Chem. Res. 2023, 32, 2269–2278. [Google Scholar] [CrossRef]
- Claudio, B.; Adriana, M.; Francesco, M.; Keith, W.; Monica, B.; Antonino, S.; Santi, S. New Coumarin-Based Anti-Inflammatory Drug: Putative Antagonist of the Integrins aLb2 and aMb2. J. Pharm. Pharmacol. 2008, 60, 1473–1479. [Google Scholar]
- Matos, M.J.; Varela, C.; Vilar, S.; Hripcsak, G.; Borges, F.; Santana, L.; Uriarte, E.; Fais, A.; Petrillo, A.D.; Pintus, F.; et al. Design and discovery of tyrosinase inhibitors based on a coumarin scaffold. RSC Adv. 2015, 5, 94227–94235. [Google Scholar] [CrossRef]
- Matin, P.; Matin, M.; Rehaman, R.; Hadda, T.B.; Almalki, F.A.; Mehmud, S.; Ghoneim, M.M.; Maha, A.; Sultan, A. Triazoles and Their Derivatives: Chemistry, Synthesis, and Therapeutic Applications. Mol. Diagn. Ther. 2022, 9, 864286. [Google Scholar] [CrossRef] [PubMed]
- Bhat, K.S.; Poojary, B.; Prasad, D.J.; Naik, P.; Holla, B.S. Synthesis and Antitumor Activity Studies of Some New Fused 1, 2, 4-triazole Derivatives Carrying 2,4-dichloro-5-fluorophenyl Moiety. Eur. J. Med. Chem. 2009, 44, 5066–5070. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, N.; Kumar, R.; Dureja, P.; Khurana, J.M. Synthesis, Antimicrobial Evaluation and QSAR analysis of Novel Nalidixic Acid Based 1, 2, 4-triazole Derivatives. Eur. J. Med. Chem. 2021, 46, 4089–4099. [Google Scholar] [CrossRef]
- Divar, M.; Tadayyon, S.; Khoshneviszadeh, M.; Pirhadi, S.; Attarroshan, M.; Mobaraki, K.; Damghani, T.; Mirfazli, S.; Edraki, N. Benzyl-Triazole Derivatives of Hydrazinecarbothiamide Derivatives as Potent Tyrosinase Inhibitors: Synthesis, Biological Evaluation, Structure-Activity Relationship and Docking Study. Chem. Sel. 2023, 8, e202203382. [Google Scholar] [CrossRef]
- Najafi, Z.; Ebadi, A.; Chehardoli, G.; Ziaei, M.; Akbarzadeh, T.; Saeedi, M.; Gholamhoseini, P.; Mahdavi, M. Design, synthesis, in vitro, and in silico studies of novel benzylidene 6-methoxy-1-tetralone linked to benzyloxy and benzyl-1, 2, 3-triazole rings as potential tyrosinase inhibitors. J. Mol. Struct. 2023, 1271, 134018. [Google Scholar] [CrossRef]
- Gao, F.; Wang, T.; Xiao, J.; Huang, G. Antibacterial Activity Study of 1, 2, 4-triazole Derivatives. Eur. J. Med. Chem. 2019, 173, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Faiz, S.; Zahoor, A.F.; Ajmal, M.; Kamal, S.; Ahmad, S.; Abdelgawad, A.M.; Elnaggar, M.E. Design, Synthesis, Antimicrobial Evaluation, and Laccase Catalysis Effect of Novel Benzofuran—Oxadiazole and Benzofuran–triazole hybrids. J. Heterocycl. Chem. 2019, 56, 2839–2852. [Google Scholar] [CrossRef]
- Akın, S.; Demir, E.A.; Colak, A.; Kolcuoglu, Y.; Yildirim, N.; Bekircan, O. Synthesis, Biological Activities and Molecular Docking Studies of Some Novel 2,4,5- trisubstituted-1,2,4-triazole-3-one Derivatives as Potent Tyrosinase Inhibitors. J. Mol. Struct. 2019, 1175, 280–286. [Google Scholar] [CrossRef]
- Landge, S.; Philp, J.; Ugboya, A.; Graves, I.; Fasusi, E.; Jordan, K.; Aiken, K.; Sittaramane, V. Evaluation of ortho-substituted Bis-Functionalized Triazoles as Tyrosinase Inhibitors: Modulating Dopamine Synthesis and Behavior in Zebrafish. Med. Chem. Res. 2024, 33, 651–662. [Google Scholar] [CrossRef]
- Wood, W.J.L.; Patterson, A.W.; Tsuruoka, H.; Jain, R.K.; Ellman, J.A. Substrate Activity Screening: A Fragment-based Method for the Rapid Identification of Non peptidic Protease Inhibitors. J. Am. Chem. Soc. 2005, 127, 15521. [Google Scholar] [CrossRef] [PubMed]
- Apperley, K.Y.P.; Roy, I.; Saucier, V.; Brunet-Filion, N.; Piscopo, S.P.; Pardin, C.; De Francesco, E.; Hao, C.; Keillor, J.W. Development of New Scaffolds as Reversible Tissue Transglutaminase Inhibitors, with Improved Potency or Resistance to Glutathione Addition. Med. Chem. Comm. 2017, 8, 338. [Google Scholar] [CrossRef] [PubMed]
- Anand, N.; Jaiswal, N.; Pandey, S.K.; Srivastava, A.K.; Tripathi, R.P. Application of Click Chemistry Towards an Efficient Synthesis of 1, 2, 3-1H-triazolyl Glycohybrids as Enzyme Inhibitors. Carbohydr. Res. 2011, 346, 16. [Google Scholar] [CrossRef]
- Pardin, C.; Pelletier, J.N.; Lubell, W.D.; Keillor, J.W. Cinnamoyl inhibitors of tissue transglutaminase. J. Org. Chem. 2008, 73, 5766–5775. [Google Scholar] [CrossRef]
- Fan, Y.L.; Ke, X.; Liu, M. Coumarin–triazole hybrids and their biological activities. J. Heterocycl. Chem. 2018, 55, 791–802. [Google Scholar] [CrossRef]
- Liu, J.; Wu, F.; Chen, L.; Zhao, L.; Zhao, Z.; Wang, M.; Lei, S. Biological Evaluation of Coumarin Derivatives as Mushroom Tyrosinase Inhibitors. Food. Chem. 2012, 135, 2872. [Google Scholar] [CrossRef]
- Fais, A.; Corda, M.; Era, B.; Fadda, M.B.; Matos, M.J.; Quezada, E.; Santana, L.; Picciau, C.; Podda, G.; Delogu, G. Tyrosinase Inhibitor Activity of Coumarin-Resveratrol Hybrids. Molecules 2009, 14, 2514–2520. [Google Scholar] [CrossRef] [PubMed]
- Nunes, J.A.; Araújo, R.S.A.d.; Silva, F.N.d.; Cytarska, J.; Łączkowski, K.Z.; Cardoso, S.H.; Mendonça-Júnior, F.J.B.; Silva-Júnior, E.F.d. Coumarin-based compounds as inhibitors of tyrosinase/tyrosine hydroxylase: Synthesis, kinetic studies, and in silico approaches. Int. J. Mol. Sci. 2023, 24, 5216. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Zhang, X.; Kang, Y.; Xiong, Z.; Zhang, K.; Xu, X.; Bai, L.; Li, H. Novel Coumarin Derivatives as Potential Tyrosinase Inhibitors: Synthesis, Binding Analysis and Biological Evaluation. Arab. J. Chem. 2023, 16, 104724. [Google Scholar] [CrossRef]
- Ivasiv, V.; Viktoriya, R.; Claudia, A.; Ana, E.; Gonçalves, M.R.; Maria, L. Molecular Hybridization as a Tool for Designing Multitarget Drug Candidates for Complex Diseases. Curr. Top. Med. Chem. 2019, 19, 1694–1711. [Google Scholar] [CrossRef] [PubMed]
- Cetinkaya, Y.; Göçer, H.; Göksu, S.; Gülçin, I. Synthesis and Carbonic Anhydrase Isoenzymes I and II Inhibitory Effects of Novel Benzylamine Derivatives. J. Enzym. Inhib. Med. Chem. 2014, 29, 168–174. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, J.V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Tran, L.M.; Stang, J.L. Induced-fit Docking Studies of the Active and Inactive States of Protein Tyrosine Kinases. J. Mol. Graph. Model. 2009, 28, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Ismaya, W.T.; Rozeboom, H.J.; Weijn, A.; Mes, J.J.; Fusetti, F.; Wichers, H.J.; Dijkstra, B.W. Crystal Structure of Agaricus Bisporus Mushroom Tyrosinase: Identity of the Tetramer Subunits and Interaction with Tropolone. Biochemistry 2011, 50, 5477–5486. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sr. No. | Target Compound | R1 | R2 | %Age Inhibition | IC50 (µM) a |
|---|---|---|---|---|---|
| 1 | 17a | H | C6H5 | 12.02 ± 0.21 | 8.13 ± 0.13 |
| 2 | 17b | 7-OMe | C6H5 | 6.30 ± 0.14 | 14.06 ± 0.17 |
| 3 | 17c | 7-OMe | 4-OMeC6H4 | 5.11 ± 0.15 | 12.98 ± 0.32 |
| 4 | 17d | 5-Br | C6H5 | 23.09 ± 0.13 | 5.89 ± 0.09 |
| 5 | 17e | 5-Br | 4-OMeC6H4 | 30.85 ± 0.15 | 0.33 ± 0.08 |
| 6 | 17f | 5-Cl | C6H5 | 36.13 ± 0.21 | 3.14 ± 0.23 |
| 7 | 17g | 5-Cl | 4-OMeC6H4 | 31.060 ± 0.19 | 3.98 ± 0.12 |
| 8 | 17h | Fused C6H4 | C6H5 | 30.62 ± 0.33 | 5.13 ± 0.05 |
| 10 | Ascorbic acid (standard) | 58.66 ± 1.00 | 11.5 ± 1.00 | ||
| 11 | Kojic acid (standard) | 6.79 ± 0.58 | 30.34 ± 0.75 |
| Interaction Study | Kojic Acid | Compound 17e | Compound 17f |
|---|---|---|---|
| Docking score (kcal/mol) | −5.25 | −6.75 | −6.29 |
| Ligand–receptor interaction | VAL-283,ALA-286, GLY-281, HIS-61, HIS-263, HIS-85, and MET-280 | GLY-281, HIS-263, HIS-259, VAL-283, HIS-85, PHE-264, and VAL-248 | VAL-268, LEU-275, VAL-283, HIS-85, HIS-259, HIS-263, GLY-281, and ARG-268 |
| Interaction types | π–π stacked, hydrogen bonding, and π–alkyl | C-H bond, alkyl, π–π T-shaped, and alkyl–π | Carbon–hydrogen bond, alkyl, π–alkyl, and π–donor hydrogen bond |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kausar, R.; Zahoor, A.F.; Tabassum, H.; Kamal, S.; Ahmad Bhat, M. Synergistic Biomedical Potential and Molecular Docking Analyses of Coumarin–Triazole Hybrids as Tyrosinase Inhibitors: Design, Synthesis, In Vitro Profiling, and In Silico Studies. Pharmaceuticals 2024, 17, 532. https://doi.org/10.3390/ph17040532
Kausar R, Zahoor AF, Tabassum H, Kamal S, Ahmad Bhat M. Synergistic Biomedical Potential and Molecular Docking Analyses of Coumarin–Triazole Hybrids as Tyrosinase Inhibitors: Design, Synthesis, In Vitro Profiling, and In Silico Studies. Pharmaceuticals. 2024; 17(4):532. https://doi.org/10.3390/ph17040532
Chicago/Turabian StyleKausar, Rukhsana, Ameer Fawad Zahoor, Hina Tabassum, Shagufta Kamal, and Mashooq Ahmad Bhat. 2024. "Synergistic Biomedical Potential and Molecular Docking Analyses of Coumarin–Triazole Hybrids as Tyrosinase Inhibitors: Design, Synthesis, In Vitro Profiling, and In Silico Studies" Pharmaceuticals 17, no. 4: 532. https://doi.org/10.3390/ph17040532
APA StyleKausar, R., Zahoor, A. F., Tabassum, H., Kamal, S., & Ahmad Bhat, M. (2024). Synergistic Biomedical Potential and Molecular Docking Analyses of Coumarin–Triazole Hybrids as Tyrosinase Inhibitors: Design, Synthesis, In Vitro Profiling, and In Silico Studies. Pharmaceuticals, 17(4), 532. https://doi.org/10.3390/ph17040532