Aptamers for Targeted Drug Delivery

Abstract
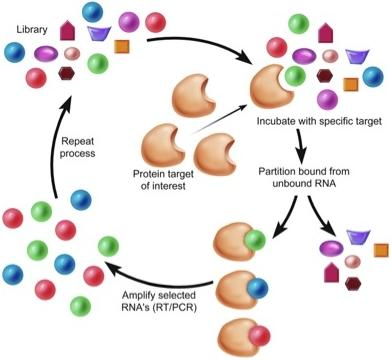
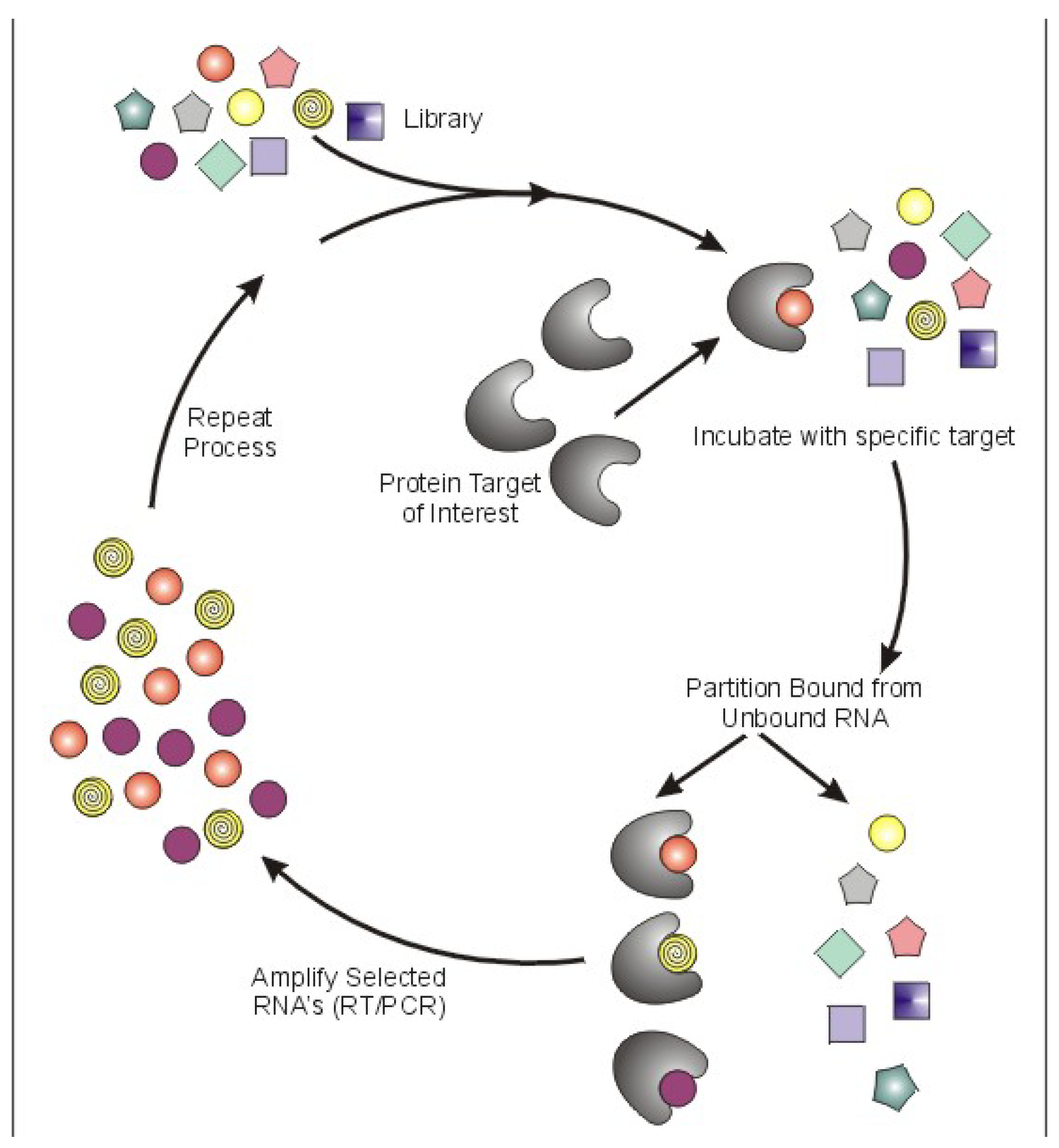
:
1. Introduction


{kind=link}
{kind=link}
{kind=link}
| Receptor Name | RNA/DNA | Selection Technique | Delivery Application |
|---|---|---|---|
| Tenascin-C (TN-C) | RNA | Purified TN-C | In vivo tumor imaging [25] |
| Nucleolin | DNA | Not applicable | Photodynamic therapy (PDT) [26], tumor imaging [27] |
| Prostate Specific Membrane Antigen (PSMA) | RNA | Purified extracellular domain of PSMA | siRNA delivery [28,29,30], cytotoxin delivery [31], Chemotherapeutic drug delivery and cellular imaging [32,33,34] |
| gp120 | RNA | Purified recombinant gp120 | siRNA delivery [35] |
| Transferrin receptor (TfR) | RNA/DNA | Purified extracellular domain of mouse TfR | Protein targeting to lysosome [13] |
| Mucin-1 (MUC-1) | DNA | Recombinant peptides | Photodynamic therapy (PDT) [36], Radionuclide delivery [37] |
| Protein tyrosine kinase-7 (PTK7) | DNA | Cell SELEX using T-cell acute lymphoblastic leukemia (ALL) cell line | Chemotherapeutic drug delivery [38] |
| Immunoglobin heavy mu chain (IGHM) | DNA | Cell SELEX using Burkitt’s lymphoma cell line (Ramos) | Micelle nanoparticles for drug delivery [39] |
| Epidermal growth factor receptor (EGFR) | RNA | Purified extracellular domain of EGFR | Nanoparticle delivery [22] |
2. Tenascin-C
3. Prostate-Specific Membrane Antigen
4. gp120
5. Nucleolin
6. Transferrin receptor (TfR)
7. Mucin-1(MUC-1)
8. Protein tyrosine kinase 7 (PTK7)
9. Immunoglobin Heavy Mu Chain
10. Epidermal Growth Factor Receptor (EGFR)
11. Summary
References
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar]
- Knudsen, S.M.; Robertson, M.P.; Ellington, A.D. In vitro selection using modified or unnatural nucleotides. Curr. Protoc. Nucleic Acid Chem. 2002, Chapter 9. Unit 9.6.. [Google Scholar]
- Sledz, C.A.; Holko, M.; de Veer, M.J.; Silverman, R.H.; Williams, B.R. Activation of the interferon system by short-interfering RNAs. Nat. Cell Biol. 2003, 5, 834–839. [Google Scholar]
- Okahira, S.; Nishikawa, F.; Nishikawa, S.; Akazawa, T.; Seya, T.; Matsumoto, M. Interferon-beta induction through toll-like receptor 3 depends on double-stranded RNA structure. DNA Cell Biol. 2005, 24, 614–623. [Google Scholar]
- Healy, J.M.; Lewis, S.D.; Kurz, M.; Boomer, R.M.; Thompson, K.M.; Wilson, C.; McCauley, T.G. Pharmacokinetics and biodistribution of novel aptamer compositions. Pharm. Res. 2004, 21, 2234–2246. [Google Scholar]
- Bunka, D.H.; Stockley, P.G. Aptamers come of age—At last. Nat. Rev. Microbiol. 2006, 4, 588–596. [Google Scholar]
- Gold, L. Oligonucleotides as research, diagnostic, and therapeutic agents. J. Biol. Chem. 1995, 270, 13581–13584. [Google Scholar] [PubMed]
- White, R.R.; Sullenger, B.A.; Rusconi, C.P. Developing aptamers into therapeutics. J. Clin. Invest. 2000, 106, 929–934. [Google Scholar]
- Nimjee, S.M.; Rusconi, C.P.; Sullenger, B.A. Aptamers: An emerging class of therapeutics. Annu. Rev. Med. 2005, 56, 555–583. [Google Scholar]
- Ng, E.W.; Shima, D.T.; Calias, P.; Cunningham, E.T., Jr.; Guyer, D.R.; Adamis, A.P. Pegaptanib, a targeted anti-VEGF aptamer for ocular vascular disease. Nat. Rev. Drug. Discov. 2006, 5, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Khati, M.; Schuman, M.; Ibrahim, J.; Sattentau, Q.; Gordon, S.; James, W. Neutralization of infectivity of diverse R5 clinical isolates of human immunodeficiency virus type 1 by gp120-binding 2'F-RNA aptamers. J. Virol. 2003, 77, 12692–12698. [Google Scholar]
- Chen, C.H.; Dellamaggiore, K.R.; Ouellette, C.P.; Sedano, C.D.; Lizadjohry, M.; Chernis, G.A.; Gonzales, M.; Baltasar, F.E.; Fan, A.L.; Myerowitz, R.; Neufeld, E.F. Aptamer-based endocytosis of a lysosomal enzyme. Proc. Natl. Acad. Sci. USA 2008, 105, 15908–15913. [Google Scholar]
- Lupold, S.E.; Hicke, B.J.; Lin, Y.; Coffey, D.S. Identification and characterization of nuclease-stabilized RNA molecules that bind human prostate cancer cells via the prostate-specific membrane antigen. Cancer Res. 2002, 62, 4029–4033. [Google Scholar]
- Santulli-Marotto, S.; Nair, S.K.; Rusconi, C.; Sullenger, B.; Gilboa, E. Multivalent RNA aptamers that inhibit CTLA-4 and enhance tumor immunity. Cancer Res. 2003, 63, 7483–7489. [Google Scholar]
- Cerchia, L.; Duconge, F.; Pestourie, C.; Boulay, J.; Aissouni, Y.; Gombert, K.; Tavitian, B.; de Franciscis, V.; Libri, D. Neutralizing aptamers from whole-cell SELEX inhibit the RET receptor tyrosine kinase. PLoS Biol. 2005, 3, e123. [Google Scholar]
- Mi, J.; Zhang, X.; Giangrande, P.H.; McNamara, J.O., 2nd; Nimjee, S.M.; Sarraf-Yazdi, S.; Sullenger, B.A.; Clary, B.M. Targeted inhibition of alphavbeta3 integrin with an RNA aptamer impairs endothelial cell growth and survival. Biochem .Biophys. Res. Commun. 2005, 338, 956–963. [Google Scholar] [CrossRef] [PubMed]
- Ohuchi, S.P.; Ohtsu, T.; Nakamura, Y. Selection of RNA aptamers against recombinant transforming growth factor-beta type III receptor displayed on cell surface. Biochimie 2006, 88, 897–904. [Google Scholar]
- McNamara, J.O.; Kolonias, D.; Pastor, F.; Mittler, R.S.; Chen, L.; Giangrande, P.H.; Sullenger, B.; Gilboa, E. Multivalent 4-1BB binding aptamers costimulate CD8+ T cells and inhibit tumor growth in mice. J. Clin. Invest. 2008, 118, 376–386. [Google Scholar]
- Dollins, C.M.; Nair, S.; Boczkowski, D.; Lee, J.; Layzer, J.M.; Gilboa, E.; Sullenger, B.A. Assembling OX40 aptamers on a molecular scaffold to create a receptor-activating aptamer. Chem. Biol. 2008, 15, 675–682. [Google Scholar]
- Mi, J.; Liu, Y.; Rabbani, Z.N.; Yang, Z.; Urban, J.H.; Sullenger, B.A.; Clary, B.M. In vivo selection of tumor-targeting RNA motifs. Nat. Chem. Biol. 2010, 6, 22–24. [Google Scholar] [PubMed]
- Li, N.; Larson, T.; Nguyen, H.H.; Sokolov, K.V.; Ellington, A.D. Directed evolution of gold nanoparticle delivery to cells. Chem. Commun. 46, 392–394.
- Thiel, K.W.; Giangrande, P.H. Therapeutic applications of DNA and RNA aptamers. Oligonucleotides 2009, 19, 209–222. [Google Scholar]
- Levy-Nissenbaum, E.; Radovic-Moreno, A.F.; Wang, A.Z.; Langer, R.; Farokhzad, O.C. Nanotechnology and aptamers: Applications in drug delivery. Trends Biotechnol. 2008, 26, 442–449. [Google Scholar]
- Hicke, B.J.; Stephens, A.W.; Gould, T.; Chang, Y.F.; Lynott, C.K.; Heil, J.; Borkowski, S.; Hilger, C.S.; Cook, G.; Warren, S.; Schmidt, P.G. Tumor targeting by an aptamer. J. Nucl. Med. 2006, 47, 668–678. [Google Scholar]
- Shieh, Y.A.; Yang, S.J.; Wei, M.F.; Shieh, M.J. Aptamer-based tumor-targeted drug delivery for photodynamic therapy. ACS Nano 2010, 4(3), 1433–1442. [Google Scholar] [PubMed]
- Hwang do, W.; Ko, H.Y.; Lee, J.H.; Kang, H.; Ryu, S.H.; Song, I.C.; Lee, D.S.; Kim, S. A nucleolin-targeted multimodal nanoparticle imaging probe for tracking cancer cells using an aptamer. J. Nucl. Med. 2009, 51, 98–105. [Google Scholar]
- Chu, T.C.; Twu, K.Y.; Ellington, A.D.; Levy, M. Aptamer mediated siRNA delivery. Nucleic Acids Res. 2006, 34, e73. [Google Scholar]
- McNamara, J.O., 2nd; Andrechek, E.R.; Wang, Y.; Viles, K.D.; Rempel, R.E.; Gilboa, E.; Sullenger, B.A.; Giangrande, P.H. Cell type-specific delivery of siRNAs with aptamer-siRNA chimeras. Nat. Biotechnol. 2006, 24, 1005–1015. [Google Scholar] [PubMed]
- Wullner, U.; Neef, I.; Eller, A.; Kleines, M.; Tur, M.K.; Barth, S. Cell-specific induction of apoptosis by rationally designed bivalent aptamer-siRNA transcripts silencing eukaryotic elongation factor 2. Curr. Cancer Drug Targets 2008, 8, 554–565. [Google Scholar]
- Chu, T.C.; Marks, J.W., III; Lavery, L.A.; Faulkner, S.; Rosenblum, M.G.; Ellington, A.D.; Levy, M. Aptamer: Toxin conjugates that specifically target prostate tumor cells. Cancer Res. 2006, 66, 5989–5992. [Google Scholar] [PubMed]
- Bagalkot, V.; Zhang, L.; Levy-Nissenbaum, E.; Jon, S.; Kantoff, P.W.; Langer, R.; Farokhzad, O.C. Quantum dot-aptamer conjugates for synchronous cancer imaging, therapy, and sensing of drug delivery based on bi-fluorescence resonance energy transfer. Nano Lett. 2007, 7, 3065–3070. [Google Scholar] [CrossRef] [PubMed]
- Farokhzad, O.C.; Cheng, J.; Teply, B.A.; Sherifi, I.; Jon, S.; Kantoff, P.W.; Richie, J.P.; Langer, R. Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proc. Natl. Acad. Sci. USA 2006, 103, 6315–6320. [Google Scholar]
- Dhar, S.; Gu, F.X.; Langer, R.; Farokhzad, O.C.; Lippard, S.J. Targeted delivery of cisplatin to prostate cancer cells by aptamer functionalized Pt(IV) prodrug-PLGA-PEG nanoparticles. Proc. Natl. Acad. Sci. USA 2008, 105, 17356–17361. [Google Scholar]
- Zhou, J.; Li, H.; Li, S.; Zaia, J.; Rossi, J.J. Novel dual inhibitory function aptamer-siRNA delivery system for HIV-1 therapy. Mol. Ther. 2008, 16, 1481–1489. [Google Scholar]
- Ferreira, C.S.; Cheung, M.C.; Missailidis, S.; Bisland, S.; Gariepy, J. Phototoxic aptamers selectively enter and kill epithelial cancer cells. Nucleic Acids Res. 2009, 37, 866–876. [Google Scholar]
- Pieve, C.D.; Perkins, A.C.; Missailidis, S. Anti-MUC1 aptamers: radiolabelling with (99 m)Tc and biodistribution in MCF-7 tumour-bearing mice. Nucl. Med. Biol. 2009, 36, 703–710. [Google Scholar]
- Huang, Y.F.; Shangguan, D.; Liu, H.; Phillips, J.A.; Zhang, X.; Chen, Y.; Tan, W. Molecular assembly of an aptamer-drug conjugate for targeted drug delivery to tumor cells. Chembiochem 2009, 10, 862–868. [Google Scholar]
- Wu, Y.; Sefah, K.; Liu, H.; Wang, R.; Tan, W. DNA aptamer-micelle as an efficient detection/delivery vehicle toward cancer cells. Proc. Natl. Acad. Sci. USA 2010, 107, 5–10. [Google Scholar]
- Jahkola, T.; Toivonen, T.; Nordling, S.; von Smitten, K.; Virtanen, I. Expression of tenascin-C in intraductal carcinoma of human breast: Relationship to invasion. Eur. J. Cancer 1998, 34, 1687–1692. [Google Scholar]
- Erickson, H.P.; Bourdon, M.A. Tenascin: An extracellular matrix protein prominent in specialized embryonic tissues and tumors. Annu. Rev. Cell Biol. 1989, 5, 71–92. [Google Scholar]
- Daniels, D.A.; Chen, H.; Hicke, B.J.; Swiderek, K.M.; Gold, L. A tenascin-C aptamer identified by tumor cell SELEX: Systematic evolution of ligands by exponential enrichment. Proc. Natl. Acad. Sci. USA 2003, 100, 15416–15421. [Google Scholar]
- Hicke, B.J.; Marion, C.; Chang, Y.F.; Gould, T.; Lynott, C.K.; Parma, D.; Schmidt, P.G.; Warren, S. Tenascin-C aptamers are generated using tumor cells and purified protein. J. Biol. Chem. 2001, 276, 48644–48654. [Google Scholar]
- Tasch, J.; Gong, M.; Sadelain, M.; Heston, W.D. A unique folate hydrolase, prostate-specific membrane antigen (PSMA): A target for immunotherapy? Crit. Rev. Immunol. 2001, 21, 249–261. [Google Scholar]
- Liu, H.; Moy, P.; Kim, S.; Xia, Y.; Rajasekaran, A.; Navarro, V.; Knudsen, B.; Bander, N.H. Monoclonal antibodies to the extracellular domain of prostate-specific membrane antigen also react with tumor vascular endothelium. Cancer Res. 1997, 57, 3629–3634. [Google Scholar]
- Dassie, J.P.; Liu, X.Y.; Thomas, G.S.; Whitaker, R.M.; Thiel, K.W.; Stockdale, K.R.; Meyerholz, D.K.; McCaffrey, A.P.; McNamara, J.O., II; Giangrande, P.H. Systemic administration of optimized aptamer-siRNA chimeras promotes regression of PSMA-expressing tumors. Nat. Biotechnol. 2009, 27, 839–849. [Google Scholar] [PubMed]
- Kim, E.; Jung, Y.; Choi, H.; Yang, J.; Suh, J.S.; Huh, Y.M.; Kim, K.; Haam, S. Prostate cancer cell death produced by the co-delivery of Bcl-xL shRNA and doxorubicin using an aptamer-conjugated polyplex. Biomaterials 2010, 31, 4592–4599. [Google Scholar]
- Barre-Sinoussi, F.; Chermann, J.C.; Rey, F.; Nugeyre, M.T.; Chamaret, S.; Gruest, J.; Dauguet, C.; Axler-Blin, C.; Vezinet-Brun, F.; Rouzioux, C.; Rozenbaum, W.; Montagnier, L. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 1983, 220, 868–871. [Google Scholar]
- Gallo, R.C.; Sarin, P.S.; Gelmann, E.P.; Robert-Guroff, M.; Richardson, E.; Kalyanaraman, V.S.; Mann, D.; Sidhu, G.D.; Stahl, R.E.; Zolla-Pazner, S.; Leibowitch, J.; Popovic, M. Isolation of human T-cell leukemia virus in acquired immune deficiency syndrome (AIDS). Science 1983, 220, 865–867. [Google Scholar]
- Dalgleish, A.G.; Beverley, P.C.; Clapham, P.R.; Crawford, D.H.; Greaves, M.F.; Weiss, R.A. The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature 1984, 312, 763–767. [Google Scholar]
- Berger, E.A.; Doms, R.W.; Fenyo, E.M.; Korber, B.T.; Littman, D.R.; Moore, J.P.; Sattentau, Q.J.; Schuitemaker, H.; Sodroski, J.; Weiss, R.A. A new classification for HIV-1. Nature 1998, 391, 240. [Google Scholar]
- Melikyan, G.B.; Markosyan, R.M.; Hemmati, H.; Delmedico, M.K.; Lambert, D.M.; Cohen, F.S. Evidence that the transition of HIV-1 gp41 into a six-helix bundle, not the bundle configuration, induces membrane fusion. J. Cell Biol. 2000, 151, 413–423. [Google Scholar]
- Dey, A.K.; Khati, M.; Tang, M.; Wyatt, R.; Lea, S.M.; James, W. An aptamer that neutralizes R5 strains of human immunodeficiency virus type 1 blocks gp120-CCR5 interaction. J. Virol. 2005, 79, 13806–13810. [Google Scholar]
- Dey, A.K.; Griffiths, C.; Lea, S.M.; James, W. Structural characterization of an anti-gp120 RNA aptamer that neutralizes R5 strains of HIV-1. RNA 2005, 11, 873–884. [Google Scholar]
- Zhou, J.; Swiderski, P.; Li, H.; Zhang, J.; Neff, C.P.; Akkina, R.; Rossi, J.J. Selection, characterization and application of new RNA HIV gp 120 aptamers for facile delivery of Dicer substrate siRNAs into HIV infected cells. Nucleic Acids Res. 2009, 37, 3094–3109. [Google Scholar]
- Orrick, L.R.; Olson, M.O.; Busch, H. Comparison of nucleolar proteins of normal rat liver and Novikoff hepatoma ascites cells by two-dimensional polyacrylamide gel electrophoresis. Proc. Natl. Acad. Sci. USA 1973, 70, 1316–1320. [Google Scholar]
- Ginisty, H.; Sicard, H.; Roger, B.; Bouvet, P. Structure and functions of nucleolin. J. Cell. Sci. 1999, 112, 761–772, (Pt 6). [Google Scholar]
- Westmark, C.J.; Malter, J.S. Up-regulation of nucleolin mRNA and protein in peripheral blood mononuclear cells by extracellular-regulated kinase. J. Biol. Chem. 2001, 276, 1119–1126. [Google Scholar]
- Christian, S.; Pilch, J.; Akerman, M.E.; Porkka, K.; Laakkonen, P.; Ruoslahti, E. Nucleolin expressed at the cell surface is a marker of endothelial cells in angiogenic blood vessels. J. Cell Biol. 2003, 163, 871–878. [Google Scholar]
- Kadomatsu, K.; Muramatsu, T. Midkine and pleiotrophin in neural development and cancer. Cancer Lett. 2004, 204, 127–143. [Google Scholar]
- Bates, P.J.; Laber, D.A.; Miller, D.M.; Thomas, S.D.; Trent, J.O. Discovery and development of the G-rich oligonucleotide AS1411 as a novel treatment for cancer. Exp. Mol. Pathol. 2009, 86, 151–164. [Google Scholar]
- Soundararajan, S.; Chen, W.; Spicer, E.K.; Courtenay-Luck, N.; Fernandes, D.J. The nucleolin targeting aptamer AS1411 destabilizes Bcl-2 messenger RNA in human breast cancer cells. Cancer Res. 2008, 68, 2358–2365. [Google Scholar]
- Ireson, C.R.; Kelland, L.R. Discovery and development of anticancer aptamers. Mol. Cancer Ther. 2006, 5, 2957–2962. [Google Scholar]
- Drecoll, E.; Gaertner, F.C.; Miederer, M.; Blechert, B.; Vallon, M.; Muller, J.M.; Alke, A.; Seidl, C.; Bruchertseifer, F.; Morgenstern, A.; Senekowitsch-Schmidtke, R.; Essler, M. Treatment of peritoneal carcinomatosis by targeted delivery of the radio-labeled tumor homing peptide bi-DTPA-[F3]2 into the nucleus of tumor cells. PLoS One 2009, 4, e5715. [Google Scholar]
- Klausner, R.D.; Van Renswoude, J.; Ashwell, G.; Kempf, C.; Schechter, A.N.; Dean, A.; Bridges, K.R. Receptor-mediated endocytosis of transferrin in K562 cells. J. Biol. Chem. 1983, 258, 4715–4724. [Google Scholar]
- Dautry-Varsat, A.; Ciechanover, A.; Lodish, H.F. pH and the recycling of transferrin during receptor-mediated endocytosis. Proc. Natl. Acad. Sci. USA 1983, 80, 2258–2262. [Google Scholar]
- Lepelletier, Y.; Camara-Clayette, V.; Jin, H.; Hermant, A.; Coulon, S.; Dussiot, M.; Arcos-Fajardo, M.; Baude, C.; Canionni, D.; Delarue, R.; Brousse, N.; Benaroch, P.; Benhamou, M.; Ribrag, V.; Monteiro, R.C.; Moura, I.C.; Hermine, O. Prevention of mantle lymphoma tumor establishment by routing transferrin receptor toward lysosomal compartments. Cancer Res. 2007, 67, 1145–1154. [Google Scholar]
- Gendler, S.J. MUC1, the renaissance molecule. J. Mammary Gland Biol. Neoplasia 2001, 6, 339–353. [Google Scholar]
- Altschuler, Y.; Kinlough, C.L.; Poland, P.A.; Bruns, J.B.; Apodaca, G.; Weisz, O.A.; Hughey, R.P. Clathrin-mediated endocytosis of MUC1 is modulated by its glycosylation state. Mol. Biol. Cell 2000, 11, 819–831. [Google Scholar]
- Muller-Tidow, C.; Schwable, J.; Steffen, B.; Tidow, N.; Brandt, B.; Becker, K.; Schulze-Bahr, E.; Halfter, H.; Vogt, U.; Metzger, R.; Schneider, P.M.; Buchner, T.; Brandts, C.; Berdel, W.E.; Serve, H. High-throughput analysis of genome-wide receptor tyrosine kinase expression in human cancers identifies potential novel drug targets. Clin. Cancer Res. 2004, 10, 1241–1249. [Google Scholar]
- Shangguan, D.; Li, Y.; Tang, Z.; Cao, Z.C.; Chen, H.W.; Mallikaratchy, P.; Sefah, K.; Yang, C.J.; Tan, W. Aptamers evolved from live cells as effective molecular probes for cancer study. Proc. Natl. Acad. Sci. USA 2006, 103, 11838–11843. [Google Scholar]
- Shangguan, D.; Cao, Z.; Meng, L.; Mallikaratchy, P.; Sefah, K.; Wang, H.; Li, Y.; Tan, W. Cell-specific aptamer probes for membrane protein elucidation in cancer cells. J. Proteome Res. 2008, 7, 2133–2139. [Google Scholar]
- Tong, G.J.; Hsiao, S.C.; Carrico, Z.M.; Francis, M.B. Viral capsid DNA aptamer conjugates as multivalent cell-targeting vehicles. J. Am. Chem. Soc. 2009, 131, 11174–11178. [Google Scholar]
- Xiao, Z.; Shangguan, D.; Cao, Z.; Fang, X.; Tan, W. Cell-specific internalization study of an aptamer from whole cell selection. Chemistry 2008, 14, 1769–1775. [Google Scholar]
- Kang, H.; O'Donoghue, M.B.; Liu, H.; Tan, W. A liposome-based nanostructure for aptamer directed delivery. Chem. Commun. 2010, 46, 249–251. [Google Scholar]
- Taghdisi, S.M.; Abnous, K.; Mosaffa, F.; Behravan, J. Targeted delivery of daunorubicin to T-cell acute lymphoblastic leukemia by aptamer. J. Drug Target 2010, 18, 277–281. [Google Scholar]
- Cambier, J.C.; Campbell, K.S. Membrane immunoglobulin and its accomplices: new lessons from an old receptor. FASEB J. 1992, 6, 3207–3217. [Google Scholar]
- Thomas, M.D.; Srivastava, B.; Allman, D. Regulation of peripheral B cell maturation. Cell Immunol. 2006, 239, 92–102. [Google Scholar]
- Mallikaratchy, P.; Tang, Z.; Kwame, S.; Meng, L.; Shangguan, D.; Tan, W. Aptamer directly evolved from live cells recognizes membrane bound immunoglobin heavy mu chain in Burkitt's lymphoma cells. Mol. Cell Proteomics 2007, 6, 2230–2238. [Google Scholar]
- Ullrich, A.; Schlessinger, J. Signal transduction by receptors with tyrosine kinase activity. Cell 1990, 61, 203–212. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ray, P.; White, R.R. Aptamers for Targeted Drug Delivery. Pharmaceuticals 2010, 3, 1761-1778. https://doi.org/10.3390/ph3061761
Ray P, White RR. Aptamers for Targeted Drug Delivery. Pharmaceuticals. 2010; 3(6):1761-1778. https://doi.org/10.3390/ph3061761
Chicago/Turabian StyleRay, Partha, and Rebekah R. White. 2010. "Aptamers for Targeted Drug Delivery" Pharmaceuticals 3, no. 6: 1761-1778. https://doi.org/10.3390/ph3061761
APA StyleRay, P., & White, R. R. (2010). Aptamers for Targeted Drug Delivery. Pharmaceuticals, 3(6), 1761-1778. https://doi.org/10.3390/ph3061761